
Abstract
Background
Alzheimer’s disease and other dementias are the fourth largest contributors to neurological disability and the second largest contributor to deaths from neurological disease. Described in the 1980s as ‘the silent epidemic’ these disorders principally, though not exclusively, affect persons 80 years or older, and in developed countries, this ‘old old’ population continues to grow. Definitive diagnosis of the underlying cause of the neurodegenerative disease relies on neuropathological evaluation.`
Aims
Herein, we review the sampling methods, analysis and interpretation of both pathological and immunocytochemical techniques in the diagnostic assessment of neurodegenerative disease.
Findings
Neurodegenerative disorders are characterised by accumulation of pathologically altered protein in the human brain, and in some cases, in the peripheral tissues. Whilst it is suggested that a comprehensive review of the patient’s clinical history, cognition and behaviour, together with a full clinical examination and radiological analysis, should lead to a high degree of confidence in the clinical diagnosis, the view persists that underlying pathology can only be predicted on clinical grounds especially in Alzheimer’s disease, vascular brain injury and diffuse Lewy body disease with only limited accuracy.
Conclusions
Neuropathological assessment of well characterised clinical cases provides accurate data on the prevalence of neurodegenerative diseases. This will aid future biomarker, neuroimaging studies and clinical trials focussed on population based cohorts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dementia, like most late-life chronic diseases, is a syndrome presenting with complex symptomatology, due to multiple aetiologies [1]. Described in the 1980s as the ‘silent epidemic’, it principally affects persons 80 years or older, and in developed countries, such as Ireland, this ‘old old’ population continues to grow. The vast majority of dementia cases, especially those occurring very late in life, tend to involve a mixture of Alzheimer’s disease (AD), vascular associated dementia (VaD) and other degenerative factors [1]. Neurological disorders are an important cause of disability and death worldwide. Alzheimer’s and other dementias are the fourth largest contributors to disability-associated life years (DALYs) but the second largest contributors to deaths from neurological disease [2]. It has been suggested that the incidence of dementia is declining in high-income countries [3] but the most striking change has been the doubling of patients who die, or are disabled from AD, and other dementias over the past 25 years [3].
Clearly an ageing demographic will drive an increase in the number of dementia cases. Clinical accuracy in the diagnosis of these cases is important; not only for therapeutic and scientific studies but also as a guide to prognosis, organising clinical care and recruitment to clinical trials. Clinical diagnostic accuracy may be high, when all features and investigations, are taken into consideration [4], but nonetheless, the view persists that underlying pathology can be predicted exclusively on clinical grounds with only limited accuracy. This is particularly true with respect to the sub classification of dementias and differing Parkinsonian phenotypes [5].
Despite the increase in sensitivity of in vivo diagnostic markers, the ‘gold standard’ for definitive diagnosis of neurodegenerative diseases (NDD’s) remains a neuropathological evaluation. This is particularly true with respect to AD, diffuse Lewy body disease (DLBD) and VaD, which are the major contributors to dementia in community- and population-based studies [6]—though even in these conditions the neuropathologist, to date, could not truly provide an absolute diagnosis, but only predict the ‘likelihood’ that AD or DLBD was the responsible entity [7]. Neuropathological evaluation may be performed as part of a neuropathology autopsy diagnostic service or may be performed in the interests of brain banking for research purposes.
NDDs are classified as dysfunction and loss of neurones, associated with the deposition of pathologically altered proteins that accumulate in the human brain but also in peripheral tissues. This distinct involvement of functional systems largely defines the clinical presentation [8]. In a process that is self-perpetuating, and likened to that observed in prion diseases, progressive seeded protein aggregate, and spread to interconnected neurones and adjacent glial cells [9]. NDDs are subdivided based on clinical presentation, protein deposition, cellular and subcellular pathology. With any neurodegenerative syndrome, it is possible to define but not necessarily explain anatomical, cellular and protein vulnerability—all of which may be altered if there is a genetic component [8].
The aim of this review is to outline how a neuropathological diagnosis is reached based on macroscopic examination of the brain, assessment of vascular brain injury (vBi) and analysis of protein deposition.
