
Opinion statement
In recent years, we have seen an increase in the study and interest of the role of the microbiome in the development of malignancies, their progression, and evasion of therapies. This has been particularly fruitful in the case of colorectal cancer; multiple investigators have described correlative observations as well as hypotheses strengthened in preclinical studies that have begun to elucidate the critical role the gut and tumoral microbiome plays in carcinogenesis. Furthermore, these landmark studies lay the groundwork in describing the microbiome’s role in carcinogenesis and provide a rich field of future study. Here, we review contemporary understandings of these observations and proposed mechanisms behind them.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Harboring 70% of the bacterial load of the gut microbiome, the colon hosts approximately 1014 bacteria comprising 103 different species [1]. Bacteria interact with local host tissues and the systemic immune system in a complex system crucial to normal gut physiology, global immunologic, and metabolic functions. Baseline physiologic gut microbiota typically varies along the gastrointestinal tract due to multiple factors including transit time, enzymatic activity, pH, and fermentation of luminal contents. The most abundant phyla of bacteria have been variously reported to be Bacteroides, Firmicutes, Proteobacteria, Actinobacteria, and Verrucomicrobia [2, 3]. Given the high variability in luminal bacterial communities, it is hypothesized that a homeostatic equilibrium exists between host and gut microbiota that supports health, and perturbations of this equilibrium can elicit local gut injury and alterations in the host immune status. To date, clear markers of a “healthy” gut microbiome remain elusive.
Disruptions in the composition and function of the native microbiota, referred to as dysbiosis, have been implicated in the pathogenesis of a variety of local and systemic inflammatory and autoimmune disorders [4,5,6,7]. Over the past decade, evidence has increasingly linked gut dysbiosis to the development and progression of cancer. To date, the gut microbiome has been shown to affect carcinogenesis via direct metabolic effects, systemic immune modulation, and modulation of the tumor immune microenvironment (TIME), both in gastrointestinal and non-GI cancers [5, 8]. New evidence suggests the gut microbiota participates in modulating response to both immune and cytotoxic cancer therapeutics [5, 9,10,11,12], opening the door for gut microbial modulation as a promising anti-cancer intervention in combination with existing therapeutics. These observations and proposed mechanisms are especially prescient in the case of colorectal cancer (CRC) given its proximity to the gut microbiome and early supported mechanistic hypotheses behind carcinogenesis, evasion of therapeutics, and progression.
Currently, colorectal cancer (CRC) is the 2nd leading cause of cancer-related death in the USA and worldwide with an increasing incidence, especially in younger patient populations [13]. The development of colonic adenocarcinoma is multifactorial, with contributions from genetic, immunologic, and environmental factors. These tumors represent a heterogenous group of cancers classically presented as four subtypes as defined by their canonical molecular subtype (CMS) [14]. These subtypes, initially identified by transcriptomic differences, are also associated with unique immune cell infiltrates. Beyond subclassifying of tumors themselves, there is an urgent need for more in depth understanding of tumor biology and interactions between the tumor microenvironment and global host response. While CMS classification greatly improves understanding of disease biology, correlations with subtype, outcome, and TIME remain incompletely understood.
Tumor immune infiltrates, which correlate with CMS, have also been shown to correlate with gut microbial and tumor microbial signatures. These associations are in part linked by a patient’s “exposome,” consisting of environmental factors such as smoking, diet, and antibiotic use, which trigger composition changes of the microbiome, pro-inflammatory pathways, and perturbations of the TIME [15, 16]. The gut microbiome plays an important role as part of and reflecting the broader exposome [17]. The mucosal interface between the colon and this exposome relies on an intricate balance of homeostatic processes. Irregularities in inflammation, immune response, gut microbiome, and host epithelial barrier can potentially result in a tumorigenic cascade. Recent evidence highlights the gut microbiotas’ interplay with this barrier function and subsequent immune infiltrates within tumors. Opportunities now exist to test clinical applications in prevention, detection, and management of CRC. Here, we review the current literature behind these associations and opportunities to leverage these results to novel therapeutics.
Gut dysbiosis and colorectal cancer carcinogenesis
Studies have aimed to identify specific bacterial taxa and their metabolites responsible for colonic mucosal injury and tumorigenesis. CRC patients have significantly lower stool bacterial diversity [18] and the fecal microbiota is predominated by pro-carcinogenic species including Fusobacterium, Bacteroides fragilis, and Escherichia [19,20,21]. Not only are there compositional differences in the gut microbiota of CRC patients, but stage-specific analyses of the gut microbiome of patients with colorectal adenomas and carcinomas have demonstrated shifts in the relative abundance of taxa between progressive stages of tumor development [22, 23]. This suggests the mechanistic influence of these bacterial communities is dynamic and intricate. In this analysis, Fusobacterium nucleatum spp., a predominantly oral symbiotic species, were found to be progressively more abundant from precursor to late stages; meanwhile, Atopobium parvulum and Actinomyces odontolyticus were only elevated in early stages [22•]. In addition to the hypothesis generating pathologic implications of these findings, this also demonstrates a potential clinical role of species abundance as a biomarker for disease and progression.
