
Opinion statement
Acute myeloid leukemia (AML) disease prognosis is poor and there is a high risk of chemo-resistant relapse for both young and old patients. Thus, there is a demand for alternative and target-specific drugs to improve the 5-year survival rate. Current treatment mainstays include chemotherapy, or mutation-specific targeting molecules including FLT3 inhibitors, IDH inhibitors, and monoclonal antibodies. Efforts to devise new, targeted therapy have included recent advances in methods for high-throughput genomic screening and the availability of computer-assisted techniques for the design of novel agents predicted to specifically inhibit mutant molecules involved in leukemogenesis. Crosstalk between the leukemia cells and the bone marrow microenvironment through cell surface molecules, such as the integrins αvβ3 and αvβ5, might influence drug response and AML progression. This review article focuses on current AML treatment options, new AML targeted therapies, the role of integrins in AML progression, and a potential therapeutic agent—integrin αvβ3 antagonist.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myeloid leukemia (AML) is a multifaceted life-threatening hematological malignancy that affects the blood and lymphoid system of the body [1]. AML is heterogeneous in nature and is characterized by abnormal proliferation and absence of differentiation of immature and abnormal blast cells. In adults, AML is one of the most common types of leukemia and accounts for about 3.5% of all cancers [2]. The prevalence of AML cases is slightly more common in men compared to women, even though the average lifetime risk of acquiring AML in both genders is around one half of 1% [3]. According to the American Cancer Society’s estimations for AML cases in the USA for 2019, there will be 21,450 new cases and 10,920 deaths from AML [3]. Among all the leukemia cases, 32% of cases in adults are due to AML [4].
The 5-year survival rate for adults suffering from AML is only about 24% [4], although recently there have been more selective and targeted immunotherapy options developed [5], which have slightly increased the 5-year survival rate. These new targeted treatments are either used as monotherapy or in combination with chemotherapy to achieve a better outcome. However, managing the side effects of these combinations of drugs can be challenging and needs more research on how to optimize the therapy for the best possible outcome, which is to ensure maximum efficacy with minimum adverse effects. Given the disease prognosis, heterogeneity, and mortality rates, the goal is to ensure and provide treatment options that are not only effective but also safe in the long run [6].
Recent clinical studies have demonstrated that heterodimeric cell surface molecules composed of α and β chains, called integrins and which can bind extracellular matrix (ECM) molecules, cell surface molecules, and variable soluble mediators [7, 8], have a role in AML progression and cessation. This has raised interest to develop therapeutic agents targeting integrin receptors. The β3 integrin (ITGB3) chain can form heterodimers only with the two α chains αIIb and αV, which play an important role in leukemogenesis and chemo-resistance in human AML.
Pathophysiology
AML is a heterogeneous disease with a complex and distinct pathogenesis that involves a wide array of molecular modifications, leading to disruption of cell growth and development. These molecular modifications comprise cellular processes like regulation of cell differentiation and proliferation, survival, self-renewal, DNA-repair, chromatin stability, cell-cycle checkpoint control, and cell distribution [9]. Molecular genetic alterations underlie the core of AML pathogenesis and disease progression, hence understanding these processes is vital to develop more disease-specific therapies.
Recently, many cytogenetic abnormalities were identified in the different subtypes of leukemia. Genetic abnormalities are present in more than 80% of AML patients and acute lymphoblastic leukemia patients, and most of them are recurrent [10]. Moreover, the heterogeneity of acute leukemia is related not only to clinical outcome but also to heterogeneity in the response to chemotherapies. For example, acute promyelocytic leukemia (APL) patients with t(15;17) and (q22;q21) are responsive to all-trans-retinoic acid (ATRA) treatment, but APL patients with t(11;17) and (q23;q21) are resistant to ATRA [11].
Evasion from programmed cell death
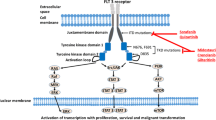
The evasion of apoptosis by cancer cells is one of the most important processes by which they develop malignancy (Fig. 1). Usually, cancer cells proliferate and attain increased cell survival probability due to the activation of protein tyrosine kinase, which in turn activates the phosphatidylinositol 3-kinase (PI3K) signaling [12]. PI3K signaling activates AKT serine/threonine kinase, which in turn helps to release the BCL-2 pro-survival molecule by phosphorylating BAD proteins [13]. In AML, survival rates and clinical response rates can be predicted by observing the expression of the BCL-2 pro-survival molecule and hence it is an important molecular target for AML treatment [14, 15].

Cancer cells’ evasion from apoptosis and metastasis. Auto-activation of tyrosine kinases by different mutations associated with activation of phosphatidylinositol 3-kinase (PI3K) signaling. PI3K signaling activates AKT serine/threonine kinase, which in turn helps to release the BCL-2 pro-survival molecule by phosphorylating BAD proteins. Another important mechanism is the destabilization of p53 protein, which can occur by different mechanisms including suppression of p14ARF expression by the RUNX1–MTG8 fusion protein, binding of the MOZ–TIF2 fusion protein to core binding protein (CBP), or mutation of nucleophosmin (NPM) protein that is important for the cytoplasmic localization of p53. On the other hand, integrin αvβ3 binds a wide range of extracellular matrix molecules with an Arg-Gly-Asp (RGD) motif, including VEGF and osteopontin. Upon binding, the cytoplasmic tail initiates a signaling cascade, including the activation of Src kinases via phosphorylation of focal adhesion kinase (FAK). These signaling pathways are involved in cell survival, proliferation, osteoclast-mediated bone resorption, and neovascularization, which are important for tumor metastasis.
Another important protein that is linked to cell-cycle regulation and apoptosis is p53 protein (Fig. 1). Patients with AML experience adverse reactions in response to chemotherapy treatment due to p53 protein mutations [16]. Leukemogenic signaling causes alterations in p53 regulation, which compromise the function of this protein in AML. Destabilization of p53 protein occurs due to the suppression of p14ARF expression by the RUNX1–MTG8 fusion protein. Furthermore, p53 transcriptional activity is debilitated indirectly due to binding of the MOZ–TIF2 fusion protein to core binding protein (CBP) [17]. The stability of p53 protein is regulated by the nucleophosmin protein (NPM), but unfortunately, about 35% of newly diagnosed AML cases are due to mutations within the NPM coding region that is linked with cytoplasmic localization of this protein [18].