Clinical information
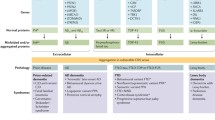
There is an increasing need to diagnose NDDs in a timely fashion. Clinical symptoms often overlap during the course of disease thus clinical classification is helpful in determining whether cognition/behavioural issues or a movement disorder is the predominant problem. This will also have directed clinical investigations and imaging studies which are helpful in some but not all conditions (Tables 1 and 2). Presentation with dementia, visual hallucinations and later bradykinesia may suggest DLBD, whereas behavioural problems may suggest a frontotemporal dementia. Involvement of organ systems other than the central nervous system (CNS) may also raise suspicion for particular disorders, e.g. anosmia and constipation in Parkinson’s disease, and cardiomyopathy in Fredric’s ataxia.
The documented neuroradiological and electrophysiological abnormalities should be interpreted specifically, with respect to the stage of the disease in which they were demonstrated [10]. In frontotemporal dementias, for example, the distribution of disease is asymmetrical at disease onset [11].
Autopsy
With respect to the autopsy, consent is a prerequisite. It is beyond the scope of this article to deal with the issues involved but emphasis should be placed on the need to retain the brain, and in some cases, the spinal cord. It is also essential to document what arrangements are to be made with the organ subsequently.
Although a detailed neuropathologic investigation is frequently performed, and a diagnosis reached without a general autopsy, the quality of the report is enhanced if these details are taken into consideration. Before slicing or fixing the brain, it is important to determine whether or not frozen samples should be taken. This is routine in brain banking but not for diagnostic purposes.
Specific sampling procedures are used for examination of the brain that includes neocortical regions, basal ganglia, thalamus, brain stem and cerebellum [8, 10]. Macroscopic abnormalities are routinely noted: the circle of Willis is carefully examined to assess the degree of atheroma affecting the vessels, and this is graded from mild to severe. Some abnormalities that may be detected on gross inspection are shown in Fig. 1.

Examples of abnormalities seen on gross pathology. a Saggital section of the brain showing frontal atrophy. b Depigmentation of the substantia nigra. c Coronal section of the brain showing putaminal discolouration
The techniques and use of the various proteins for diagnostic purposes have been extensively reviewed [8, 10]. In our laboratory, we routinely cut 4 μm sections and stain them with haematoxylin and eosin (H&E). Following examination of the H&E sections, the following immunocytochemical stains are carried out where appropriate: Polyclonal TAU (Innu genetics 1:100), 3R-Tau (clone and dilution), 4R-Tau (clone and dilution) Amyloid-β protein (Dako 1:100), alpha Synuclein (Inviterogen 1:500), GFAP (Dako 1:1500), Polyclonal Ubiquitin (Leica 1:250), Transactive response DNA binding protein 43 (TDP-43) (Proteintec 1:1500), Fused in Sarcoma (FUS) (Sigma 1:600), Polyclonal P62 (Santa Cruz, 1:1000), CD68 (clone and dilution) and Luxol fast blue/H + E (clone and dilution).
Vascular brain injury
This is not infrequent in the brains of the elderly but its impact on cognition is less clear. Whilst prevalence rates are high in clinical studies, it is rarely found to be the neuropathological correlate of clinical dementia in post mortem studies [12]. The three vascular lesions that contribute to vascular cognitive impairment are the following: (i) atherosclerosis (most commonly the basilar artery and the circle of Willis); (ii) small vessel disease (which includes small vessel atherosclerosis, lipohyalinosis and arteriolosclerosis); and (iii) cerebral amyloid angiopathy. These can cause infarcts—ischaemic or haemorrhagic or white matter lesions.
Cerebrovascular disease may result in pure VaD without widespread neurodegenerative pathology such as AD or DLBD. It can be classified into three major forms—multi-infarct, strategic or subcortical vascular encephalopathy. The distinction between AD, VaD and mixed AD/VaD remains controversial, and it has been suggested that the oligaemia produced by atherosclerosis may increase accumulation of β-amyloid, which in turn aggravates the oligaemia [13, 14].
Proteins
Protein is deposited either extracellular (which is the case with β- amyloid (Aβ) and prion protein, or intracellular (Tau, α-Synuclein, TDP-43, FUS), and those associated with trinucleotide repeat disorders or rare hereditary diseases (Figs. 2–4 and Table 3). Our approach to prion disease has been discussed in previous publications [15, 16].