A causal role of dysbiosis on CRC development and progression has been demonstrated in murine models through the administration of stool from CRC patients leading to higher rates of high-grade dysplasia and polyp formation [24]. Its impact on the host immune status was evidenced by upregulation of inflammatory pathways and intestinal recruitment of T helper cells. Furthermore, in animal models with induced colorectal carcinogenesis, germ-free (GF) rats grew fewer and smaller tumors when compared to rats with conventional gut microbiota. The interplay between gut dysbiosis and the host mucosa is multifaceted, but suspected mechanisms include genotoxin-driven DNA injury, inflammation from digestion-derived metabolites, and immunity dysregulation [25], which in combination, tip the scales towards a colonic environment favoring dysplastic phenotypes.
To better understand a potentially causal link between the presence of specific bacterial taxa and CRC oncogenesis, metabolomic studies have demonstrated pro-tumorigenic or anti-tumorigenic environments of the colonic mucosa can largely be described by specific bacterial metabolomic signatures. Specific food habits correlate with gut microbiota signatures, and the resulting microbial byproducts can impact the host immune system [26, 27]. This provides a potential mechanistic link with observational data that has long supported associations of CRC with certain dietary factors including red and processed meats [28, 29], and multiple toxins generated from microbial metabolism.
Bacterial production of the short chain fatty acid, butyrate, provides the primary energy source for colonocytes and has been shown to demonstrate anti-inflammatory and immunomodulatory properties. Butyrate can inhibit release of inflammatory cytokines and drive the differentiation of regulatory T cells in vitro and in vivo, creating an anti-inflammatory local environment [30]. Fiber-fermenting bacteria, including Fusobacterium nucleatum, can be butyrate producers, and may be involved in normal gut homeostasis. While butyrate is generally considered anti-tumorigenic, given its high levels in at risk populations (African-Americans) and low levels in low risk populations (Native Americans) [29], in vitro studies have demonstrated butyrate’s ability to drive colonic epithelial proliferation [31], suggesting the need for homeostasis within the host, bacteria, and metabolite interaction.
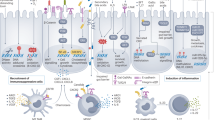
Beyond metabolomic interactions, the bacterial driver-passenger theory helps to describe the series of microbial events that drive colorectal carcinogenesis. In this model, colonization of the colonic mucosa by pathogenic bacterial species results in epithelial DNA damage, leading to driver mutations initiating carcinogenesis [32]. Activation of inflammatory pathways allows opportunistic passenger bacteria to proliferate, outcompete native species, and support disease progression, resulting in a remodeled, dysregulated TIME [33]. The result is a milieu of chronic inflammation, immune dysregulation, and microbial dysbiosis, leading to an accumulation of mutations and tumor progression.
Two microbial oncogenic drivers of CRC are enterotoxigenic Bacteroides fragilis and colibactin-producing Escherichia coli [34, 35]. B. fragilis’ enterotoxin has been shown to directly damage DNA strands through the production of reactive oxidative species. This further stimulates a cascade, characterized by TH17 recruitment, IL-17 production, and NF-κB signaling, creating a pro-inflammatory setting featuring immature myeloid cells [34]. The B. fragilis enterotoxin also compromises the protective mucus layer of the colonic epithelium, allowing adhesion of pathogenic opportunistic bacteria [36]. Similarly, colibactin production by E. coli is directly genotoxic and has been shown to cause chromosomal instability in murine models [37]. Similar findings have been described across numerous bacterial species (Table 1). Initial work by Cougnoux et al. demonstrated potential therapeutic strategies targeting bacterial genotoxins. Utilizing an inhibitory molecule to the bacterial colibactin-producing enzyme resulted in suppression of DNA damage and cell proliferation in CRC murine models [38].
Chronic inflammation is a common and well-known driver of CRC development. Murine models of colonic epithelial injury have shown that bacterial byproducts and toxins drive IL-23 and IL-17 pathways that lead to tumorigenesis [39]. Similarly, colonic microbiota have been linked to tumorigenesis via activation of Th17 cell responses [40]. Ultimately, gut dysbiosis as well as specific bacterial pathogens has been correlated with intestinal inflammation and epithelial injury that results in gut permeability and immune activation, leading to the chronic inflammation, proliferation, invasion, and cancer development. Given the complexity of these systems, specific mechanisms have been proposed but a unifying cause has remained elusive.
