
Abstract
For half a century, it has been known that the mast cell is the cell responsible for the majority of anaphylactic events. Its mediators, taken as a whole, are capable of producing all of the clinical manifestations of these events. With the discovery of immunoglobulin E (IgE), it was originally felt that the vast majority of anaphylactic episodes were due to antigen coupling with two cell-bound IgE molecules. More recently it has been learned that many episodes are produced by direct activation of mast cells, not involving antigen binding to IgE, and that monomeric IgE under certain conditions can also cause degranulation. Of note—in regard to antigen independent degranulation—are recent reports that the human G-protein-coupled receptor, MRGPRX2, may be the receptor for many drugs and cationic proteins capable of producing direct mast cell degranulation and anaphylactic events.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Today, it is common knowledge that the mast cell and anaphylaxis are intimately related. One can hardly speak of one without the mention of the other. But their relationship was originally far from obvious, and the sequence of scientific discoveries that elucidated the relationship between the two followed a circuitous path. It is interesting to note that in Robert A. Cooke’s classic text “Allergy in Theory and Practice”, published in 1946, there is no mention of the mast cell although anaphylaxis was a well-recognized entity discussed in detail in this text [1].
Mention of the mast cell came first. It was discovered by Paul Ehrlich in 1877 [2]; however, Ehrlich did not discern the function of the mast cell. Anaphylaxis came later, in 1901, through the work of Charles Richet and Paul Portier who were attempting to induce prophylaxis to the venom of the sea anemone and accidentally caused a phenomenon that they characterized as the opposite (“ana”, Greek for “opposite”) of the desirable goal, and so coined the term [3]. However, a link between the mast cell and anaphylaxis was not established until 1952 when J. F. Riley and G. B. West found that the mast cell was a major source of histamine [4]. It was just a short step thereafter, nonetheless over 50 years after Richet and Portier’s seminal work, to link the mast cell, with its abundance of histamine and other mediators, to anaphylactic events in animal models [5].
In this article, we plan to explore the now well established role of the mast cell in the production of anaphylactic events.
The Mast Cell
Origin, Maturation and Function of the Mast Cell
Because of the role of the mast cell in the production of allergic events, it is easy to lose sight of its participation in homeostasis and protection. But the mast cell is also a “good” cell and is now considered an immune cell with multiple functions. These activities are involved with homeostasis and tissue repair, angiogenesis, innate immunity, adaptive immunity, and immune tolerance [6]. The contents of mast cells (Table 1) are known to be important biological mediators which are released in response to various threats to survival. They are thus purveyors of “helpful inflammation” as well as healing.
All human mast cells are derived from a common pluripotent hematopoietic progenitor originating in the bone marrow [7•]. This forbearer cell enters the systemic circulation bearing the cell-surface marker, CD34. Thus, even though mature tissue mast cells express different neutral granule proteases, dependent upon their final tissue of residence after leaving the circulation, their circulating progenitor is of a single cell type [7•].
The major growth cell factor for human mast cells is stem cell factor (SCF). SCF is the ligand for CD117, a tyrosine-kinase transmembrane receptor located on the surface of all mast cells and other cells as well. SCF (also known as KIT-ligand because CD117 is encoded by the c-Kit gene) induces dimerization of CD117. This results in enhanced survival, growth, cell migration, and effector functions of mast cells. The binding of stem cell factor induces mast cell progenitors to migrate from the bone marrow to the systemic circulation and hence to the tissues. This process is probably aided and abetted by other mast cell chemo-attractants as well. And in these tissues they assume long-term residence. They settle in areas of the body which face the external environment. Thus, they are found in the highest quantities on mucosal surfaces and in connective tissue. Therefore, the tissues with the highest quantity are the skin, lung, gastrointestinal tract, and uterus. When they enter these tissues, they acquire new phenotypes, a process induced by their local microenvironment [8••]. These micro-environs foster phenotypic changes which effect function, contents, structure and the response to endogenous stimuli and drugs.