a Pick bodies. Haematoxylin and eosin x 60. b Pick bodies. 3 R –tau x 60. c Globose tangles – PSP . Haematoxylin and eosin x 60. d Tufted astrocytes (arrowhead) and globose tangles. 4R-tau x 60
Tau
With respect to intracellular deposition, we will deal first with tau. Tau, a microtubule-associated protein functions in assembly and stabilisation of the neuronal microtubule network. Abnormal aggregates of hyperphosphorylated tau protein occur in NDDs [17]. These tauopathies are characterised by the deposition of abnormal tau protein in neurones or glial cells (astrocytes or oligiodendrocytes) in the brain. They are classified as primary; where tau is the only abnormal protein. Primary taupathies include Pick’s disease (PiD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD), primary age-related tauopathy (PART) previously known as neurofibrillary tangle-only dementia (NFT-dementia) and a recently described entity globular glial tauopathy (GGT) [8]. AD is regarded as a secondary tauopathy as Aβ is deposited in addition to tau.
Human tau proteins are encoded by a single gene consisting of 16 exons on chromosome 17q21 and the CNS isoforms (3-repeat [R] and 4-repeat [R]-tau) are generated by alternative mRNA splicing of these exons. In AD, both 3R- and 4R-tau are present, whereas in PiD only 3R-tau is present, and in PSP, CBD and AGD 4R-tau aggregate [17].
Neuronal tau deposits include pretangles, NFT (Fig. 3) and Pick Bodies (Fig. 2). Astrocytes show a variety of inclusions including tufted astrocytes (PSP) and astrocytic plaques (CBD). Oligodendrocyte inclusions include coiled bodies and globular oligodendrocytes inclusions. These tau pathologies may be observed as secondary phenomena in a wide range of other neurodegenerative diseases, e.g. in Huntington’s disease brains [18].

a Neuronal inclusions cortico-basal degeneration. Haematoxylin and eosin x 60. b 4 R-tau inclusions parenchymal (arrowhead) and neuronal (arrow) CBD x 60 c Neurofibrillary tangles (arrowhead) and neuritic plaques (arrow). Tau x 60. d Amyloid angiopathy (arrow) and amyloid plaques (arrowhead) BA4 x 60
Chronic traumatic encephalopathy (CTE), a recently described tauopathy is a progressive NDD, triggered by repetitive mild traumatic injury. CTE is distinguished from other tauopathies by a distinctive pattern of tau deposition; beginning in the superficial cortex and spreading eventually to the medial temporal lobe, diencephalon and brain stem [19].
AD, a secondary tauopathy, is the most common NDD. It is characterised by the extracellular deposition of Aβ fibrils and the intracellular accumulation of abnormally phosphorylated tau protein. Aβ may deposit in the parenchyma in the form of plaques and in the vessel walls as cerebral amyloid angiopathy (CAA). The characteristic lesions of AD include NFTs and senile plaques (SP). NFTs are observed in limbic regions early in the disease and follow a characteristic staging system [20, 21]. Senile plaques on the other hand are extracellular deposits of Aβ. These may take the form of neuritic plaques (NP), with dystrophic neurites at the centre, or diffuse plaques (DP) without. NPs are considered to be most closely related to neuronal injury. Assessment of AD neuropathological change is made using an ABC score that incorporates histological assessment of Aβ deposits, staging of neurofibrillary tangles and scoring of NPs [20,21,22,23,24,25].
Whilst AD is the commonest cause of dementia, and may exist in a ‘pure ‘form, it may co-exist with DLBD, VaD and hippocampal sclerosis (HS) as well as AGD and TDP-43- inclusions [25]. Whilst it may be difficult to assess how these have contributed to the cognitive decline in a given case, it is nonetheless important to document their presence.