Hypotheses of carcinogenesis and progression
Fusobacterium nucleatum is seen in high abundance in the gut microbiome of colorectal cancer patients and has been associated with chemoresistance in CRC [22, 49]. F. nucleatum within colorectal tumor itself appears to be critical to tumor development and progression [18, 50]. Multiple hypotheses have been suggested for the mechanism of F. nucleatum tumorigenesis, suggesting this is likely a multifactorial effect. This microbe utilizes a virulent adhesin, Fap2, which permits epithelial adhesion to a CRC polysaccharide and invasion [43]. This generates a FadA adhesion complex that stimulates wnt/β-catenin signaling and oncogenic transcription profiles [43, 44]. This mechanism has been established in preclinical murine models, providing mice F. nucleatum results in inflammatory and TIME alterations mimicking those seen in F.nucleatum associated CRC [50, 51]. Similarly, colorectal cell lines incubated with F. nucleatum and injected into mice result in xenograft tumor growth increased in size and rate compared to controls [44, 52]. Among colorectal cancer patients, a high F. nucleatum level corresponds with predominantly right sided cancers and a decreased cancer-specific survival when compared to those with low or undetectable levels of F. nucleatum [49, 53, 54].
In addition to its role in tumor development, recent evidence has suggested that the microbiota, specifically F. nucleatum, is involved in tumor metastasis. Studies have demonstrated the presence of F. nucleatum in lymph nodes and liver metastases from F. nucleatum-positive primary colorectal tumors, suggesting bacteria travel with cancer cells [55, 56]. In murine models, antibiotic treatment resulted in decreased F. nucleatum loads, as well as cancer cell proliferation and tumor cell growth [55]. These results highlight a link between the development of CRC metastases and the tumor microbiome, as well as the potential for microbiota modulation as a treatment strategy for CRC.
Potential mechanisms for the microbiota’s involvement in metastasis involve injury to the gut vascular barrier (GVB) and the development of the pre-metastatic niche (PMN) within sites of metastatic spread. Enteric microbes can invade the mucosal barrier and directly injure the GVB, allowing circulatory access for pathogens [57]. Once in circulation, microbes can induce environments rich in innate immune cells and pro-inflammatory signaling, which fosters the settling of migrating cancer cells [58, 59]. Bertocchi et al. recently demonstrated that GVB injury was associated with significantly more bacteria within hepatic metastases, suggesting that bacterial translocation occurs when the GVB is compromised [60••]. Furthermore, in antibiotic-treated mice, hepatic PMN formation was significantly diminished. These telling findings indicate bacterial dissemination to the liver may be responsible for the promotion of this pro-tumorigenic pre-metastatic niche.
Role of the virome in CRC
The bacterial contribution to carcinogenesis has been the large focus of research into the microbiome and cancer, yet nonbacterial entities are known to be involved as well. Human viruses have been shown to be critical to the development of a variety cancers, including hepatocellular, nasopharyngeal, cervical, and some gastrointestinal cancers, typically with long-standing infection. The host virome, consisting of both eukaryotic viruses and bacteriophages, influences host cellular function and bacterial community composition [61]. In a recent study of shotgun metagenomic analyses of viromes from fecal samples, patients with CRC demonstrated higher diversity of gut bacteriophages [62, 63] suggesting bacteriophages indirectly affect carcinogenesis by altering the composition of gut bacteria. By altering commensal bacterial communities, opportunistic passenger bacteria can migrate in and proliferate. Moreover, others theorize that bacteriophages play a role in biofilm production, which supports proliferation and invasion of opportunistic bacteria [63]. Bacteriophages have also been shown to cross epithelial cell layers in the gut allowing for the possibilities that they play a role in CRC invasiveness [64].
Gut microbiota and cancer therapy
Role of the gut microbiota in checkpoint blockade
Immune checkpoint blockade (ICB) is an effective therapeutic strategy for a subset of CRC patients. This therapy targets the inhibitory signals to anti-tumor T cell activation, enabling appropriate anti-tumor immune responses[65]. Over the last decade, clinically relevant discoveries have been made demonstrating the gut microbiomes’ role ICB response across cancer types [9, 11]. Among those receiving ICB, gut microbiota signatures vary between responder and nonresponders to ICB [66]; species shown to be associated with ICB response include Akkermansia municiphila, B. fragilis, Bifidobacterium spp., Eubacterium liomosum, and Faecalibacterium spp. [9,10,11, 67]. Further investigation demonstrated higher abundance of Faecalibacterium spp. in anti-PD1 responders, correlating with more robust tumor immune infiltrates, consisting of higher levels of anti-tumor T cells and lower levels of regulatory T cells [67]. To study the role of the gut microbiome in these observations, preclinical melanoma models have demonstrated that responder or non-responder phenotypes could be altered among GF or antibiotic-treated mice through treatment with fecal microbiota transplantation (FMT) or gavage of specific bacterial species [9, 11]. Similarly, in mouse models of colon and melanoma tumors, antibiotic-treated or GF mice did not respond to CTLA-4 blockade, whereas gavage with B. fragilis restored response to therapy.[10] The underlying mechanism attributing to this immunologic effect and ICB response is through presentation of bacterial components to antigen-presenting cells (APCs) and innate effectors, ultimately inducing an adaptive immune response [5]. Globally, this is believed to cause a stronger anti-tumor effect, heightening the action of ICB.
