
Abstract
Future direct-fired supercritical CO2 power cycles require steels resistant to oxidation/corrosion in high-temperature CO2 environments containing various impurities. Herein we studied the oxidation behavior of 14 candidate steels in a simulated direct-fired CO2 power cycle environment consisting of 95% CO2, 4% H2O, 1% O2 with/without 0.1% SO2 at 1 atm and 550 °C, 600 °C, 650 °C for up to 2500 h. Steels with ≥ 11.5 wt% Cr exhibited at least partial coverage by Cr-rich oxide scales leading to a significant decrease in the oxidation rates in both gases. While SO2 had little effect on low-Cr steels that formed Fe-rich oxides, it generally worsened performance of high-Cr (> 11.5 wt%) steels by hindering the establishment of a protective Cr-rich oxide. This effect was most pronounced at the lowest temperature of 550 °C, which was attributed to strong preferential adsorption of sulfur-containing species within the oxide at relatively low temperatures.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Supercritical CO2 (sCO2) power cycles represent a potentially transformative technology for next-generation high-efficiency power plants. Indirect sCO2 cycles, which rely on a closed loop of (relatively) pure CO2, are amenable to virtually any heat source and are thus being considered for future power generation via fossil, nuclear and concentrated solar energy.1 Alternatively, a direct (aka direct-fired) sCO2 cycle requires fossil fuels as the heat source, as it utilizes the CO2-rich stream produced by a pressurized oxy-fuel combustion process as the working fluid of the system.2
A primary consideration for materials selection in both indirect and direct sCO2 cycles is chemical compatibility with the circulating CO2-rich fluid. This is especially true of the primary heat exchanger, where compact designs are needed to enable high recuperation efficiencies. Such designs utilize thin metal sections and narrow flow channels (≈ 0.5 mm) and therefore have minimal tolerance for oxidation/corrosion. As such, many research groups have reported within the last 10 years on the oxidation performance of candidate structural alloys in pure CO2 at the temperatures (550–750 °C) and pressures (20–30 MPa) of interest for the high temperature portion of the primary heat exchanger in an indirect sCO2 power cycle.3,4,5,6,7,8,9,10,11,12 This ongoing work, combined with extensive previous work evaluating alloy behavior in CO2 at lower pressures relevant to gas-cooled nuclear reactors and oxy-fuel combustion environments, is beginning to yield a consistent picture of the oxidation performance that can be expected for various categories of alloys in an indirect sCO2 power cycle.13
The oxidation performance of candidate alloys in the impure sCO2 environments characteristic of a direct sCO2 power cycle is less clear. Specifically, streams containing 85–95% CO2, with the balance being primarily H2O and O2, are expected via pressurized oxy-combustion of fossil fuels, while impurity levels (≤ 0.1%) of SO2 might also be present for the case of coal syngas.12 Initial work has shown a complex pattern of behavior for candidate alloys in these environments, where the effects of impurities have ranged from minimal to considerable, depending on the precise exposure conditions.3,12,14,15,16,17,18,19,20,21,22,23,24,25,26 In particular, small amounts of SO2 can significantly affect the oxidation behavior of steels in CO2-rich environments.27,28 This paper summarizes ongoing work at the National Energy Technology Laboratory (NETL) to understand the oxidation behavior of candidate steels in impure CO2-rich environments to inform materials selection for the intermediate-to-high temperature portions of direct sCO2 power cycles.
Experimental
Table I summarizes the compositions of the alloys tested in this study. The alloys are separated as ferritic/martensitic steels and austenitic steels and listed in order of Cr content in each category. Most of the alloys are commercially available, while JMP3, JMP4 and CPJ7 are experimental steels being developed at NETL. Samples were machined from the plate or sheet form of the alloy in the service-ready condition to dimensions of 19 mm × (8–13 mm) × (0.9–2.9 mm), then surface finished using 600 grit (CAMI designation) SiC paper. Either 2 or 3 samples of each steel were included in each exposure. The 550 °C tests were the first conducted and the number of steels evaluated expanded in the subsequent tests. Therefore, not all steels were included in the 550 °C test.
Samples were cleaned in acetone and/or alcohol and weighed using a microbalance (0.01 mg precision) prior to loading in a horizontal flowing tube furnace. The samples were heated to test temperature (550, 600, 650 °C) under a constant gas flow that corresponded to a linear rate of 25 cm/min at temperature. The gas consisted of only pure (99.999%) CO2 during an initial purge and during heating and cooling, while impurities were introduced upon reaching the exposure temperature to achieve a gas composition of (mol%) 95% CO2, 4% H2O, 1% O2 or the same gas containing 0.1% (1000 ppm) SO2. Liquid water was introduced via syringe pump into a hot section of the furnace. The gas mixture was passed through a Pt-Rh catalyst mesh to achieve the equilibrium composition of the O2/SO2 components (i.e., to catalyze the reaction SO2 + ½O2 = SO3). The samples were cooled and weighed every 500 h. The above procedure was repeated until a maximum total exposure time of 2500 h. The final 500 h exposure was interrupted for the SO2 test at 550 °C, resulting in a total time of 2250 h for these samples. The reported mass changes represent the average and error bars represent one standard deviation. The reported rate constants (kp) represent the average obtained for the individual samples for a given steel/exposure condition. The rate constants were derived by fitting a linear function to the mass change vs square root of time. Select data points were removed at our discretion when it significantly improved the fit. In particular, the first one or two data points (t = 0 and t = 500 h) corresponding to the transient oxidation period were typically removed, while those corresponding to obvious spallation (reflected by significant mass loss and confirmed by visual inspection of the samples) were also removed.
Cross-sections were prepared for several of the steels by sectioning and mounting the sample followed by grinding and polishing to a 1-µm diamond finish. The sample cross-sections were analyzed by scanning electron microscopy (SEM) and energy-dispersive x-ray spectroscopy (EDS) using an FEI Inspect F50 operating at 20 kV. Images were collected in backscattered electron (BSE) mode. Select samples were also analyzed by electron probe microanalysis (EPMA) using a JXA-8530FPlus HyperProbe.
Results and Discussion
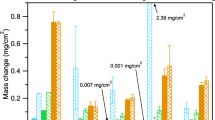
Figure 1 summarizes the mass change of all steels and conditions after a total exposure time ranging from 2250 to 2500 h, while plots of the average mass change vs time for each steel are included in Fig. S1 of the supplementary information. The steels in Fig. 1 are arranged in order of increasing Cr content, which is listed above the mass change for each steel. The plots reveal a clear transition from high to low mass gains (note the broken y-axis) for steels with Cr content between 10.5 wt% (SAVE12) and 11.5 wt% (409) at temperatures of 600 and 650 °C, regardless of SO2. A similar transition is seen at 550 °C; however, the fewer steels at this temperature prevented as precise determination of the Cr content where this occurred. Figure 1 does not reveal a significant effect of SO2 on the mass gains of most of the low-Cr steels, with two exceptions. The first is Grade 22 at 650 °C, where the mass change indicates significant spallation in the absence of SO2, whereas little or none occurred in the presence of SO2 (Fig. S1). This spallation behavior of Grade 22 was confirmed by visual inspection of the samples. The second exception was Grade 91 at 650 °C, where SO2 produced notably lowers mass gains compared with the SO2-free condition (Fig. S1). The mechanism responsible for this behavior will be described in a subsequent paper. Unlike for low-Cr steels, Fig. 1 indicates a notable effect of SO2 for high-Cr steels for at least some exposure temperatures. In particular, the mass gains imply a clear negative effect of SO2 at the lowest temperature 550 °C.15 While less obvious, slightly higher mass gains and large error bars also suggest a negative effect of SO2 at the highest temperature of 650 °C. Alternatively, SO2 appeared to have little effect on high-Cr steels at 600 °C.