α-synuclein
Parkinson’s disease (PD), dementia with Lewy bodies and multiple systems atrophy (MSA) are the major diseases associated with the deposition of α-Synuclein. This presynaptic protein leaves its binding site in presynaptic boutons to aggregate as Lewy body neurites or Lewy bodies (Fig. 4) [26]. Deposition of α-synuclein may be neuronal or glial. The former includes DLBD and PD whereas glial inclusions are characteristic of MSA. Clinical data (movement disorder or cognitive decline as early symptoms) and pathological distribution of Lewy body and neurite distribution are required to classify Lewy body diseases. The clinical distinction between PD and DLBD rests on the time interval between the onset of motor symptoms and dementia with a minimum 1 year interval being required for a diagnosis of PD as opposed to DLBD [27]. In PD, the earliest involvement is seen in the medulla and from there involves lower brain stem nuclei with eventual involvement of the cerebral cortex [27]. Non-motor symptoms such as anosmia may antedate the motor symptoms of PD by some years, and this is thought to be due to α-synuclein deposition in olfactory structures [26, 28]. Lewy body deposition in the brain is assessed on a semi-quantitative stage as brain stem, limbic or neocortical [29].

a Lewy bodies in the substantia nigra. Haematoxylin and eosin × 60. b Lewy bodies in the substantia nigra. Alpha-syneuclin × 60. c Haematoxylin and eosin x 60. d Glial cytoplasmic inclusion. Alpha-syneuclin × 60
MSA, the rarest of the syneuclinopathies differs from PD and DLBD in its clinical presentation (aggressive with either cerebellar [MSA–C] or Parkinsonian features [MSA-P] and its pathology (putaminal discolouration [Fig. 1c] and glial cytoplasmic inclusions [Fig. 4c, d]) [30].
TDP-43
This is a major component of tau negative, ubiquitin-positive inclusion that characterise amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). ALS is the most frequent motor neurone disease and FTLD is, after AD, the commonest cause of dementia under the age of 65. TDP-43 belongs to the group of RNA-binding domain proteins and is involved in multiple cellular processes. Familial forms of FTLD are associated with various genes such as granulin (GRN), valosin-containing protein (VCP) TARDBP and chromosome 9 open reading frame 72 (C9orf72)—all now collectively referred to as TDP-proteinopathy [31]. The spectrum of inclusions includes both neuronal (cytoplasmic, intranuclear and dystrophic neurites) and cytoplasmic (glial cell inclusions—mostly in oligodendrocytes). TDP-43 pathology may also co-exist with other pathologies such as AD, DLBD and HS. Four subtypes are recognised (A-D) based on the predominance and distribution of neuronal inclusions. Characteristic patterns of TDP-43 pathology progression have been described in both behavioural variant FTD (bv-FTD) and ALS [8].
FUS
Fused in sarcoma protein also belongs to a family of RNA-binding proteins. In post mortem brains of ALS and FTD patients, FUS or TDP-43 appear to be partially lost from the nucleus in neuronal and glial cells and aggregate both in the cytoplasm and rarely in the nucleus [32].
Discussion
Neurodegenerative diseases represent a spectrum of disorders characterised by accumulation of abnormal proteins and are subdivided based on clinical presentation, protein deposition, cellular and sub-cellular pathology. AD, vBI and DLBD are the major contributors to dementia in population-based studies [6]. Although we have come a long way in understanding these disorders, there still exist cases where, even with hindsight, the pathology is unexpected [4]. Likewise, the particular molecular mechanisms that trigger the initial conversion of normal protein into pathological aggregates are still unknown [9].
In a previous study [5], we found strong concordance (86%) between the clinical diagnosis and final pathological diagnosis supporting the observation that pathology can be predicted on clinical grounds with a high degree of accuracy [4]. Most discordant cases were in the dementia group, in particular AD may mimic a range of other disorders. Discordance mainly arises due to phenotypic variation in patients sharing a common pathology and is exacerbated when the time interval between clinical diagnosis and death is protracted. These phenotypic variations are particularly common in AD—the commonest cause of dementia. Neuroimaging may be helpful, and even predictive in many cases of dementia, and in particular AD, but currently there are no validated neuroimaging or biomarker protocols for the common dementias such as vBI or LBD [6, 33].
Whilst a comprehensive discussion is beyond the scope of this article, a number of in vivo biomarkers are currently in development. These include (but are not limited to) CSF and blood biomarkers in AD [34]. These are primarily used to estimate tau, Aβ and neurofilament light chains. In PD, other modalities such as skin biopsy and CSF, in combination with both neuroimaging and genetics, are in development but are not presently routinely implemented in clinical practice [35]. In pathology, we have widely accepted protocols for assessing disease progression in both AD and DLB but no widely accepted criteria for assessing vBI [12].