Leukemia cell propagation
Leukemia cells’ dissemination from the bone marrow to other cells and tissues has not been clearly understood until recently; however, it can be assumed that there is a role for RUNX1 fusion protein in regulating expression of cell surface proteins [19]. In AML, high levels of selectin expression on the surface of the leukemic cells is a detrimental predictive marker that is increased by the secretion of other cytokines and tumor necrosis factors. This can lead to increased expression of adhesion proteins like selectins, integrins, and cadherin on vascular endothelium, augmenting leukemic cell adhesion. Monoclonal antibodies like anti-CD33 could be a possible therapeutic option because it can interfere with leukemic cell adhesions with cell surface proteins, diminishing cancer cell growth and proliferation [20].
The process of invasion and metastasis depends mainly on degradation of the ECM. Integrins, a family of cell adhesion molecules, are involved in a wide range of cell–ECM and cell–cell interactions. Integrin αvβ3 binds a wide range of ECM molecules with an Arg-Gly-Asp (RGD) motif, including vascular endothelial growth factor (VEGF), fibronectin, fibrinogen, vitronectin, osteopontin, and laminin [21]. Upon binding, the cytoplasmic tail initiates a signaling cascade, including the activation of Src kinases (a family of non-receptor tyrosine kinases) via phosphorylation of focal adhesion kinase (FAK) [22]. Activation of Src kinases enhances activation of different signaling pathways including PI3K/AKT and mitogen-activated protein kinases (MAPK) [23]. Therefore, the αvβ3 signaling pathway plays an important role not only in cell proliferation but also in osteoclast-mediated bone resorption and neovascularization, which are important for tumor metastasis (Fig. 1).
Conventional AML therapies
Treatment of AML is dependent on the subtype of AML present and may be different than treatment used in other forms of leukemia. In addition, prognostic factors including patient age, overall health status, and presence of genetic or chromosomal abnormalities are important in determining which treatment will lead to the safest and most effective outcome for each patient. The following are some of the cornerstones used in current treatment of AML [10, 24].
Chemotherapy
According to the American Cancer Society, intravenous chemotherapy is the mainstay treatment for most patients affected by AML due to its usefulness in reaching all areas of the body, that is, where leukemia has also reached. The two chemotherapy drugs most often used are a combination of cytarabine plus daunorubicin, or cytarabine plus idarubicin [25]. The standard induction regimen of these drugs for adult patients is a continuous infusion of cytarabine for 7 days, plus daunorubicin or idarubicin injections for the first 3 days, also commonly referred to as the 7 + 3 regimen [3, 26,27,28]. Cytarabine’s main mechanism of action involves cytotoxicity on the dividing cells specifically in the S-phase. Cytarabine also gets metabolized to cytarabine-triphosphate (Ara-CTP), which ultimately inhibits DNA strand elongation and repair by incorporating into tumor cellular DNA [29]. The anthracyclines, daunorubicin and idarubicin, work via similar mechanisms by incorporating themselves between DNA base pairs, causing a DNA helix conformational change. This transformation in shape inhibits DNA and RNA polymerases, thereby interfering with strand elongation and protein synthesis of the tumor cells [30]. Table 1 shows a list of currently approved chemotherapies for AML and investigational drugs in the pipeline.
Typically, chemotherapy is given in two phases for adults under the age of 60. The first, called induction, is a short but intense treatment used to completely eliminate the leukemic myoblasts from the bloodstream and reduce the number of myoblasts in the bone marrow to below the normal 5% threshold [31]. When induction has been completed after approximately 1 week and the patient has had the chance to recover, the second phase, known as consolidation or post-remission therapy, is initiated. Consolidation is given in cycles and is used to eliminate the small number of leukemic cells that are known to be still present in the patient, although not necessarily detectable. A subsequent phase known as maintenance or post-consolidation is given months to years after the completion of phase II for a APL, but is rarely utilized for most other types of AML [3, 32].
New AML-targeted therapies
Despite using the combination of cytosine arabinoside (Ara-C) and anthracycline as the main pillar of AML treatment for almost 40 years, this regimen is associated with a high relapse rate. Different new agents have been developed to improve survival and decrease the relapse rate [33]. A summary of new trials against AML is given in Table 2.
FMS-related tyrosine kinase 3
FMS-related tyrosine kinase 3 (FLT3) is one of the most frequent mutations in AML, and it represents about 25–30% of mutations in AML patients [34•]. Different generations of FLT3 inhibitors have been developed. The first-generation FLT3 inhibitors include sorafenib (multikinase inhibitor) [35], sunitinib [36], lestaurtinib (a bioavailable indolocarbazole alkaloid compound derived from the bacterial) [37], and midostaurin (a small-molecule tyrosine kinase inhibitor (TKI) approved by the US FDA in 2017) [38, 39]. Second-generation FLT3 inhibitors include quizartinib (a selective and highly potent second-generation class III receptor TKI even in a low dose, 1 mg/kg) [40] and crenolanib (a selective inhibitor of FLT3/wild type mutations (WT), FLT3/internal tandem duplication mutations (ITD), FLT3-tyrosine kinase domain mutations (TKD), platelet-derived growth factor receptor mutations (PDGFRα/β), stem cell factor receptor gene mutations (KIT), and FLT3/aspartic acid codon 835 mutations (D835)) [41].
Several mechanisms can explain the efficacy of FLT3 inhibitors besides blocking of FLT3 phosphorylation, including (1) secretion of interleukin (IL)-15 by FLT3/ITD-mutated leukemic cells [42], (2) maintaining the low blast level in bone marrow by enhancing CD3+/CD8+ cell invasion in the bone marrow with high expression of IL-12 [43], and (3) downregulation of apoptosis regulatory proteins’ expression (e.g., Mcl-1 and Bcl-2) by blocking Src kinase-mediated STAT3 phosphorylation [44, 45].
The different FLT3 inhibitors such as lestaurtinib have been used in clinical trials involving patients with refractory AML, showing a great ability to induce rapid clearance of blasts from peripheral and bone marrow without associated normal cell toxicity. Combined therapy of FLT3 inhibitors with conventional chemotherapy was associated with ~90% complete remission [46,47,48,49].