Among colorectal cancers, there is a dichotomous response to ICB based on a tumor’s microsatellite instability (MSI) status, with MSI-high (MSI-H) tumors having typically robust responses to anti-PD1 therapy [68, 69]. Phenotypic differences exist between MSI-H and microsatellite stable tumors (MSI-S), with significantly more prominent cytotoxic T cells and TH1 cell infiltrates in MSI-H and upregulated expression of immune checkpoints [70, 71]. Based on this rationale, a phase 2 trial for metastatic or recurrent MSI-H CRC tumors treated with PD-1 inhibition demonstrated improved and durable responses with prolonged survival compared to the expected survival of MSI-H metastatic cancer patients [72]. There is need for future investigations into how MSI-H associated microbiota signatures contribute mechanistically and into the potential therapeutic role of modulating gut microbe populations to enhance treatment responses.
The gut microbiome plays an important role in priming host immunity. Certain microbial species can stimulate T cell activation and differentiation, although the resulting immune response can vary based on the colonic local environment. Helicobacter hepaticus (Hhep) colonization in a healthy colon induces T cell differentiation into regulatory and follicular helper T cells, while in immunodeficient models stimulates Th1 and Th17 cells. In the context of modern anti-cancer strategies, understanding this process could help close an important knowledge- gap, as most anti-tumor immunotherapy relies on activating T cells. In a recent study, Hhep colonic colonization in a CRC-induced mouse model limited tumor burden and increased the tumor’s immune infiltrate. Furthermore, investigators identified Hhep-specific follicular T helper cell (TFH) activation and TFH-induced tertiary lymphoid structures as necessary for this anti-tumor immunity and tied to response to ICB in other histologies [73•].
The impact of the gut microbiota on immunotherapy toxicity, particularly autoimmune colitis, has also been explored. Evaluation of microbiota signatures in patients treated with anti-CTLA4 therapy revealed that increased abundance of Bacteroides spp. is protective against autoimmune colitis [74, 75]. Meanwhile, higher abundance of Firmicutes correlated with higher rates of anti-PD1-induced colitis [74]. In our experience, immunotherapy-induced colitis was successfully treated in two patients with FMT, by reconstituting the gut microbiome, which led to increased infiltration of regulatory T cells into the colonic mucosa [76]. Ultimately future validation of this work in CRC is needed.
The role of the gut microbiome in chemotherapeutic response
In addition to associations with ICB response, evidence exists demonstrating that the microbiome’s influence on the immune system may tailor responses to other forms of cancer therapy as well [77]. In preclinical murine models, cyclophosphamide, an alkylating chemotherapeutic, not only altered gut microbiota composition, but also resulted in migration of Gram positive bacteria into mesenteric lymph nodes where TH17 cell stimulation and memory TH1 cell response were visualized [77, 78]. This process resulted in a systemic anti-tumor effect. In this same study, antibiotic treatment suppressed bacterial invasion and immune response, resulting in resistance to cyclophosphamide. Platinum-based chemotherapeutics have similarly been described to have microbiome-dependent responses. Response to oxaliplatin in CRC and lymphoma murine models was diminished in GF and antibiotic-treated mice [79]. Moreover, oxaliplatin response depended on microbe-induced inflammation and ROS production in the TIME.
Specific to CRC, intratumoral F. nucleatum has been shown to promote chemoresistance to oxaliplatin and 5-FU, via activation of autophagy pathways by targeting TLR4 and MyD88 receptors for innate immune signaling [49]. In CRC models, Gammaproteobacteria have been linked to oxaliplatin and gemcitabine resistance as it harbors an inactivating enzyme, cytidine deaminase [80]. As the immune system’s role in response to cytotoxic therapies continues to be elucidated, the role of tumoral and gut microbes in modulating tumor response to cytotoxics should not be ignored.
Role of gut microbiota on radiation therapy
Radiation therapy (RT) plays a key role in the management of rectal adenocarcinoma. In preclinical models, RT has been shown to alter the normal gut microbiota [81]. Specifically, RT response has been associated with a reduction in Firmicutes abundance and increase in Proteobacteria. The microbiota’s effect on radiosensitivity of the intestinal endothelium is highlighted by a preclinical study that demonstrated that GF mice developed less radiation-induced enteritis and less lymphocytic infiltration [82].