In addition to biomarkers, genetic studies have provided additional insights into a number of NDDs. These have included a greater understanding of the genetics of FUS [36], which relates to both FTD and ALS. In addition, previous cases of Huntington’s disease were considered to be ‘gene negative’ but recent evidence shows that the C9orf72 mutation may account for the phenotype [37]. In other more common NDDs, a number of additional risk loci have been reported such as in PD [38] and AD [39]. These and other genetic mutations that may be tested may improve the diagnosis of NDDs. In addition, post mortem tissue may be used for genetic analysis [40], facilitating genetic testing in tissue from the brain bank.
Clinicopathological discordance underscores the fact that patients sharing a similar pathology are not clinically homogenous [4]. In some AD patients, memory impairment may be the initial and dominant clinical feature. In other NDDs, memory disorders may emerge as part of cognitive disturbance that includes difficulties with language, calculation, etc. [4]. Memory problems may even be absent in some AD cases. In these atypical variants, the diagnostic boundaries may be blurred. Likewise, some patients although clinically well, may have a pathological burden of both α-amyloid and tau—a burden that without knowledge of the patient’s cognitive state may lead to an erroneous autopsy diagnosis of AD. In these situations, it has been suggested that larger brains and larger hippocampal volumes are associated with preserved cognitive function during life; despite a high burden of AD neuropathologic abnormalities at autopsy [41]. AD with a rapidly progressive course may even be confused with sCJD [42].
The phenotypes of AD and DLBD have numerous overlapping features. The presence of AD neuropathologic abnormalities modifies the clinical features of DLBD, making it harder to distinguish DLBD from AD during life [43]. Both diseases can have the ‘core’ features of DLBD depending on the stage of clinical dementia; so at various points in the natural history the two diseases may be clinically indistinguishable [43]. Possible explanations for this are that patients with DLBD and AD (high Braak stages) display a pattern of clinical and behavioural changes so typical of AD, that the core features of DLBD are essentially masked. It is also possible that the extent of α-syneuclin pathology may be less extensive in patients with high Braak scores [43]. Consensus criteria for the diagnosis of DLBD were published in 1996 and revised in 2005 to include dopamine transporter imaging (DAT) [27, 44]. Good correlation between clinical diagnosis including DAT scanning and autopsy diagnosis have been reported even with a gap of nearly 3 years between scan and autopsy [45]. This may be better than clinical diagnosis alone, which may misclassify patients [46]. The recommendations have been updated again and distinguish between clinical features and diagnostic biomarkers [47]. These remain to be validated by autopsy studies.
Cerebrovascular disease is increasingly recognised as a cause of cognitive impairment and dementia in later years, either alone or in combination with AD, or other pathologies [48]. Vascular diseases encompass a heterogenous group of disorders producing different types of cerebrovascular lesions contributing to cognitive decline, and later, development of dementia. Whilst prevalence rates are high in clinical studies, VaD is rarely found to be the neuropathological correlate of cognitive decline [12]. The terminology has varied over the years but is generally now referred to as vascular cognitive impairment (VCI) or vascular-associated dementia (VaD) [48]. VCI/VaD may share many risk factors with AD including increasing prevalence with age, and in many patients they co-exist [33]. Imaging evidence of cerebrovascular disease is required before making a clinical diagnosis of VaD. Absence of vascular damage on magnetic resonance imaging (MRI) makes it an unlikely cause of cognitive impairment, by corollary; extensive vascular changes are likely to produce significant effects. Clinical diagnostic difficulties arise mainly in determining whether mild or moderate vascular changes are sufficient to explain the clinical picture. This is particularly difficult in older individuals where mixed vascular and neurodegenerative diseases are common [33].