Recently, the FDA approved gilteritinib (ASP2215), a novel dual FLT3/AXL inhibitor, for the treatment of patients who have relapsed or have refractory AML with a FLT3 mutation [34•, 50]. It targets mainly the FLT3/ITD-positive leukemia cells, decreasing their colony-forming capacity [51] by blocking the phosphorylation of FLT3 and its downstream targets [51, 52]. Approval of gilteritinib was based on a clinical trial that included 252 adult patients with relapsed or refractory AML having a FLT3 ITD, D835, or I836 mutation [53]. Gilteritinib was well tolerated and the response rate was more than 50% in patients with FLT3 mutation at doses ≥80 mg/day. No major side effects were reported (less than 5% experienced neutropenia). Mild thrombocytopenia, muscle pain with increased creatine phosphokinase enzyme, diarrhea, nausea, headache, and vomiting were the most common associated side effects [53].
Isocitrate dehydrogenase (IDH)
IDH inhibitors (IDH-i) are used in patients with AML who have mutations in either IDH1 or IDH2 genes, causing abnormal maturation patterns in white blood cells, thus leading to leukemia [54]. Two targeted IDH-i, ivosidenib and enasidenib, work by blocking the proteins IDH1 and IDH2, respectively. This inhibition allows for the normal maturation and differentiation of what would have been leukemic white blood cells, thereby reducing immature blast counts and increasing the percentage of mature myoblasts [55, 56]. While quite efficacious, a safety concern with IDH-i is the possible side effect known as differentiation syndrome, brought on by the release of inflammatory cytokines from cancerous promyelocytes, sometimes referred to as the “cytokine storm”. Differentiation syndrome is serious and potentially fatal, with complications ranging from hypoxemia, hypotension, and respiratory distress to renal and hepatic dysfunction, but may be reversed by stopping the offending agent [47].
Aurora kinases
Aurora kinases are a family of mitotic serine/threonine kinases that are essential for cytokinesis during cell division. Aurora A (AURKA), Aurora B (AURKB), and Aurora C (AURKC) kinases are important for both mitosis (especially the process of chromosomal segregation) as well as for meiosis process [57]. Deletion of Aurora kinases leads to cell division arrest, while the overexpression of Aurora kinases has been associated with a number of cancers such as lung, colorectal, and melanoma. Recent studies have demonstrated that inhibition of Aurora kinases could potentiate the effect of chemotherapies, especially in AML [57, 58].
An Aurora kinase inhibitor was tested against different AML cell lines (NB4 and KG1) and also primary cells obtained from AML patients, and it was associated with increased apoptosis (~3-fold) [59]. Recently, different Aurora kinases inhibitors were tested in clinical trials including AURKA inhibitors (e.g., MLN8054, phase I [60]; ENMD-2076, phase II [61]; MLN8237, phase III [62, 63]) and AURKB inhibitors (e.g., AZD1152, phase II/III [64, 65]; PHA-680632, preclinical [66]).
Beta cell lymphoma-2 (BCL-2)
Venetoclax is a recently approved (November 2018) agent to treat AML based on its extensive response rates as a selective small-molecule inhibitor of beta cell lymphoma-2 (BCL-2) protein [67]. It is another alternative for newly diagnosed patients of age 75 years and older who are incapable of sustaining high-intensity induction chemotherapy. The BCL-2 protein is responsible for antagonizing cellular apoptosis and has been associated with chemotherapy resistance. Venetoclax’s role in binding to BCL-2 with high affinity allows for inhibition of the anti-apoptotic protein and, ultimately, leukemic cell death. Prescribers should remain aware of the serious side effect profile of venetoclax, including the potential for tumor lysis syndrome, which may lead to renal failure, the need for dialysis, or death [68, 69].
NF-κB inhibitors
NF-κB is an important transcription factor that plays a role for cell growth and apoptotic activity. The significant expression of NF-κB within leukemic cells is one of the promising targets for selective leukemic cell eradication [70].
Several studies are focused on targeting NF-κB, including using the proteasome inhibitor bortezomib, which enhances the anti-NF-κB effect by blocking the degradation of inhibitor of κB (IκB) [71, 72]. NF-κB can also be inhibited in leukemic cells by parthenolide (PTL), a naturally occurring sesquiterpene lactone, which occurs naturally in the plant feverfew (Tanacetum parthenium). PTL can stimulate the apoptotic pathways through inhibition of NF-κB, activation of p53, and the increase of reactive oxygen species, leading to decreased engraftment of leukemic cells into NOD/SCID mice [73]. In vitro studies were done to compare the effects of PTL against AML versus normal specimens, and the viability of AML CD34 cells was markedly suppressed by more than 70% in comparison to normal CD34 controls [74].
According to our preliminary data, both surface molecule CD44 and intracellular molecule NF-κB were broadly expressed in our AML patients in comparison to normal individuals (~25-fold). Our team designed a nano-encapsulating PTL to improve bioavailability and targeted delivery to leukemic cells. The preliminary data from 103 AML patients’ bone marrow samples showed the correlation of high expression of NF-κB with a poor prognosis. Also, our targeting strategy using nano-antiCD44 encapsulating PTL (7.5 μm) was associated with a significant ~50% decrease in the cell proliferation.
BMI-1 small molecule inhibitor (PTC-209)
BMI-1 (polycomb complex protein) is a transcription factor that is essential for the regulation of cell division, especially in highly dividing cells. Our previous work showed that BMI-1 was markedly increased in AML patients, especially FAB (M1, M5, and M7) [75]. Other studies have reported that the forced expression of BMI-1 led to a marked expansion of both multi-potential progenitors and hematopoietic stem cells (HSCs) in vivo [76, 77]. On the other hand, the absence of BMI-1 is associated with a marked defect in HSCs’ self-renewal [78].
PTC-209 interferes with post-transcriptional regulation of BMI-1; however, the actual mechanism has not been clear until recently. In our previous study, we demonstrated the high expression of BMI-1 in AML cell lines, which confirms the role of BMI-1 in leukemogenesis and the possibility to be used as a new target for AML therapy. In vitro cell proliferation assay using different AML cell lines showed that the lowest concentration of anti-BMI-1 small molecule inhibitor PTC-209 (2 mM) decreased proliferation by about ~50–70% [75]. Kreso et al. have reported the effectiveness of PTC-209 against BMI-1 in human colorectal HCT116 and human fibrosarcoma HT1080 tumor cells [79].