Mass change of steels after 2250-h or 2500-h exposure to 95% CO2, 4% H2O, 1% O2 without/with 0.1% SO2 at 1 atm and temperatures of (a) 550 °C, (b) 600 °C and (c) 650 °C. The steels are shown in order of increasing Cr content, which is listed above the bars in wt%. Note the broken y-axis for the lower-Cr steels.
Several of the steels were cross-sectioned and analyzed by various techniques to understand the oxide scales that formed in these environments. Unsurprisingly, the low-Cr (≤ 10.5 wt%) steels, which showed high mass gains, formed Fe-rich oxide scales. Figure 2 shows an example of this scale for Grade 91 exposed to the SO2-free environment at 550 °C for 2500 h. The analysis reveals a scale constitution that is similar to that which has been observed frequently for relatively low-Cr steels in pure CO2.13 That is, a duplex oxide scale consisting of an outward-growing layer of Fe3O4 covered by outermost Fe2O3, an inward-growing layer of Fe3-xCrxO4 spinel and an internal oxidation zone consisting of Cr-rich oxide precipitates dispersed in the alloy matrix. That this scale structure/composition is very similar to what is observed in pure CO2 implies that the impurities did not have a significant effect on the oxidation behavior of these low-Cr steels. It is well established that growth of these Fe-rich oxide scales in high-temperature CO2 is accompanied by significant carbon uptake by the steel,13 and indeed this remains true for the impurities tested herein.15 While carburization is an important form of degradation in high-temperature CO2, it is not considered further in the present paper, which is principally focused on the oxidation behavior of the steels.