The prevalence of VCI/VaD varies in clinical population-based series. The prevalence rates of VaD are unlikely to be accurate, because even the best clinical diagnostic criteria show only moderate sensitivity and variable specificity [12, 48]. These should also be interpreted cautiously due to referral bias. In autopsy series, the prevalence of VaD also varies tremendously, and this may reflect the lack of internationally accepted consensus criteria for the diagnosis of VaD [12, 48]. In autopsy studies of elderly people, the prevalence of pure VaD (without other pathologies) varied from 5 to 78% in the oldest-old. AD alone was present in some, whilst mixed cases, i.e. AD + VaD or DLBD, were present in 74–93% and 9–28%, respectively [48]. VaD and VCI are potentially treatable diseases, so studies focussing on its prevalence and risk factors are important to guide public health policies. Standardised neuropathological criteria including post mortem MRI would be helpful, in order to correlate the pathological findings with the imaging abnormalities ante mortem.
Characteristic patterns of protein distribution occur in the various neurodegenerative diseases over time and are associated with increasing severity of the clinical disease [9]. This has enabled staging patterns for these diseases. This pattern was first established with tau protein in AD, where aggregates first appear in the locus coeruleus of the pons followed by the temporal lobe and finally the neocortex. In other tau diseases such as CTE, tau pathology originates in perivascular spaces within the depths of the sulci and subsequently spreads to large areas of the neocortex, basal ganglia, brainstem and spinal cord. In contrast to tau, Aβ plaques in AD begin in the cortex and later spread to the brainstem. Observations from in vitro and in vivo studies suggest that Aβ may drive tau accumulation [49] and that tau may induce Aβ toxicity [50].
In contrast to tau, α-synuclein, the key protein PD and DLBD is first detected in ventral areas such as the olfactory bulb and nucleus and dorsal motor nucleus of the vagus in the medulla. It then spreads to the pons, midbrain and eventually to the neocortex [51]. Only in more advanced stages of PD does α-synuclein aggregation cause the loss of midbrain dopaminergic neurones in the pars compacta of the midbrain. Loss of neurones in this has been linked to the classic motor symptoms of PD such as tremor, rigidity and bradykinesia [9]. Olfactory involvement is clinically reflected by hyposmia. Deposition of α-syneuclin has also been found outside the CNS in neurones of Meissner’s and Auerbach’s plexi of the enteric nervous system and could explain early symptoms, such as constipation [52].
Four sequential stages of TDP-43 accumulation in ALS have been described, whereby the protein appears to spread from the neocortex towards the spinal cord and brain stem [9]. TDP aggregation has also been described in AD and CTE. It is thought to spread by different pathways in different neurodegenerative diseases.
In conclusion, although we know that neurodegenerative diseases are associated with accumulation of abnormal protein in various sites, and we can stage disease progression, the molecular triggers that result in protein aggregation remain unknown. Lastly, although clinicopathologic concordance is good in the common neurodegenerative disorders such as AD, VaD and DLBD, there are still cases, which even with hindsight prove difficult to achieve an accurate clinical diagnosis, highlighting the need for neuropathological evaluation in any uncertain cases.
References
Larson EB, Yaffe K, Langa KM (2013) New insights into the dementia epidemic. NEJM 369(24):2275–2277. https://doi.org/10.1056/NEJMp1311405
GBD 2015 Neurological Disorders Collaborator Group (2017) Global, regional, and national burden of neurological disorders during 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. The Lancet Neurology 16:877–897
Prince M, Ali GC, Guerchet M et al (2016) Recent global trends in the prevalence and incidence of dementia, and survival with dementia. Alzheimers Res Ther 8(1):23. https://doi.org/10.1186/s13195-016-0188-8
Snowden JS, Thompson JC, Stopford CL et al (2011) The clinical diagnosis of early-onset dementias: diagnostic accuracy and clinicopathological relationships. Brain 134:2478–2492