The main obstacle for BMI-1 is that it is essential for both normal and leukemic cells’ survival. So, BMI-1 might need to be selectively targeted to the leukemic cells in order to avoid damage to normal cells, which can be achieved by using targeted nano-encapsulation [80].
Monoclonal antibody
The novel agent gemtuzumab ozogamicin, an antibody–toxin conjugate that causes disintegration of DNA by binding at CD33 epitope and that can be given with or without chemotherapy, may also act as a low-intensity targeted agent for patients who are unable to tolerate high-intensity chemotherapy induction [54, 81]. This monoclonal antibody is administered via intravenous infusion and works by binding to CD33 protein present on most AML tumor cells. It functions as a signaling molecule that can enter malignant cells and destroy them during cell division. Safety issues arise because CD33 protein is also expressed on normal myeloid cells, causing potentially profound cytopenia as a serious side effect of the drug [81].
Thyrointegrin αvβ3 antagonist
The roles of thyroid hormones in cancer metastasis, cell proliferation, anti-apoptosis, and angiogenesis associated with cancer have been found to be interrelated based on pre-clinical studies [82••, 83,84,85,86,87] and clinical trials [88,89,90,91].
Integrins, transmembrane receptors that enable cell-ECM adhesion, are largely expressed by malignant cells and help in the dissemination of endothelial cells that are associated with cancer [92]. The extracellular domain of αvβ3 integrin has a thyroid hormone receptor for tetraiodothyroacetic acid (tetrac), a deaminated analogue of thyroid hormone l-thyroxine (T4), which regulates cancer cell proliferation and the anti-apoptotic process of the cells [92,93,94]. The crosstalk between leukemia cells and the bone marrow microenvironment through integrins plays an important role in leukemic cell proliferation and engraftment. Some studies have reported the role of integrins in the modulation of drug response and the survival of AML [95, 96].
Other studies have demonstrated that the T4 receptor of αvβ3 integrin is associated with migration of malignant cells via various molecular mechanisms [97,98,99]. However, recently the T4 antagonism effects of tetrac at the αvβ3 integrin [92] were reported, making it a promising therapeutic agent in AML treatment. In addition, polyethylene glycol (PEG) can be covalently bonded to tetrac, which prolongs the action of tetrac molecules at the αvβ3 receptor. Studies have demonstrated that incorporation of tetrac with PEG has removed breast cancer xenografts that have already metastasized to bone [100, 101••], making it a potential therapeutic agent for AML patients.
There are various complex mechanisms by which thyroid hormones can lead to migration of the malignant cells including gene expression of matrix metalloproteinase (MMP), angiogenesis, microRNAs (miRs), epithelial–mesenchymal transition (EMT), transforming growth factor β (TGFβ), EGF receptor (EGFR), and tumor-associated macrophages [102].
Malignant cells’ migration and engraftment have been linked with MMP-2 and MMP-9 gene expression [103,104,105], which have solubilizing activity of the ECM. Nano-diamino-tetrac (NDAT), a nano-pharmaceutical formulation of tetrac, can downregulate MMP-2 and MMP-9 gene expression, which verifies that NDAT has potential anticancer activity [94, 106]. On the other hand, thyroid hormone is associated with increased activity and gene expression of MMP-2 and MMP-9 [104].
The primary goal of the formulation for thyrointegrin αvβ3 antagonist is to ensure its components specifically target the extracellular domain of the αvβ3 integrin, sparing intracellular receptors and thereby reducing adverse effects. Because αvβ3 expression is substantial in tumor cells (especially in AML) compared to non-malignant cells, normal cells should be spared from the effects due to thyrointegrin αvβ3 antagonist. Blockade of thyrointegrin αvβ3 receptor leads to various anti-metastatic events, which include broad-spectrum anti-angiogenesis activities [107], reversal of resistance to chemotherapy and radiation therapy [108], upstream and downstream regulation of tumor suppressor genes, downstream regulation of tumor survival genes, PD-1/PDL-1 inhibition [109], and regulation of cancer stem cell signaling pathways.
Because the blockade of thyrointegrin αvβ3 receptor provides a unique way to block numerous pathways linked with cancer cell proliferations and differentiations, a logical step was to assess the efficacy and safety of this novel therapeutic agent for AML treatment. A macromolecule was made by covalently binding PEG with two bi-triazole tetrac molecules, called P-bi-TAT [110]. It augments the potency and broadens the anticancer properties of tetrac for the thyrointegrin αvβ3 target. P-bi-TAT obliterates the transcriptional makeup of the life-support structure of cancer cells, providing a unique and effective way of treating AML. P-bi-TAT has promising future aspects in AML treatment based on assessing its efficacy and safety profile in murine models [111]
Summary
Here, we have summarized current treatment options for AML and discussed potential targets for AML treatment that are hopeful for improved treatment outcomes and increased survival. After about four decades without any new approved pharmacologic agent for AML treatment, new classes of drugs were approved by the FDA in 2017, such as midostaurin and enasidinib (IDH2 inhibitors) and gemtuzumab ozogamicin (monoclonal antibody), which are used in combination with chemotherapy for AML treatment. Even so, the 5-year survival rate for AML patients has not significantly increased. There is a need for further well-designed clinical trials targeting other possible pathways by which AML progression and dissemination occurs. Targeting the αvβ3 integrin receptor is another promising target in treating AML. However, clinical trials in humans need to be carried out to ensure safety and efficacy of αvβ3 integrin receptor antagonists.