In human beings, mast cells exist in two subtypes based on the phenotypic changes occurring in the respective tissue of residence. The subtypes are referred to as mucosal (MCT) and connective tissue (MCTC) mast cells. This nomenclature is based on the fact that MCTC cells contain tryptase and chymase as well as carboxypeptidase and cathepsin, whereas MCT cells characteristically contain only one of these, tryptase. MCT cells in general reside in mucosal tissue whereas MCTC cells settle in connective tissue as well as smooth muscle. Both cell types contain heparin. They both manufacture interleukins and other cytokines [9–13]. In general, MCTC cells produce high amounts of IL-4 and IL-13. Lower levels are produced by MCT cells which make higher amounts of IL-5 and IL-6. However, there is a greater heterogeneity than expressed by these classically described phenotypes with some cells; for example, containing carboxypeptidase and chymase but no tryptase [14, 15].
These phenotypes also differ in their shape as well as in their response to endogenous and exogenous stimuli provoking degranulation. Both can be activated by IgE-antigen union whereas only MCTC cells respond to complement split products (C5a) and substance P, whereas platelet activating factor only stimulates MCT cells. Opiate receptors are found only on MCTC cells. Each of the two phenotypes also differs in their response to agents designed to prevent degranulation. For example, sodium cromoglycate appears to only prevent degranulation of MCT cells.
Degranulation of the Mast Cell
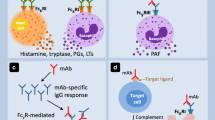
The definitive conclusion that IgE and antigen union results in degranulation of mast cells awaited the discovery and purification of IgE in 1966 and the ability to culture mast cells as well as the identification of the structure of the high affinity IgE receptor which occurred in 1989 [16•]. Originally, it was thought that the vast majority of anaphylactic reactions were related to IgE-antigen binding. However, it is now clearly known that there are many other existing pathways that can result in mast cell degranulation and thus produce events clinically identical to those caused by the union of antigen and IgE (Fig. 1). For example, oversulfated chondroitin sulfate contamination of heparin resulted in events very similar to those produced by mast cell degranulation, but appear rather to be related to direct activation of the contact system and/or complement with perhaps no involvement of mast cells whatsoever [17, 18]. However, it should be noted that activation of the mast cell can clearly result in the recruitment and activation of the complement system as well as the contact system, and can induce intravascular coagulation as well. Thus other inflammatory pathways can further escalate the clinical manifestations occurring during anaphylactic events [19–21].

Triggers for mast cell degranulation
Furthermore, a single drug can produce anaphylactic events through more than one pathway. An example of this is heparin which causes anaphylactic incidents through activation of the contact system as well as via an IgE-mediated pathway [22•, 23•].
Because of the multiple mechanisms underlying mast cell degranulation, knowledge of receptors on the mast cell is of importance. Triggers for mast cell degranulation (Fig. 1) other than IgE-antigen binding include neuropeptides such as substance p, cytokines, many drugs, some contents of insect venom, physical factors such as heat and cold, polycationic molecules such as 48/80, stem cell factor, and other agents. Of particular importance in this regard is the recent discovery of the cell surface mouse receptor, MRGPR2, the orthologue of the human G-protein-coupled receptor MRGPRX2 [24••, 25••]. McNeil, et al. [24••], found that mice connective tissue mast cells possess MRGPRB2 and that this moiety serves as the mast cell receptor for neurokinins such as substance P, basic secretagogues such as 40/80, and a number of peptidergic drugs that have been shown to produce anaphylactic events. These include fluoroquinolones, the bradykinin receptor antagonist icatibant, and several neuromuscular blocking drugs known for their propensity to induce histamine release. Of note is that this mouse receptor was present only on MCTC, connective tissue mast cells. The loss of the receptor in knockout mice did not alter reactions to IgE-mediated release, but did prevent histamine release to basic secretagogues. Downstream signaling from the receptor appears to involve activation of the phospholipase C-gamma pathway ending in the release of both preformed and granule-stored mediators, and the de novo synthesis of eicosanoids [24••, 25••]. Thus a single mast cell receptor can be responsible for mast cell degranulation due to multiple agents, many of which are drugs known to produce anaphylactic episodes. The clinical implication of course is that blockade of this receptor might be useful in preventing such episodes.