Brett F, Beausang A, Chalissery A et al (2016) Clinicopathologic concordance in the Dublin Brain Bank 2008-2016. J Alzheimer’s Parkinsonism Dementia 2016;
Sonnen JA, Santa Cruz K, Hemmy LS et al (2011) Ecology of the aging human brain. Arch Neurol 68(8):1049–1056. https://doi.org/10.1001/archneurol.2011.157
Swerdlow RH, Newell KL (2012) “Untangling” the relationship between Alzheimer disease and dementia with Lewy bodies. Neurology 79(19):1938–1939. https://doi.org/10.1212/WNL.0b013e3182735ecf
Kovacs GG (2016) Molecular pathological classification of neurodegenerative diseases: turning towards precision medicine. Int J Mol Sci 17(2). https://doi.org/10.3390/ijms17020189
Brettschneider J, Del Tredici K, Lee VM et al (2015) Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 16(2):109–120. https://doi.org/10.1038/nrn3887
Love S (2004) Post mortem sampling of the brain and other tissues in neurodegenerative disease. Histopathology 44(4):309–317. https://doi.org/10.1111/j.1365-2559.2004.01794.x
Broe M, Hodges JR, Schofield E et al (2003) Staging disease severity in pathologically confirmed cases of frontotemporal dementia. Neurology 60(6):1005–1011. https://doi.org/10.1212/01.WNL.0000052685.09194.39
McAleese KE, Alafuzoff I, Charidimou A et al (2016) Post-mortem assessment in vascular dementia: advances and aspirations. BMC Med 14(1):129. https://doi.org/10.1186/s12916-016-0676-5
Roher AE, Esh C, Kokjohn TA et al (2003) Circle of willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol 23(11):2055–2062. https://doi.org/10.1161/01.ATV.0000095973.42032.44
Iadecola C (2003) Atherosclerosis and neurodegeneration: unexpected conspirators in Alzheimer’s dementia. Arterioscler Thromb Vasc Biol 23:1951–1953
Brett FM, Looby S, Chalissery A et al (2017) Brain biopsies requiring Creutzfeldt-Jakob disease precautions in the Republic of Ireland 2005–2016. Ir J Med Sci. https://doi.org/10.1007/s11845-017-1673-1
Loftus T, Chen D, Looby S et al (2017) CJD surveillance in the Republic of Ireland from 2005 to 2015: a suggested algorithm for referrals. Clin Neuropathol 36(07):188–194. https://doi.org/10.5414/NP301016
Yoshida M (2006) Cellular tau pathology and immunohistochemical study of tau isoforms in sporadic tauopathies. Neuropathology 26(5):457–470. https://doi.org/10.1111/j.1440-1789.2006.00743.x
Fernández-Nogales M, Cabrera JR, Santos-Galindo M et al (2014) Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat Med 20(8):881–885. https://doi.org/10.1038/nm.3617
McKee AC, Stein TD, Kiernan PT et al (2015) The neuropathology of chronic traumatic encephalopathy. Brain Pathol 25(3):350–364. https://doi.org/10.1111/bpa.12248
Braak H, Braak E (1995) Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 16(3):271–278. https://doi.org/10.1016/0197-4580(95)00021-6
Braak H, Braak E (1991) Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 82(4):239–259. https://doi.org/10.1007/BF00308809
Thal DR, Rüb U, Orantes M et al (2002) Phases of a beta-deposition in the human brain and its relevance for the development of AD. Neurology 58(12):1791–1800. https://doi.org/10.1212/WNL.58.12.1791
Mirra SS, Heyman A, McKeel D et al (1991) The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41(4):479–486. https://doi.org/10.1212/WNL.41.4.479
Hyman BT, Phelps CH, Beach TG et al (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8(1):1–13. https://doi.org/10.1016/j.jalz.2011.10.007
Montine TJ, Phelps CH, Beach TG et al (2012) National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 123(1):1–11. https://doi.org/10.1007/s00401-011-0910-3
Braak H, Del Tredici K, Rüb U et al (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24(2):197–121. https://doi.org/10.1016/S0197-4580(02)00065-9
McKeith IG, Dickson DW, Lowe J et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65(12):1863–1872. https://doi.org/10.1212/01.wnl.0000187889.17253.b1
Attems J, Walker L, Jellinger KA (2014) Olfactory bulb involvement in neurodegenerative diseases. Acta Neuropathol 127(4):459–475. https://doi.org/10.1007/s00401-014-1261-7
McKeith IG, Galasko D, Kosaka K et al (1996) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 47(5):1113–1124. https://doi.org/10.1212/WNL.47.5.1113