This association is strengthened by the observation that antibiotics modulate response to radiation therapy and radiation toxicity. The addition of vancomycin to RT enhanced local and distant RT-induced anti-tumor effects [83]. Furthermore, in melanoma murine models, total body irradiation enhanced intestinal bacterial translocation into the mesenteric LNs leading to stronger anti-cancer response [84]. This may be, in part, due to the abscopal effect, the phenomenon wherein tumor irradiation results in anti-tumor immune activation, thus allowing anti-cancer activity beyond the radiated field. High-dose RT generates tumor cell death, exposing antigens to the innate immune system, which subsequently activate Th1 and cytotoxic T cells which drive anti-cancer activity. It is hypothesized that through this same process, radiation can prime the TIME, and lead to stronger response from immune CPB [85].
Dietary effects
The gut microbiome is shaped by numerous environmental exposures, particularly diet and medication use. Given the microbiomes role in disease development and response to therapy, a new frontier in cancer research revolves around harnessing microbial modulation, in an effort to alter host physiology and support favorable outcomes. Personalized nutrition, in the form of dietary intervention or recommendation, has emerged as an exciting strategy to provide individualized care in various clinical contexts [86] (Figure 1). Dietary habits affect the microbiome’s structure and function, but the interaction is complex, and the effects of a particular diet can vary significantly between individuals. Nonetheless, numerous studies have demonstrated successful modulation of the gut microbiome and consequential changes in host metabolism and immune function through changes in dietary inputs, including dietary fiber and fermented foods [26, 87, 88]. In a recent study of melanoma patients treated with ICB, higher fiber diets were associated with significantly improved progression-free survival [89•]. This was recapitulated in preclinical models in which mice treated with low-fiber diets or probiotics had impaired ICB responses, and a less robust cytotoxic T cell infiltration in the TIME. Clinical trials involving gut microbiota alteration through diet intervention are ongoing and will be crucial for understanding the safety and efficacy of this strategy in the context of ICB treatment. Another strategy for microbiome modulation is live biotherapeutics, a new class of microbiome-derived therapeutics under development. These are distinct from over the counter probiotics in that they are subjected to rigorous clinical testing and regulatory approval. While evidence exists to support probiotic-induced alterations in host metabolism and inflammatory pathways, preclinical models and controlled trials in colitis and colorectal cancer have yielded mixed results [90, 91]. The use of probiotics, especially in the cancer patient, should be undertaken with caution given preliminary findings that they may be deleterious in patients treated with ICB. Ultimately, the composition of probiotics is variable, largely homogenous, and has been inadequately studied. Further development of individualized and targeted, live biotherapeutics are necessary.

Potential applications for enhanced screening modalities and improved outcomes in response to immunotherapy and chemoradation. Created with BioRender.com
Future directions
Prior work has clearly demonstrated the role of microbes in normal gut function, immune response, and therapeutic anti-cancer therapies. Based on these findings, efforts are underway to establish preventative or therapeutic anti-cancer interventions through the modulation of the gut microbiota. FMT has demonstrated significant efficacy in treatment-resistant C. difficile infections [92], and in benign disease, an excellent safety profile has been described [93]. A number of studies are underway investigating how FMT may be used in the context of cancer therapy, including its use for modulating responsiveness to immunotherapy and treatment-related toxicities [94, 95]. Also in production are CRISPR-based antimicrobials that exploit phage delivery of CRISPR-Cas systemics to bacteria leading to genome editing and elimination of specific bacteria at a strain level [96]. Given the challenges of antibiotic resistance and the known effect of antibiotics on the gut microbiome, this development in synthetic biology may impact our ability of targeting deleterious organisms without inducing global gut microbial dysbiosis. This targeted therapy can eradicate pathogenic bacteria, while leaving the rest of the gut microbiota intact, which could potentially be utilized for the treatment of a variety of microbiome-related diseases including infectious diseases, autoimmune disorders, and cancer.
Beyond treatment of CRC, early detection is a cornerstone of management, complicated by the rise in early-onset cancers. Fecal immunochemical tests (FIT) have been posited as an option to screen fecal samples for blood with a sensitivity of 69-86%,but have poor sensitivity for adenomatous precursors [97]. Detection of CRC in early stages is associated with excellent treatment outcomes, thus supporting the need for improved biomarkers for early CRC screening. Microbial signatures associated with various stages of CRC may represent the basis for potential screening strategies. In a metagenomic analysis of patients with CRC, a signature of 20 microbial gene markers was identified that differentiated CRC from normal controls; two of these genes were enriched at early stages, highlighting its potential as a screening biomarker [19]. F. nucleatum’s abundance in CRC has been employed in biomarker studies, and its use in conjunction with the FIT test has demonstrated improved sensitivity and specificity compared to FIT alone [98]. As previously described, certain microbial metabolic byproducts are associated with CRC and may also serve as biomarkers for cancer detection.