Another mechanism involved in the activation and degranulation of mast cells is perhaps less well-known, but still intriguing. This mechanism involves activation of the mast cell via monomeric immunoglobulin E-dependent activity. As mentioned previously, classical mast cell degranulation is caused by the union of two dimeric IgE molecules by antigens with multiple epitopes causing the subsequent dimerization of the high affinity IgE receptor [26]. More recently, it has been demonstrated that mast cell mediator release can be initiated by monomeric IgE [27]. This release appears to be enhanced by the presence of SCF. The release also seems to be dependent on free IgE. It is thought that monomeric-induced IgE mediator release also works through aggregation of the high affinity IgE receptor, but the exact mechanism has not been elucidated. Also, the clinical significance of this phenomenon remains unknown, but it is interesting to speculate that, since both monomeric IgE as well as stem cell factor are found in the lung, monomeric release may be important in asthma. This is even of greater interest since monomeric IgE is found in the lungs of “intrinsic” or nonallergic asthmatics as well as the allergic variety [28].
Control and Prevention of Mast Cell Degranulation
It naturally follows that one of the major goals of understanding the factors that result in the pathologic degranulation of mast cells is to assist in developing agents that can prevent the pathologic release of mediators.
The best understood mechanism accounting for mast cell degranulation is that which participates in IgE-antigen-dependent activation and thus efforts have been concentrated on inhibiting different steps of this process. As mentioned, the union of two dimeric molecules of IgE affixed to the high affinity receptor results in the dimerization of this receptor. Downstream signaling activates a series of tyrosine kinases beginning with Lyn which phosphorylates tyrosine residues in the intracellular portion of the IgE receptor molecule. Shortly thereafter, a second protein tyrosine kinase, Syk, is recruited and further phosphorylates this chain. This begins a series of activities that produce activation of inositol triphosphate (IP3). The production of IP3 results in intracellular calcium mobilization and the influx of extracellular calcium into the cell. Subsequently granules are extruded from the cell, and the de novo synthesis of mediators such as leukotriene occurs. Theoretically interruption at any point of this cascade could prevent the pathophysiologic degranulation of these cells.
Attempts have been made to utilize spleen tyrosine-kinase (Syk) inhibitors in the treatment of rhinitis [29] and other tyrosine kinase inhibitors such as imatinib, dasatinib, and midostaurin in the treatment of mastocytosis and mast cell activation disorders [30••].
Probably the first agent employed to prevent mast cell degranulation was cromolyn sodium. It was the first “mast cell stabilizer.” It was followed shortly thereafter by nedocromil. These drugs appeared to stabilize mucosal (MCT) but not connective tissue (MCTC) cells. The action of cromolyn and nedocromil appears to be mediated through a G-protein-coupled receptor (G-protein-coupled receptor 35) [31]. Both drugs have fallen out of favor because of the need for frequent administration and the perception of lack of potency. However, the oral form of cromolyn is still used to treat mast cell activating syndromes in certain circumstances.
Conceptually, treatment to prevent degranulation might be successful by stimulating mast cell receptors that contain immunoreceptor tyrosine-based inhibitory motifs (ITIMs). The binding of these receptors to their ligand recruits phosphatases that are capable of suppressing cell activation by dephosphorylating signaling molecules. Examples of such receptors are CD72, Siglec-8, and CD300a [32].
Ketotifen, a drug which has both antihistamine and mast cell stabilizing properties, has also been used to treat mast cell activating disorders, specifically in patients who have experienced episodes of idiopathic anaphylaxis [33].
Application of our Knowledge of the Mast Cell to the Patient Experiencing Anaphylactic Episodes
The realization that abnormalities of mast cells can be responsible for anaphylaxis [34••], in particular for episodes previously thought of as idiopathic, has profoundly altered our approach to patients. Specifically, it has lowered our threshold for the performance of bone marrow biopsies in patients without a known cause of their episodes and intensified a search for mutations in Kit. The seminal article establishing a relationship between abnormal mast cells and idiopathic anaphylaxis was published in Blood in 2007 [35].
In this article, patients with a diagnosis of idiopathic anaphylaxis were evaluated. From a cohort of 72 consecutive patients, the authors reported 12 patients with recurrent anaphylaxis who on bone marrow biopsy showed some findings suggesting mastocytosis. But the biopsies did not meet the criteria established by the World Health Organization cited as necessary to establish a diagnosis of this disorder. However, some of the patients did demonstrate one or more minor criteria for mastocytosis, such as exhibiting the 816D > V (D816V) c-KIT mast cell activating mutation. Only 2 of the 12 patients had baseline serum tryptase concentrations over 20 ng/mL and the traditional cutoff value of 20 ng/mL used to establish an elevated level of serum tryptase may be too high. Since that publication, a number of other studies have confirmed the presence of D816V c-KIT mutations in patients with idiopathic anaphylaxis and even in patients where an allergen was identified. These investigations have fostered a change in the classification of anaphylactic events [36].