Barker RA, Williams-Gray CH (2016) Review: the spectrum of clinical features seen with alpha synuclein pathology. Neuropathol Appl Neurobiol 42(1):6–19. https://doi.org/10.1111/nan.12303
Arai T (2014) Significance and limitation of the pathological classification of TDP-43 proteinopathy. Neuropathology 34(6):578–588. https://doi.org/10.1111/neup.12138
Ederle H, Dormann D (2017) TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett 591(11):1489–1507. https://doi.org/10.1002/1873-3468.12646
Harper L, Barkhof F, Scheltens P et al (2014) An algorithmic approach to structural imaging in dementia. J Neurol Neurosurg Psychiatry 85(6):692–698. https://doi.org/10.1136/jnnp-2013-306285
Olsson B, Lautner R, Andreasson U et al (2016) CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. Lancet Neurol 15(7):673–684. https://doi.org/10.1016/S1474-4422(16)00070-3
Delenclos M, Jones DR, McLean PJ et al (2016) Biomarkers in Parkinson’s disease: advances and strategies. Parkinsonism Relat Disord 22(Suppl 1):S106–S110. https://doi.org/10.1016/j.parkreldis.2015.09.048
Deng H, Gao K, Jankovic J (2014) The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol 10(6):337–348. https://doi.org/10.1038/nrneurol.2014.78
Hensman Moss DJ, Poulter M et al (2014) C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology 82(4):292–299. https://doi.org/10.1212/WNL.0000000000000061
Nalls MA, Pankratz N, Lill CM et al (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46(9):989–993. https://doi.org/10.1038/ng.3043
Lambert JC, Ibrahim-Verbaas CA et al (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 45(12):1452–1458. https://doi.org/10.1038/ng.2802
Goelz SE, Hamilton SR, Vogelstein B (1985) Purification of DNA from formaldehyde fixed and paraffin embedded human tissue. Biochem Biophys Res Commun 130(1):118–126. https://doi.org/10.1016/0006-291X(85)90390-0
Erten-Lyons D, Woltjer RL, Dodge H et al (2009) Factors associated with resistance to dementia despite high Alzheimer disease pathology. Neurology 72(4):354–360. https://doi.org/10.1212/01.wnl.0000341273.18141.64
Mead S, Rudge P (2017) CJD mimics and chameleons. Pract Neurol 17(2):113–121. https://doi.org/10.1136/practneurol-2016-001571
Merdes AR, Hansen LA, Jeste DV et al (2003) Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology 60(10):1586–1590. https://doi.org/10.1212/01.WNL.0000065889.42856.F2
McKeith IG, Dickson DW, Lowe J et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 65:1863–1872
Walker Z, Jaros E, Walker RW et al (2007) Dementia with Lewy bodies: a comparison of clinical diagnosis, FP-CIT single photon emission computed tomography imaging and autopsy. J Neurol Neurosurg Psychiatry 78(11):1176–1181. https://doi.org/10.1136/jnnp.2006.110122
Skogseth R, Hortobágyi T, Soennesyn H et al (2017) Accuracy of clinical diagnosis of dementia with Lewy bodies versus neuropathology. J Alzheimers Dis 59(4):1139–1152. https://doi.org/10.3233/JAD-170274
McKeith IG, Boeve BF, Dickson DW et al (2017) Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology 89(1):88–100. https://doi.org/10.1212/WNL.0000000000004058
Jellinger KA (2013) Pathology and pathogenesis of vascular cognitive impairment—a critical update. Front Aging Neurosci 5:1–19
De Felice FG, Wu D, Lambert MP et al (2008) Alzheimer's disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging 29(9):1334–1347. https://doi.org/10.1016/j.neurobiolaging.2007.02.029
Roberson ED, Scearce-Levie K, Palop JJ et al (2007) Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316(5825):750–754. https://doi.org/10.1126/science.1141736
Ubeda-Bañon I, Saiz-Sanchez D, de la Rosa-Prieto C et al (2014) α-Synuclein in the olfactory system in Parkinson’s disease: role of neural connections on spreading pathology. Brain Struct Funct 219(5):1513–1526. https://doi.org/10.1007/s00429-013-0651-2
Mrabet S, Ben Ali N, Achouri A et al (2016) Gastrointestinal dysfunction and Neuropathologic correlations in Parkinson disease. J Clin Gastroenterol 50(9):e85–e90. https://doi.org/10.1097/MCG.0000000000000606
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Brett, F.M., Kearney, H. Neuropathology correlates of cognitive assessments. Ir J Med Sci 187, 835–844 (2018). https://doi.org/10.1007/s11845-017-1733-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11845-017-1733-6