Conclusion
Over the past decades, enormous strides have been made in understanding the effects of the gut microbiome on normal health-promoting functions, as well as variety of benign and malignant disease processes. Microbial patterns and mechanisms have now been described that clearly associate with cancer development and treatment response. As we continue to treat colorectal cancer in the future, our ability to leverage these observations will be determined by continued engagement by the research community, directed study of mechanisms behind these observations, and early clinical trials to test the translation of these findings to patients at population scale.
Change history
05 April 2022
A Correction to this paper has been published: https://doi.org/10.1007/s11864-022-00980-2
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Sekirov I, et al. Gut microbiota in health and disease. Physiol Rev. 2010;90(3):859–904.
Hillman ET, et al. Microbial ecology along the gastrointestinal tract. Microbes Environ. 2017;32(4):300–13.
Kang M, Martin A. Microbiome and colorectal cancer: unraveling host-microbiota interactions in colitis-associated colorectal cancer development. Semin Immunol. 2017;32:3–13.
Hansen JJ, Sartor RB. Therapeutic manipulation of the microbiome in IBD: current results and future approaches. Curr Treat Options Gastroenterol. 2015;13(1):105–20.
Helmink BA, et al. The microbiome, cancer, and cancer therapy. Nat Med. 2019;25(3):377–88.
Irrazábal T, et al. The multifaceted role of the intestinal microbiota in colon cancer. Mol Cell. 2014;54(2):309–20.
Kim D, Zeng MY, Núñez G. The interplay between host immune cells and gut microbiota in chronic inflammatory diseases. Exp Mol Med. 2017;49(5):e339.
Rajagopala SV, et al. The human microbiome and cancer. Cancer Prev Res (Phila). 2017;10(4):226–34.
Gopalakrishnan V, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103.
Vétizou M, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079–84.
Matson V, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104–8.
Galan-Ros J, Ramos-Arenas V, Conesa-Zamora P. Predictive values of colon microbiota in the treatment response to colorectal cancer. Pharmacogenomics. 2020;21(14):1045–59.
Bray F, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Guinney J, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350.
Boursi B, et al. Recurrent antibiotic exposure may promote cancer formation--another step in understanding the role of the human microbiota? Eur J Cancer. 2015;51(17):2655–64.
Dahm CC, et al. Dietary fiber and colorectal cancer risk: a nested case-control study using food diaries. J Natl Cancer Inst. 2010;102(9):614–26.
Abegunde AT, et al. Environmental risk factors for inflammatory bowel diseases: evidence based literature review. World J Gastroenterol. 2016;22(27):6296–317.
Ahn J, et al. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. 2013;105(24):1907–11.
Yu J, et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. 2017;66(1):70–8.
Feng Q, et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun. 2015;6:6528.
Chen W, et al. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 2012;7(6):e39743.
• Yachida S, et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med. 2019;25(6):968–76 In this 2019 study, fecal metagenomic and metabolomic studies were performed on samples from adenomas, intramucosal carcinomas and more advanced lesions. F. nucleatum abundance was elevated throughout disease stages, while shifts in other species occurred across disease stages. These shifts in microbiome across stages of CRC development carry important implications for both etiology and possible clinical applications.
Nakatsu G, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun. 2015;6:8727.
Wong SH, et al. Gavage of fecal samples from patients with colorectal cancer promotes intestinal carcinogenesis in germ-free and conventional mice. Gastroenterology. 2017;153(6):1621–1633.e6.
Tilg H, et al. The intestinal microbiota in colorectal cancer. Cancer Cell. 2018;33(6):954–64.
• Wastyk HC, et al. Gut-microbiota-targeted diets modulate human immune status. Cell. 2021;184(16):4137–4153.e14 This study highlights the ability to modulate the gut microbiome and immune system through dietary intervention. Whether microbiome modulation through dietary intervention can enhance immunotherapy treatment response is an exciting prospect, which is being explored in trials across multiple cancer types.
Flint HJ, Duncan SH, Louis P. The impact of nutrition on intestinal bacterial communities. Curr Opin Microbiol. 2017;38:59–65.
Alexander DD, et al. Meta-analysis of prospective studies of red meat consumption and colorectal cancer. Eur J Cancer Prev. 2011;20(4):293–307.
O’Keefe SJ. Diet, microorganisms and their metabolites, and colon cancer. Nat Rev Gastroenterol Hepatol. 2016;13(12):691–706.
Furusawa Y, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504(7480):446–50.
Belcheva A, et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell. 2014;158(2):288–99.
Tjalsma H, et al. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012;10(8):575–82.
Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10(10):717–25.
Boleij A, et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin Infect Dis. 2015;60(2):208–15.
Goodwin AC, et al. Polyamine catabolism contributes to enterotoxigenic bacteroides fragilis-induced colon tumorigenesis. Proc Natl Acad Sci U S A. 2011;108(37):15354–9.
Dejea CM, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science. 2018;359(6375):592–7.
Dalmasso G, et al. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes. 2014;5(5):675–80.
Cougnoux A, et al. Small-molecule inhibitors prevent the genotoxic and protumoural effects induced by colibactin-producing bacteria. Gut. 2016;65(2):278–85.
Grivennikov SI, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491(7423):254–8.
Wu S, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15(9):1016–22.
Buc E, et al. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One. 2013;8(2):e56964.
Huycke MM, Abrams V, Moore DR. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis. 2002;23(3):529–36.
Abed J, et al. Fap2 mediates fusobacterium nucleatum colorectal adenocarcinoma enrichment by binding to tumor-expressed gal-GalNAc. Cell Host Microbe. 2016;20(2):215–25.
Rubinstein MR, et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14(2):195–206.
Ikegami A, Chung P, Han YW. Complementation of the fadA mutation in fusobacterium nucleatum demonstrates that the surface-exposed adhesin promotes cellular invasion and placental colonization. Infect Immun. 2009;77(7):3075–9.
Shmuely H, et al. Relationship between helicobacter pylori CagA status and colorectal cancer. Am J Gastroenterol. 2001;96(12):3406–10.
Tsoi H, et al. Peptostreptococcus anaerobius induces intracellular cholesterol biosynthesis in colon cells to induce proliferation and causes dysplasia in mice. Gastroenterology. 2017;152(6):1419–1433.e5.
Abdulamir AS, et al. Investigation into the controversial association of streptococcus gallolyticus with colorectal cancer and adenoma. BMC Cancer. 2009;9:403.
Yu T, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell. 2017;170(3):548–563 e16.
Kostic AD, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14(2):207–15.
Yu YN, et al. Berberine may rescue fusobacterium nucleatum-induced colorectal tumorigenesis by modulating the tumor microenvironment. Oncotarget. 2015;6(31):32013–26.
Yang Y, et al. Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor-κB, and up-regulating expression of MicroRNA-21. Gastroenterology. 2017;152(4):851–866.e24.
Mima K, et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut. 2016;65(12):1973–80.
Mima K, et al. Fusobacterium nucleatum in colorectal carcinoma tissue according to tumor location. Clin Transl Gastroenterol. 2016;7(11):e200.
Bullman S, et al. Analysis of fusobacterium persistence and antibiotic response in colorectal cancer. Science. 2017;358(6369):1443–8.
Yu J, et al. Invasive fusobacterium nucleatum may play a role in the carcinogenesis of proximal colon cancer through the serrated neoplasia pathway. Int J Cancer. 2016;139(6):1318–26.
Spadoni I, et al. Gene expression profile of endothelial cells during perturbation of the gut vascular barrier. Gut Microbes. 2016;7(6):540–8.
Costa-Silva B, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17(6):816–26.
Seubert B, et al. Tissue inhibitor of metalloproteinases (TIMP)-1 creates a premetastatic niche in the liver through SDF-1/CXCR4-dependent neutrophil recruitment in mice. Hepatology. 2015;61(1):238–48.
•• Bertocchi A, et al. Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell. 2021;39(5):708–724.e11 This exciting new study demonstates that the colorectal tumor microbiome is involved in metasasis. Specifically, they show E. Coli’s ability to injure the gut vascular barrier, and subsequently demonstrate bacterial dissemination from the CRC primary to the liver where they support the formation of a pre-metastatic niche.
Emlet C, Ruffin M, Lamendella R. Enteric virome and carcinogenesis in the gut. Dig Dis Sci. 2020;65(3):852–64.
Nakatsu G, et al. Alterations in enteric virome are associated with colorectal cancer and survival outcomes. Gastroenterology. 2018;155(2):529–541.e5.
Hannigan GD, et al. Diagnostic potential and interactive dynamics of the colorectal cancer virome. mBio. 2018;9(6).
Nguyen S, et al. Bacteriophage transcytosis provides a mechanism to cross epithelial cell layers. mBio. 2017;8(6).
Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–86.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61.
Sivan A, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350(6264):1084–9.
Goldstein J, et al. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H). Ann Oncol. 2014;25(5):1032–8.
•• Overman MJ, et al. Durable clinical benefit with nivolumab plus ipilimumab in dna mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36(8):773–9 In this clinical trial, MSI-H colorectal cancer demonstrated high response rates and encouraging progression-free survival and overall survival with treatment with combination nivolumab and ipilimumab. Further investigation into the microbiome’s role in the development of CMS subtype and response to immunotherapy is needed.