This proposed classification suggests that disorders of mast cells, some of which predispose to anaphylaxis, be classified into three distinct categories:
-
1.
Mastocytosis and mast cell activating syndromes (MCAS).
-
2.
IgE-mediated anaphylactic events.
-
3.
Idiopathic anaphylactic episodes.
Mast cell activating syndrome (MCAS) resembles mastocytosis and can cause anaphylaxis, but individuals with this disorder lack sufficient bone marrow findings to make a diagnosis of mastocytosis according to the criteria established by the World Health Organization [37]. Such patients have some of the bone marrow findings seen in mastocytosis and also can have gain in function mutations in KIT.
The importance of the establishment that mastocytosis and MCAS can be the cause of idiopathic anaphylaxis lies in the fact that mast cell activating disorders can, on occasion, be controlled with tyrosine-kinase inhibitors. Baseline elevations in serum tryptase, plasma histamine, 24-hour urinary histamine metabolites, or prostaglandin D2 suggest these conditions. Mastocytosis and MCAS can be present in patients with lower levels of serum tryptase. A study of patients who had hymenoptera anaphylaxis found a level of 11.7 ng/mL was a marker for underlying mastocytosis [38].
A screening test performed on peripheral blood to detect the 816 V mutation found in the majority of cases of mastocytosis can now be performed [39]. Using an analysis with high sensitivity has been suggested especially in patients with serum tryptase <20 ng/mL. Still the most definitive way to make a diagnosis of mastocytosis and MCAS is to do a bone marrow biopsy. Thus one is faced with the decision as to whether or not to do a bone marrow biopsy in patients in whom no cause for anaphylaxis has been determined. When to do so remains a cause of debate but suggested algorithms for the management of such patients are available in the literature. However, there is growing importance regarding making such a diagnosis since as noted some mast cell activating syndromes and some cases of mastocytosis which are 816 V-negative can be treated with available tyrosine-kinase inhibitors [40].
Another clinically significant result of our basic research into mast cell biology is the application of the measurement of mast cell contents to assist in the diagnosis of anaphylaxis and its causes with specific reference to mastocytosis. Of particular importance in this regard is the measurement of serum tryptase. Tryptase is of particular importance because levels do not peak until approximately 60 to 90 min after the onset of symptoms in most patients, and occasionally levels can remain elevated for 5 h and sometimes even longer in protracted cases [41, 42]. Thus tryptase is more useful than plasma histamine which begins to rise within 5 to 10 min after symptoms, but it remains elevated for only 30 to 60 min. Nonetheless, urinary histamine metabolites can also be useful and are elevated for a longer period after the onset of symptoms.
It should be noted that disparities between histamine and tryptase levels can occur. In an emergency department evaluation of patients presenting with acute allergic reactions, plasma histamine concentration was elevated in 42 of 97 adult patients, but serum tryptase was elevated in only 20. Histamine levels correlated better with clinical signs than tryptase. Patients with elevated plasma histamine levels were more likely to have urticaria, more extensive erythema, wheezing, and abnormal abdominal findings [43]. Thus, both mediators can be of use in establishing a diagnosis of anaphylaxis. More recently, measurement of urinary prostaglandin D2 levels have been shown to correlate with anaphylactic events, and occasionally do so when there is no elevation of serum tryptase [44, 45]. All three of these mediators can be measured commercially.
Serum tryptase determinations are also useful in another context. Elevations of baseline serum tryptase levels are a marker for mastocytosis. Recently, the upper limits of normal for baseline serum tryptase have been revised from a level of 20 ng/mL to a level of 11.4 ng/mL based on studies of anaphylactic reactions to hymenoptera stings [46]. Analyzing reactions to stings, it was demonstrated that a serum tryptase level of greater than 11.4 was a significant risk factor for anaphylaxis. In evaluating patients who had stings with levels above 11.4 it was shown that such patients were likely to have systemic mastocytosis.