Abbreviations
- AML:
-
Acute myeloid leukemia
- BCL-2:
-
B-cell lymphoma gene 2
- CAM:
-
Chick egg chorioallantoic membrane
- CDK:
-
Cyclin-dependent kinase
- c-KIT:
-
C-kit is a type of receptor tyrosine kinase, also called CD117
- c-FMS:
-
Proto-oncogene that codes for the MCSF (CSF1) receptor
- EGF:
-
Epidermal growth factor
- EGFR:
-
EGF receptor
- EMT:
-
Epithelial–mesenchymal transition
- FAK:
-
Focal adhesion kinase
- FGF:
-
Basic fibroblast growth factor
- FLT3:
-
FMS-like tyrosine kinase 3
- IDH:
-
Isocitrate dehydrogenase
- JAK-2:
-
Janus kinase-2
- LSD:
-
Lysine-specific demethylase
- MAPK:
-
Mitogen-activated protein kinase
- MDM2:
-
Mouse double minute chromosome 2
- miR:
-
MicroRNAs
- NDAT:
-
Nano-diamino-tetrac
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NPM:
-
Nucleophosmin
- P-bi-TAT:
-
Polyethylene glycol (PEG) covalently bonded with two bi-tri-azole tetraiodothyroacetic acid molecules
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed death-ligand 1
- PDGF:
-
Platelet-derived growth factor
- PEG:
-
Polyethylene glycol
- PI3K:
-
Phosphatidylinositol 3-kinase
- STAT:
-
Signal transducer and activator of transcription
- Tetrac:
-
Tetraiodothyroacetic acid
- TGFβ:
-
Transforming growth factor β
- TK:
-
Tyrosine kinase
- T4:
-
l-Thyroxine
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Greenberg PL, Gordeuk V, Issaragrisil S, Siritanaratkul N, Fucharoen S, Ribeiro RC. Major hematologic diseases in the developing world—new aspects of diagnosis and management of thalassemia, malarial anemia, and acute leukemia. Hematology Am Soc Hematol Educ Program. 2001:479–98.
Gocek E, Marcinkowska E. Differentiation therapy of acute myeloid leukemia. Cancers (Basel). 2011;3:2402–20.
American Cancer Society. Key statistics for acute myeloid leukemia (AML). 2019. https://www.cancer.org/cancer/acute-myeloid-leukemia/about/key-statistics.html. Accessed 1 May 2019.
Deschler B, Lubbert M. Acute myeloid leukemia: epidemiology and etiology. Cancer. 2006;107:2099–107.
Estey EH. Treatment of acute myeloid leukemia. Haematologica. 2009;94:10–6.
Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, et al. Combination therapy in combating cancer. Oncotarget. 2017;8:38022–43.
Johansen S, Brenner AK, Bartaula-Brevik S, Reikvam H, Bruserud Ø. The possible importance of β3 integrins for leukemogenesis and chemoresistance in acute myeloid leukemia. Int J Mol Sci. 2018;19:251.
Raab-Westphal S, Marshall JF, Goodman SL. Integrins as therapeutic targets: successes and cancers. Cancers (Basel). 2017;9:110.
Licht JD, Sternberg DW. The molecular pathology of acute myeloid leukemia. Hematology Am Soc Hematol Educ Program. 2005:137–42.
De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6:e441.
Lagunas-Rangel FA, Chávez-Valencia V, Gómez-Guijosa MÁ, Cortes-Penagos C. Acute myeloid leukemia—genetic alterations and their clinical prognosis. Int J Hematol Oncol Stem Cell Res. 2017;11:328–39.
Zenonos K, Kyprianou K. RAS signaling pathways, mutations and their role in colorectal cancer. World J Gastrointest Oncol. 2013;5:97–101.
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74.
Montero J, Letai A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018;25:56–64.
Gibson CJ, Davids MS. BCL-2 antagonism to target the intrinsic mitochondrial pathway of apoptosis. Clin Cancer Res. 2015;21:5021–9.
Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;2:a001107-a.
Kindle KB, Troke PJ, Collins HM, Matsuda S, Bossi D, Bellodi C, et al. MOZ-TIF2 inhibits transcription by nuclear receptors and p53 by impairment of CBP function. Mol Cell Biol. 2005;25:988–1002.
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–66.
Liu S, Shen T, Huynh L, Klisovic MI, Rush LJ, Ford JL, et al. Interplay of RUNX1/MTG8 and DNA methyltransferase 1 in acute myeloid leukemia. Cancer Res. 2005;65:1277–84.
Stucki A, Rivier AS, Gikic M, Monai N, Schapira M, Spertini O. Endothelial cell activation by myeloblasts: molecular mechanisms of leukostasis and leukemic cell dissemination. Blood. 2001;97:2121–9.
Liu Z, Wang F, Chen X. Integrin αvβ3-targeted cancer therapy. Drug Dev Res. 2008;69:329–39.
Guo W, Giancotti FG. Integrin signaling during tumor progression. Nat Rev Mol Cell Biol. 2004;5:816–26.
Song X, Wei Z, Shaikh ZA. Requirement of ERα and basal activities of EGFR and Src kinase in Cd-induced activation of MAPK/ERK pathway in human breast cancer MCF-7 cells. Toxicol Appl Pharmacol. 2015;287:26–34.
Saif A, Kazmi SFA, Naseem R, Shah H, Butt MO. Acute myeloid leukemia: is that all there is? Cureus. 2018;10:e3198.
American Cancer Society. Chemotherapy for acute myeloid leukemia (AML). 2019. https://www.cancer.org/cancer/acute-myeloid-leukemia/treating/chemotherapy.html. Accessed 1 May 2019.
American Cancer Society. Risk factors for acute myeloid leukemia (AML). 2019. https://www.cancer.org/cancer/acute-myeloid-leukemia/causes-risks-prevention/risk-factors.html. Accessed 1 May 2019.
O’Donnell MR, Tallman MS, Abboud CN, Altman JK, Appelbaum FR, Arber DA, et al. Acute myeloid leukemia, version 3.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw. 2017;15:926–57.
Medinger M, Lengerke C, Passweg J. Novel prognostic and therapeutic mutations in acute myeloid leukemia. Cancer Genomics Proteomics. 2016;13:317–29.
Elsevier Inc. Clinical pharmacology: Cytarabine. 2019. https://www.clinicalkey.com/pharmacology/monograph/161?n=Cytarabine, ARA-C. Accessed 1 May 2019.
Elsevier Inc. Clinical pharmacology: Idarubicin. 2019. https://www.clinicalkey.com/pharmacology/monograph/305?n=Idarubicin. Accessed 1 May 2019.
Maurillo L, Buccisano F, Del Principe MI, Sarlo C, Di Caprio L, Ditto C, et al. Treatment of acute myeloid leukemia with 20–30% bone marrow blasts. Mediterr J Hematol Infect Dis. 2013;5:e2013032-e.