Llosa NJ, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5(1):43–51.
Giannakis M, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;15(4):857–65.
Overman MJ, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–91.
•• Overacre-Delgoffe AE, et al. Microbiome-specific T follicular helper cells drive tertiary lymphoid structures and anti-tumor immunity against colorectal cancer. bioRxiv. 2021:2021.01.27.428417 In this 2021 study, introduction of Helicobacter hepaticus (Hhep) in a mouse model of CRC increased tumor immune infiltration that inhibited tumor growth. Furthemore, Hhep colonization-induced specific follicular helper T cells and supported tumor-adjacent tertiary lymphoid structures that were necessary to anti-tumor immunity. This exciting new study demonstrates a possible role for immunogenic bacterial introduction to support more robust immune infiltrates and anti-tumor responses, which could enhance treatment of CRC.
Chaput N, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2017;28(6):1368–79.
Andrews MC, et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat Med. 2021;27(8):1432–41.
Wang Y, et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med. 2018;24(12):1804–8.
Viaud S, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342(6161):971–6.
Viaud S, et al. Cyclophosphamide induces differentiation of Th17 cells in cancer patients. Cancer Res. 2011;71(3):661–5.
Iida N, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342(6161):967–70.
Villéger R, et al. Intestinal microbiota: a novel target to improve anti-tumor treatment? Int J Mol Sci. 2019;20(18).
Kim YS, Kim J, Park SJ. High-throughput 16S rRNA gene sequencing reveals alterations of mouse intestinal microbiota after radiotherapy. Anaerobe. 2015;33:1–7.
Crawford PA, Gordon JI. Microbial regulation of intestinal radiosensitivity. Proc Natl Acad Sci U S A. 2005;102(37):13254–9.
Uribe-Herranz M, et al. Gut microbiota modulate dendritic cell antigen presentation and radiotherapy-induced antitumor immune response. J Clin Invest. 2020;130(1):466–79.
Paulos CM, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. 2007;117(8):2197–204.
Tonneau M, et al. The role of the gut microbiome on radiation therapy efficacy and gastrointestinal complications: a systematic review. Radiother Oncol. 2021;156:1–9.
Kolodziejczyk AA, Zheng D, Elinav E. Diet-microbiota interactions and personalized nutrition. Nat Rev Microbiol. 2019;17(12):742–53.
Michalak L, et al. Microbiota-directed fibre activates both targeted and secondary metabolic shifts in the distal gut. Nat Commun. 2020;11(1):5773.
Leshem A, Segal E, Elinav E. The gut microbiome and individual-specific responses to diet. mSystems. 2020;5(5).
• Spencer CN, et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science. 2021;374(6575):1632–40 In this study, dietary fiber was shown to help shape the gut microbiome, which in turn, led to enhanced immunotherapy responses in melanoma. Dietary intervention may develop into a promising adjunct to anti-cancer therapy.
Suez J, et al. The pros, cons, and many unknowns of probiotics. Nat Med. 2019;25(5):716–29.
Arthur JC, et al. VSL#3 probiotic modifies mucosal microbial composition but does not reduce colitis-associated colorectal cancer. Sci Rep. 2013;3:2868.
Gough E, Shaikh H, Manges AR. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Dis. 2011;53(10):994–1002.
Kelly CR, et al. Fecal microbiota transplant for treatment of Clostridium difficile infection in immunocompromised patients. Am J Gastroenterol. 2014;109(7):1065–71.
Baruch EN, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. 2020.
Davar D, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. 2021;371(6529):595–602.
Ramachandran G, Bikard D. Editing the microbiome the CRISPR way. Philos Trans R Soc Lond Ser B Biol Sci. 2019;374(1772):20180103.
Lee JK, et al. Accuracy of fecal immunochemical tests for colorectal cancer: systematic review and meta-analysis. Ann Intern Med. 2014;160(3):171.
Wong SH, et al. Quantitation of faecal fusobacterium improves faecal immunochemical test in detecting advanced colorectal neoplasia. Gut. 2017;66(8):1441–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Samuel Cass, Nadim Ajami, and Michael White each declare no potential conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Lower Gastrointestinal Cancers
The original online version of this article was revised: The degrees of the authors Michael White and Nadim Ajami were interchanged. It should be Michael White, MD MSc, and Nadim Ajami, PhD (as shown above) instead of Michael White, PhD and Nadim Ajami, MD MSc.
Rights and permissions
About this article

Cite this article
Cass, S.H., Ajami, N.J. & White, M.G. The Microbiome: the Link to Colorectal Cancer and Research Opportunities. Curr. Treat. Options in Oncol. 23, 631–644 (2022). https://doi.org/10.1007/s11864-022-00960-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11864-022-00960-6