Conclusions
The vast majority of anaphylactic events, but not all, are due to systemic mast cell degranulation. This degranulation process is in many instances IgE-mediated, but in a significant number of episodes direct mast cell degranulation independent of IgE occurs. Of note in this regard are recent reports that the human G-protein-coupled receptor, MRGPRX2, may be the receptor for many drugs and cationic proteins capable of producing direct mast cell degranulation and anaphylactic events.
The basic knowledge of the events involved in mast cell degranulation has permitted us to understand mast cell pathology that results in a predisposition for anaphylactic events in disorders such as mastocytosis and MCAS. There have been advances in controlling these conditions using tyrosine-kinase inhibitors which have improved the outcomes in these disorders.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Allergy in theory and practice. In: RA Cooke, ed. W. B. Saunders Company, Philadelphia, 1947.
Avenberg KM, Harper DS, Larsson BL. Footnotes on allergy. Uppsala: Published by Pharmacia AB; 1980. p. 84–5.
Samter M. Excerpts from classics in allergy (edited for the 25th Anniversary Committee of the American Academy of Allergy, Asthma, and Immunology). Published by Ross Laboratories, Columbus; copyright 1969. Library of Congress Catalog Number 70-77908, 32–33.
Riley JF, West GB. Histamine in tissue mast cells. J Physiol. 1952;117(4):72P–3.
Mota I. Action of anaphylactic shock and anaphylatoxin on mast cells and histamine in rats. Brit J Pharmacol. 1957;12:453–7.
Zayas E, da Silva M, Jamur MC, Oliver C. Mast cell function: a new vision of an old cell. J Histochem Cytochem. 2014;62(10):698–738. doi:10.1369/0022155414545334.
Maaninka K, Lappalainen J, Kovanen PT. Human mast cells arise from a common circulating progenitor. J Allergy Clin Immunol. 2013;132(2):463–9. These investigators demonstrated conclusively that mast cells that reside in the mucosal tissue (known as MC mast cells) and mast cells that reside in the connective tissue (known as MCTC mast cells) have a common progenitor. They demonstrate that these cells assume their phenotypic profiles only upon reaching the tissue in which they reside after exiting the bone marrow.
Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373:163–72. doi:10.1056/NEJMra1409760. A superb and comprehensive review on the role of mast cells in the production of anaphylactic events with an emphasis on mast cell activation disorders such as systemic mastocytosis, urticaria pigmentosa, and other mast cell activating syndromes.
Bradding P, Feather IH, Wilson S, Bardin PG, Heusser CH, Holgate ST, et al. Immunolocalization of cytokines in the nasal mucosa of normal and perennial rhinitic subjects. The mast cell as a source of IL-4, IL-5, and IL-6 in human allergic mucosal inflammation. J Immunol. 1993;151(7):3853–65.
Qu Z, Liebler JM, Powers MR, Galey T, Ahmadi P, Huang XN, et al. Mast cells are a major source of basic fibroblast growth factor in chronic inflammation and cutaneous hemangioma. Am J Pathol. 1995;147(3):564.
Gordon JR, Galli SJ. Mast cells as a source of both preformed and immunologically inducible TNF-alpha/cachectin. Nature. 1990;346(6281):274.
Jaffe JS, Glaum MC, Raible DG, Post TJ, Dimitry E, Govindarao D, et al. Human lung mast cell IL-5 gene and protein expression: temporal analysis of upregulation following IgE-mediated activation. Am J Respir Cell Mol Biol. 1995;13(6):665.
Bradding P, Saito H. Biology of mast cells and their mediators. In: Middleton’s Allergy: Principles and Practice, Ed 8, 228-251, 2014 (edited by N. Franklin Adkinson Jr., M.D., Bruce S. Bochner, M.D.).
Weidner N, Horan RF, Austen KF. Mast-cell phenotype in indolent forms of mastocytosis. Ultrastructural features, fluorescence detection of avidin binding, and immunofluorescent determination of chymase, tryptase, and carboxypeptidase. Am J Pathol. 1992;140(4):847–57.
Bradding P, Okayama Y, Howarth PH, Church MK, Holgate ST. Heterogeneity of human mast cells based on cytokine content. J Immunol. 1995;155:297–307.