American Cancer Society. Cancer facts & Figs. 2017. 2017. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2017/cancer-facts-and-figures-2017.pdf. Accessed 1 May 2019.
Fathi AT, Karp JE. New agents in acute myeloid leukemia: beyond cytarabine and anthracyclines. Curr Oncol Rep. 2009;11:346–52.
• Lee LY, Hernandez D, Rajkhowa T, Smith SC, Raman JR, Nguyen B, et al. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood. 2017;129:257–60 The inhibitory activity of gilteritinib against different forms of FLT3 mutations in leukemia cells was studied with immunoblotting. Gilteritinib showed significant inhibitor activity against different FLT3 mutations including the resistant mutations.
Liu T, Ivaturi V, Sabato P, Gobburu JV, Greer JM, Wright JJ, et al. Sorafenib dose recommendation in acute myeloid leukemia based on exposure-FLT3 relationship. Clin Transl Sci. 2018;11:435–43.
Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–37.
Levis M, Allebach J, Tse K-F, Zheng R, Baldwin BR, Smith BD, et al. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002;99:3885–91.
Wu M, Li C, Zhu X. FLT3 inhibitors in acute myeloid leukemia. J Hematol Oncol. 2018;11:133.
Levis M. Midostaurin approved for FLT3-mutated AML. Blood. 2017;129:3403–6.
Kampa-Schittenhelm KM, Heinrich MC, Akmut F, Dohner H, Dohner K, Schittenhelm MM. Quizartinib (AC220) is a potent second generation class III tyrosine kinase inhibitor that displays a distinct inhibition profile against mutant-FLT3, -PDGFRA and -KIT isoforms. Mol Cancer. 2013;12:19.
Kampa-Schittenhelm KM, Frey J, Haeusser LA, Illing B, Pavlovsky AA, Blumenstock G, et al. Crenolanib is a type I tyrosine kinase inhibitor that inhibits mutant KIT D816 isoforms prevalent in systemic mastocytosis and core binding factor leukemia. Oncotarget. 2017;8:82897–909.
Mathew NR, Baumgartner F, Braun L, O’Sullivan D, Thomas S, Waterhouse M, et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat Med. 2018;24:282.
Lange A, Jaskula E, Lange J, Dworacki G, Nowak D, Simiczyjew A, et al. The sorafenib anti-relapse effect after alloHSCT is associated with heightened alloreactivity and accumulation of CD8+ PD-1+(CD279+) lymphocytes in marrow. PLoS One. 2018;13:e0190525.
Zhao W, Zhang T, Qu B, Wu X, Zhu X, Meng F, et al. Sorafenib induces apoptosis in HL60 cells by inhibiting Src kinase-mediated STAT3 phosphorylation. Anti-Cancer Drugs. 2011;22:79–88.
Feldmann F, Schenk B, Martens S, Vandenabeele P, Fulda S. Sorafenib inhibits therapeutic induction of necroptosis in acute leukemia cells. Oncotarget. 2017;8:68208.
Wander SA, Levis MJ, Fathi AT. The evolving role of FLT3 inhibitors in acute myeloid leukemia: quizartinib and beyond. Ther Adv Hematol. 2014;5:65–77.
DiNardo CD, Stone RM, Medeiros BC. Novel therapeutics in acute myeloid leukemia. Am Soc Clin Oncol Educ Book. 2017;37:495–503.
Sutamtewagul G, Vigil CE. Clinical use of FLT3 inhibitors in acute myeloid leukemia. Onco Targets Ther. 2018;11:7041–52.
Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7:165–76.
U.S. Food and Drug Administration. Novel drug approvals for 2018. 2018. https://www.fda.gov/drugs/developmentapprovalprocess/druginnovation/ucm592464.htm. Accessed 1 May 2019.
Cucchi DG, Denys B, Kaspers GJ, Janssen JJ, Ossenkoppele GJ, de Haas V, et al. RNA-based FLT3-ITD allelic ratio is associated with outcome and ex vivo response to FLT3 inhibitors in pediatric AML. Blood. 2018;131:2485–9.
Mori M, Kaneko N, Ueno Y, Yamada M, Tanaka R, Saito R, et al. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Investig New Drugs. 2017;35:556–65.
Short NJ, Kantarjian H, Ravandi F, Daver N. Emerging treatment paradigms with FLT3 inhibitors in acute myeloid leukemia. Ther Adv Hematol. 2019;10:2040620719827310.
Stone RM. Which new agents will be incorporated into frontline therapy in acute myeloid leukemia? Best Pract Res Clin Haematol. 2017;30:312–6.
Myers RA, Wirth S, Williams S, Kiel PJ. Enasidenib: An oral IDH2 inhibitor for the treatment of acute myeloid leukemia. J Adv Pract Oncol. 2018;9:435–40.
Abou Dalle I, DiNardo CD. The role of enasidenib in the treatment of mutant IDH2 acute myeloid leukemia. Ther Adv Hematol. 2018;9:163–73.
Tang A, Gao K, Chu L, Zhang R, Yang J, Zheng J. Aurora kinases: novel therapy targets in cancers. Oncotarget. 2017;8:23937–54.
Willems E, Dedobbeleer M, Digregorio M, Lombard A, Lumapat PN, Rogister B. The functional diversity of Aurora kinases: a comprehensive review. Cell Div. 2018;13:7.
Kim S-J, Jang JE, Cheong J-W, Eom J-I, Jeung H-K, Kim Y, et al. Aurora A kinase expression is increased in leukemia stem cells, and a selective Aurora A kinase inhibitor enhances Ara-C-induced apoptosis in acute myeloid leukemia stem cells. Korean J Hematol. 2012;47:178–85.
Yang Y, Shen Y, Li S, Jin N, Liu H, Yao X. Molecular dynamics and free energy studies on Aurora kinase A and its mutant bound with MLN8054: insight into molecular mechanism of subtype selectivity. Mol BioSyst. 2012;8:3049–60.
Wang X, Sinn AL, Pollok K, Sandusky G, Zhang S, Chen L, et al. Preclinical activity of a novel multiple tyrosine kinase and aurora kinase inhibitor, ENMD-2076, against multiple myeloma. Br J Haematol. 2010;150:313–25.
Melichar B, Adenis A, Lockhart AC, Bennouna J, Dees EC, Kayaleh O, et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol. 2015;16:395–405.
Barr PM, Li H, Spier C, Mahadevan D, LeBlanc M, Ul Haq M, et al. Phase II intergroup trial of alisertib in relapsed and refractory peripheral T cell lymphoma and transformed mycosis fungoides: SWOG 1108. J Clin Oncol. 2015;33:2399–404.
Lowenberg B, Muus P, Ossenkoppele G, Rousselot P, Cahn JY, Ifrah N, et al. Phase 1/2 study to assess the safety, efficacy, and pharmacokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leukemia. Blood. 2011;118:6030–6.
Kantarjian HM, Martinelli G, Jabbour EJ, Quintas-Cardama A, Ando K, Bay JO, et al. Stage I of a phase 2 study assessing the efficacy, safety, and tolerability of barasertib (AZD1152) versus low-dose cytosine arabinoside in elderly patients with acute myeloid leukemia. Cancer. 2013;119:2611–9.
Soncini C, Carpinelli P, Gianellini L, Fancelli D, Vianello P, Rusconi L, et al. PHA-680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin Cancer Res. 2006;12:4080–9.
U.S. Food and Drug Administration. FDA approves venetoclax in combination for AML in adults. 2018. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm626499.htm. Accessed 1 May 2019.
Elsevier Inc. Clinical pharmacology: Venetoclax. 2019. https://www.clinicalkey.com/pharmacology/monograph/4844?n=Venetoclax. Accessed 1 May 2019.
AbbVie Inc. Highlights of prescribing information (venclexta). 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208573s000lbl.pdf. Accessed 1 May 2019.
Park MH, Hong JT. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells. 2016;5:15.
Annaloro C, Onida F, Saporiti G, Lambertenghi DG. Cancer stem cells in hematological disorders: current and possible new therapeutic approaches. Curr Pharm Biotechnol. 2011;12:217–25.
Shahshahan MA, Beckley MN, Jazirehi AR. Potential usage of proteasome inhibitor bortezomib (Velcade, PS-341) in the treatment of metastatic melanoma: basic and clinical aspects. Am J Cancer Res. 2011;1:913–24.
Guzman ML, Rossi RM, Karnischky L, Li X, Peterson DR, Howard DS, et al. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005;105:4163–9.
Ji Q, Ding Y-H, Sun Y, Zhang Y, Gao H-E, Song H-N, et al. Antineoplastic effects and mechanisms of micheliolide in acute myelogenous leukemia stem cells. Oncotarget. 2016;7:65012–23.
Darwish NH, Sudha T, Godugu K, Elbaz O, Abdelghaffar HA, Hassan EE, et al. Acute myeloid leukemia stem cell markers in prognosis and targeted therapy: potential impact of BMI-1, TIM-3 and CLL-1. Oncotarget. 2016;7:57811–20.
van Kemenade FJ, Raaphorst FM, Blokzijl T, Fieret E, Hamer KM, Satijn DP, et al. Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B cell non-Hodgkin lymphoma. Blood. 2001;97:3896–901.
Mihara K, Chowdhury M, Nakaju N, Hidani S, Ihara A, Hyodo H, et al. Bmi-1 is useful as a novel molecular marker for predicting progression of myelodysplastic syndrome and patient prognosis. Blood. 2006;107:305–8.
Iwama A, Oguro H, Negishi M, Kato Y, Morita Y, Tsukui H, et al. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 2004;21:843–51.
Kreso A, van Galen P, Pedley NM, Lima-Fernandes E, Frelin C, Davis T, et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat Med. 2014;20:29–36.
Srinivasan M, Bharali DJ, Sudha T, Khedr M, Guest I, Sell S, et al. Downregulation of Bmi1 in breast cancer stem cells suppresses tumor growth and proliferation. Oncotarget. 2017;8:38731–42.
Elsevier Inc. Clinical pharmacology: Gemtuzumab ozogamicin. 2019. https://www.clinicalkey.com/pharmacology/monograph/2471?n=Gemtuzumab Ozogamicin. Accessed 1 May 2019.
•• Chin YT, Wei PL, Ho Y, Nana AW, Changou CA, Chen YR, et al. Thyroxine inhibits resveratrol-caused apoptosis by PD-L1 in ovarian cancer cells. Endocr Relat Cancer. 2018;25:533–45 Authors demonstrated the potential impact of thyroid hormones on cancer apoptosis. T4 inhibited COX-2-dependent apoptosis in ovarian cancer cells by retaining inducible COX-2 with PD-L1 in the cytoplasm. These findings provide new insights into the effect of T4 antagonizing factors on cancer properties.
Cremaschi GA, Cayrol F, Sterle HA, Diaz Flaque MC, Barreiro Arcos ML. Thyroid hormones and their membrane receptors as therapeutic targets for T cell lymphomas. Pharmacol Res. 2016;109:55–63.
Davis PJ, Sudha T, Lin HY, Mousa SA. Thyroid hormone, hormone analogs, and angiogenesis. Compr Physiol. 2015;6:353–62.
Lin HY, Chin YT, Yang YC, Lai HY, Wang-Peng J, Liu LF, et al. Thyroid hormone, cancer, and apoptosis. Compr Physiol. 2016;6:1221–37.
Pinto M, Soares P, Ribatti D. Thyroid hormone as a regulator of tumor induced angiogenesis. Cancer Lett. 2011;301:119–26.
Shinderman-Maman E, Cohen K, Weingarten C, Nabriski D, Twito O, Baraf L, et al. The thyroid hormone-αvβ3 integrin axis in ovarian cancer: regulation of gene transcription and MAPK-dependent proliferation. Oncogene. 2016;35:1977–87.
Bailey EB, Tantravahi SK, Poole A, Agarwal AM, Straubhar AM, Batten JA, et al. Correlation of degree of hypothyroidism with survival outcomes in patients with metastatic renal cell carcinoma receiving vascular endothelial growth factor receptor tyrosine kinase inhibitors. Clin Genitourin Cancer. 2014;ed2015:e131–7.
Cristofanilli M, Yamamura Y, Kau SW, Bevers T, Strom S, Patangan M, et al. Thyroid hormone and breast carcinoma. Primary hypothyroidism is associated with a reduced incidence of primary breast carcinoma. Cancer. 2005;103:1122–8.