Saito H, Ishizaka T, Ishizaka K. Mast cells and IgE: from history to today. Allergol Int. 2013;62(1):3–12. doi:10.2332/allergolint.13-RAI-0537. A comprehensive history of the relationship between IgE and the mast cell with a detailed description of the necessity for a divalent antigen bridging to adjacent cell-bound IgE molecules to produce mast cell degranulation.
Kishimoto TK, Viswanathan K, Ganguly T, Elankumaran S, Smith S, Pelzer K, et al. Contaminated heparin associated with adverse clinical events and activation of the contact system. N Engl J Med. 2008;358:2457–67.
Schwartz LB. Heparin comes clean. N Engl J Med. 2008;358:2505–9.
Kaplan AP, Hunt KJ, Sobotka AK, et al. Human anaphylaxis: a study of mediator systems. Clin Rev. 1977;25:361.
Van der Linden PW, Hack CE, Kerckhaert J, et al. Preliminary report: complement activation in wasp-sting anaphylaxis. Lancet. 1990;336:904–6.
Van Hagge-Hamsten M, Hack CE, Eerenberg AJ, et al. Contact system activation and angioedema in insect-sting anaphylaxis. J Allergy Clin Immunol. 1993;91:283.
Sala-Cunill A, Björkqvist J, Senter R, Guilarte M, Cardona V, Labrador M, et al. Plasma contact system activation drives anaphylaxis in severe mast cell-mediated allergic reactions. J Allergy Clin Immunol. 2015;135(4):1031–43. Demonstration that clinical events resembling IgE-induced classical anaphylactic reactions can occur through the activation of other pathways, in this case the contact system.
Phan C, Vial-Dupuy A, Autegarden JE, Amsler E, Gaouar H, Abuaf N, et al. A study of 19 cases of allergy to heparins with positive skin testing. Ann Dermatol Venereol. 2014;141(1):23–9. Agents which can activate the contact system are potentially capable of producing anaphylactic events through an IgE-mediated mechanism as well.
McNeil BD, Pundir P, Meeker S, Han L, Undem BJ, Kulka M, et al. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature. 2015;519(7542):237–41. doi:10.1038/nature14022. The authors describe, for the first time, a cell surface receptor, Mrgprb2, in mice which is the orthologue of the human G-protein-coupled receptor MRGPRX2. They demonstrate that this receptor, in mice, is the receptor for multiple molecules including cationic proteins and many drugs that are known to produce anaphylactic events in the human. They cite examples of the secretagogues including neuromuscular blocking drugs, fluoroquinolones such as ciprofloxacin, and 40/80 inducing mast cell activation.
Grimbaldeston MA. Mast cell-MrgprB2: sensing secretagogues or a means to overreact? Immunol Cell Biol. 2015;10.1038/icb.2015.10:221–3. In this manuscript, Dr. Grimbaldeston comments on the potential clinical importance of the previous reference (McNeil, et al.) noted above and comments on the potential clinical importance of their discovery. They also describe the downstream signaling pathway initiated by the binding of drugs such as fluoroquinolones, peptidergic drugs as Icatibant, and neuromuscular blocking drugs to MrgprB2 involving the phospholipase C-gamma pathway.
Ishizaka T, Ishizaka K. Triggering of histamine release from rat mast cells by divalent antibodies against IgE-receptors. J Immunol. 1978;120(3):800–5.
Cruse G, Kaur D, Yang W, et al. Activation of human lung mast cells by monomeric immunoglobulin E. Eur Resp J. 2005;25:858–63.
Ying S, Humbert M, Meng Q, Pfister R, Menz G, Gould HJ, et al. Local expression of epsilon germline gene transcripts and RNA for the epsilon heavy chain of IgE in the bronchial mucosa in atopic and nonatopic asthma. J Allergy Clin Immunol. 2001;107(4):686–92.
Meltzer EO, Berkowitz RB, Grossbard EB. An intranasal Syk-kinase inhibitor (R112) improves the symptoms of seasonal allergic rhinitis in a park environment. J Allergy Clin Immunol. 2005;115(4):791–6.
Arock M, Akin C, Hermine O, Valent P. Current treatment options in patients with mastocytosis: status in 2015 and future perspectives. Eur J Haematol. 2015;94(6):474–90. A discussion of the treatment modalities available for the management of mastocytosis and mast cell activating disorders.