Nelson M, Hercbergs A, Rybicki L, Strome M. Association between development of hypothyroidism and improved survival in patients with head and neck cancer. Arch Otolaryngol Head Neck Surg. 2006;132:1041–6.
Schmidinger M, Vogl UM, Bojic M, Lamm W, Heinzl H, Haitel A, et al. Hypothyroidism in patients with renal cell carcinoma: blessing or curse? Cancer. 2011;117:534–44.
Davis PJ, Goglia F, Leonard JL. Nongenomic actions of thyroid hormone. Nat Rev Endocrinol. 2016;12:111–21.
Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139–70.
Davis PJ, Glinsky GV, Lin H-Y, Leith JT, Hercbergs A, Tang H-Y, et al. Cancer cell gene expression modulated from plasma membrane integrin αvβ3 by thyroid hormone and nanoparticulate tetrac. Front Endocrinol (Lausanne). 2015;5:240.
Yi H, Zeng D, Shen Z, Liao J, Wang X, Liu Y, et al. Integrin alphavbeta3 enhances β-catenin signaling in acute myeloid leukemia harboring Fms-like tyrosine kinase-3 internal tandem duplication mutations: implications for microenvironment influence on sorafenib sensitivity. Oncotarget. 2016;7:40387–97.
Gruszka AM, Valli D, Restelli C, Alcalay M. Adhesion deregulation in acute myeloid leukemia. Cells. 2019;8:66.
Shi S, Zhou M, Li X, Hu M, Li C, Li M, et al. Synergistic active targeting of dually integrin alphavbeta3/CD44-targeted nanoparticles to B16F10 tumors located at different sites of mouse bodies. J Control Release. 2016;235:1–13.
Weingarten C, Jenudi Y, Tshuva RY, Moskovich D, Alfandari A, Hercbergs A, et al. The interplay between epithelial–mesenchymal transition (EMT) and the thyroid hormones–αvβ3 axis in ovarian cancer. Horm Cancer. 2018;9:22–32.
Zhang P, Chen L, Song Y, Li X, Sun Y, Xiao Y, et al. Tetraiodothyroacetic acid and transthyretin silencing inhibit pro-metastatic effect of L-thyroxin in anoikis-resistant prostate cancer cells through regulation of MAPK/ERK pathway. Exp Cell Res. 2016;347:350–9.
Abou-El-Naga AM, Mutawa G, El-Sherbiny IM, Mousa SA. Activation of polymeric nanoparticle intracellular targeting overcomes chemodrug resistance in human primary patient breast cancer cells. Int J Nanomedicine. 2018;13:8153–64.
•• Sudha T, Bharali DJ, Yalcin M, Darwish NH, Debreli Coskun M, Keating KA, et al. Targeted delivery of paclitaxel and doxorubicin to cancer xenografts via the nanoparticle of nano-diamino-tetrac. Int J Nanomedicine. 2017;12:1305–15 Authors discuss the potential impact of therapies that target integrin αvβ3. They demonstrated the feasibility of chemotherapy delivery using a nanoparticle system that achieved higher intratumoral concentrations and improved antitumor efficacy of chemotherapies than via the conventional administration route of these agents.
Mousa SA, Glinsky GV, Lin HY, Ashur-Fabian O, Hercbergs A, Keating KA, et al. Contributions of thyroid hormone to cancer metastasis. Biomedicines. 2018;6:89.
Cai X, Zhu H, Li Y. Pkczeta, MMP2 and MMP9 expression in lung adenocarcinoma and association with a metastatic phenotype. Mol Med Rep. 2017;16:8301–6.
Hong M, Cheng H, Song L, Wang W, Wang Q, Xu D, et al. Wogonin suppresses the activity of matrix metalloproteinase-9 and inhibits migration and invasion in human hepatocellular carcinoma. Molecules. 2018;23:384.
Tauro M, Lynch CC. Cutting to the chase: how matrix metalloproteinase-2 activity controls breast-cancer-to-bone metastasis. Cancers (Basel). 2018;10:185.
Cohen K, Flint N, Shalev S, Erez D, Baharal T, Davis PJ, et al. Thyroid hormone regulates adhesion, migration and matrix metalloproteinase 9 activity via αvβ3 integrin in myeloma cells. Oncotarget. 2014;5:6312–22.
Bridoux A, Khan RA, Chen C, Cheve G, Cui H, Dyskin E, et al. Design, synthesis, and biological evaluation of bifunctional thyrointegrin inhibitors: new anti-angiogenesis analogs. J Enzyme Inhib Med Chem. 2011;26:871–82.
Mousa SA, Mousa AS. Angiogenesis inhibitors: current & future directions. Curr Pharm Des. 2004;10:1–9.
Davis PJ, Mousa SA, Lin HY. Tetraiodothyroacetic acid (tetrac), integrin αvβ3 and disabling of immune checkpoint defense. Future Med Chem. 2018;10:1637–9.
Mousa SA, Rajabi M, inventors. Non-cleaveable polymer conjugated with αvβ3 integrin thyroid antagonists. USA patent 10, 201,616. 2019.
Rajabi M, Godugu K, Sudha T, Bharali DJ, Mousa SA. Triazole modified tetraiodothyroacetic acid conjugated to polyethylene glycol: High affinity thyrointegrin αvβ3 antagonist with potent anticancer activities in glioblastoma multiforme. Bioconj Chem. 2019;30:3087-97.
Funding
There is no funding to report.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Shaheedul A. Sami declares that he has no conflict of interest.
Noureldien H. E. Darwish declares that he has no conflict of interest.
Amanda N. M. Barile declares that she has no conflict of interest.
Shaker A. Mousa was issued a patent that is owned by NanoPharmaceuticals LLC, and he owns stock in NanoPharmaceuticals LLC, which is developing anticancer drugs.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Leukemia
Rights and permissions
About this article

Cite this article
Sami, S.A., Darwish, N.H.E., Barile, A.N.M. et al. Current and Future Molecular Targets for Acute Myeloid Leukemia Therapy. Curr. Treat. Options in Oncol. 21, 3 (2020). https://doi.org/10.1007/s11864-019-0694-6
Published:
DOI: https://doi.org/10.1007/s11864-019-0694-6