Yang Y, Lu JY, Wu X, Summer S, Whoriskey J, Saris C, et al. G-protein-coupled receptor 35 is a target of the asthma drugs cromolyn disodium and nedocromil sodium. Pharmacology. 2010;86(1):1–5.
Karra L, Levi-Schaffer F. Down-regulation of mast cell responses through ITIM containing inhibitory receptors. Adv Exp Med Biol. 2011;716:143–59.
Patterson R, Fitzsimons EJ, Choy AC, Harris KE. Malignant and corticosteroid-dependent idiopathic anaphylaxis: successful responses to ketotifen. Ann Allergy Asthma Immunol. 1997;79(2):138–44.
Akin C. Mast cell activation syndromes presenting as anaphylaxis. Immunol Allergy Clin North Am. 2015;35(2):277–85. doi:10.1016/j.iac.2015.01.010. A thorough review and classification of mast cell disorders and their relation to anaphylaxis.
Akin C, Scott LM, Kocabas CN, Kushnir-Sukhov N, Brittain E, Noel P, et al. Demonstration of an aberrant mast-cell population with clonal markers in a subset of patients with “idiopathic” anaphylaxis. Blood. 2007;110:2331–3.
Valent P, Akin C, Arock M, Brockow K, Butterfield JH, Carter MC, et al. Definitions, criteria and global classifications of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal. Int Archives Allergy Immunol. 2012;157:215–25.
Horny HP, Metcalfe DD, Bennett JM, Bain BJ, Akin C, Escribano L, et al. Mastocytosis. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., editors. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press; 2008. p. 54–63.
Bonadonna P, Perbellini O, Passalacqua G, Caruso B, Colarossi S, Dal Fior D, et al. Systemic reactions after hymenoptera sting and raised serum tryptase strongly suggest clonal mast cells disorders. J Allergy Clin Immunol. 2009;123(2 Supplement):S242.
Schumacher JA, Alentoba-Johnson KS, Lim MS. Detection of the c-KIT D816V in SM patients. J Clin Pathol. 2008;61(1):109–14.
Paul C, Sans B, Suarez F, Casassus P, Barete S, Lanternier F, et al. Masitinib for the treatment of systemic and cutaneous mastocytosis with handicap: a phase 2a study. Am J Hematol. 2010;85(12):921–5.
Shanmugam G, Schwartz LB, Khan DA. Prolonged elevation of serum tryptase in idiopathic anaphylaxis. J Allergy Clin Immunol. 2006;117(4):950–1.
Vinuya RZ, Simon MR, Schwartz LB. Elevated serum tryptase levels in a patient with protracted anaphylaxis. Ann Allergy. 1994;73(3):232–4.
Lin RY, Schwartz LB, Curry A, Pesola GR, Knight RJ, Lee HS, et al. Histamine and tryptase levels in patients with acute allergic reactions: an emergency department-based study. J Allergy Clin Immunol. 2000;106:65–71.
Butterfield JH, Weiler CR. Prevention of mast cell activation disorder-associated clinical sequelae of excessive prostaglandin D(2) production. Int Arch Allergy Immunol. 2008;147:338.
Rank MA, Kita H, Li JT, Butterfield JH. Systemic reactions to allergen immunotherapy: a role for measuring a PGD2 metabolite? Ann Allergy Asthma Immunol. 2013;110(1):57–8. doi:10.1016/j.anai.2012.10.009.
Bonadonna P, Perbellini O, Passalacqua G, Caruso B, Colarossi S, Dal Fior D, et al. Clonal mast cell disorders in patients with systemic reactions to Hymenoptera stings and increased serum tryptase levels. J Allergy Clin Immunol. 2009;123:680–6.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Lieberman reports that he is a speaker for Mylan and Teva. Dr. Garvey declares no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Anaphylaxis and Drug Allergy
Rights and permissions
About this article

Cite this article
Lieberman, P., Garvey, L.H. Mast Cells and Anaphylaxis. Curr Allergy Asthma Rep 16, 20 (2016). https://doi.org/10.1007/s11882-016-0598-5
Published:
DOI: https://doi.org/10.1007/s11882-016-0598-5