Qualitative EPMA analysis of Grade 91 exposed at 550 °C and 1 atm to 95% CO2, 4% H2O, 1% O2 for 2250 h. (a) BSE reference image, (b–d) x-ray maps showing elements of interest.
As implied by the mass gains, the oxide scales formed on high-Cr (≥ 11.5 wt%) steels showed a more complex pattern of behavior as a function of both temperature and SO2. Figure 3 shows cross-sectional SEM images of 347H (17.3 wt% Cr)—an example of a “high-Cr” steel without an exceptionally high Cr content—at all exposure conditions. Figure 3a shows that a thin, Cr-rich oxide scale formed at 550 °C in the absence of SO2 (compositions annotated on the SEM images were determined by EDS). Figure 3b shows that similar (albeit slightly thicker) Cr-rich oxide scales covered a portion of the steel surface in SO2; however, these were interrupted by frequent Fe-rich oxide nodules, consistent with the higher mass gains (Fig. 1). In addition, small sulfide particles, which were usually enriched in Mn, were present beneath both the thin Cr-rich oxide scales and thicker Fe-rich oxide nodules. Alternatively, at 600 °C, thin Cr-rich oxide scales formed regardless of SO2. Finally, at 650 °C the steel showed similar behavior to the SO2 exposure at 550 °C (i.e., partial coverage by Cr-rich oxide scale with frequent Fe-oxide formation) regardless of SO2.

Cross-sectional SEM images of 347H exposed for 2250 or 2500 h to 95% CO2, 4% H2O, 1% O2 without/with 0.1% SO2 at 1 atm and temperatures of (a–b) 550 °C (c–d) 600 °C (e–f) 650 °C.
While the presence of SO2, or a sufficiently high exposure temperature (i.e., 650 °C), promoted the formation of Fe-rich oxides for 347H, the mass gains and oxide thicknesses were still significantly lower than the low-Cr steels characterized by uniform Fe-rich oxide scales. Figure 4 illustrates the reason for this discrepancy, using the SO2 exposure at 550 °C as an example. The x-ray maps confirm that the thin oxide is indeed Cr-rich, while the thicker nodule is Fe-rich. However, Fig. 4c also shows that a continuous layer of Cr-rich oxide exists at the bottom of the nodule. Hence, the region of Fe-rich oxide growth (caused in this case by the presence of SO2) underwent a recovery process, whereby a layer of Cr-rich oxide formed at the base of the nodule and subsequently limited oxide growth by diffusion through this layer. Interestingly, 347H specimens exposed to the same environment that were thinner (0.6 mm) compared with the present study (2.0 mm) were incapable of this recovery process and were therefore more severely oxidized (and carburized).29 Figure 4e and g also suggest the presence of Mn-rich sulfide particles beneath the scale, as described above. Indeed, quantitative EPMA point analysis of similar particles (not shown) confirmed significant enrichment of Mn and S.

Qualitative EPMA analysis of 347H exposed at 550 °C and 1 atm to 95% CO2, 4% H2O, 1% O2, 0.1% SO2 for 2250 h. (a) BSE reference image, (b–g) x-ray maps showing elements of interest.
Figure 5 shows cross-sectional SEM images of 310S (25.0 wt% Cr)—an example of a steel with a very high Cr content—at all exposure conditions. Figure 5a and b reveal a similar pattern of behavior compared with 347H at the lowest temperature of 550 °C, where SO2 resulted in a thicker Cr-rich oxide scale, interrupted by frequent Fe-oxide formation. However, compared with 347H, the extent of Fe-oxide growth was more limited prior to initiation of the aforementioned recovery process, resulting in less prominent nodules for 310S (note the different scale bars in Figs. 3 and 5). The behavior of 310S at 600 °C was likewise similar to 347H, where SO2 had essentially no effect on the oxidation behavior. Alternatively, 310S showed a different pattern of behavior at the highest temperature of 650 °C, where it formed a uniform Cr-rich oxide scale both with and without SO2, where the scale was slightly thicker for exposure in SO2.

Cross-sectional SEM images of 310S exposed for 2250 or 2500 h to 95% CO2, 4% H2O, 1% O2 without/with 0.1% SO2 at 1 atm and temperatures of (a–b) 550 °C (c–d) 600 °C (e–f) 650 °C.
Alloy oxidation rates are commonly controlled by diffusion through the scale,30 resulting in parabolic mass gain behavior according to
where Δm is the mass change (per exposed surface area), t is the exposure time and kp is the parabolic rate constant. While not all the steels showed clear parabolic behavior (Fig. S1), such kp values are nevertheless a useful means of visualizing the approximate oxidation rates of large numbers of samples and exposure conditions. Figure 6 summarizes these values graphically for all steels and conditions, while the tabulated values are included in Table S1 of the supplementary information. Figure 6 is also annotated with the reaction products identified through post-exposure characterization, i.e., purely Fe-rich oxide growth and purely Cr-rich oxide growth, while the regions intermediate to these two regimes reflect the instances of periodic Fe-oxide formation and recovery as described above. Figure 6a shows that exposures done without SO2 exhibited a relatively distinct transition between these two regimes, where 409 (and 347H at 650 °C), were the only steels that did not conform to either regime. Furthermore, Fig. 6a shows that most steels exhibited the expected temperature dependence, where kp increased with increasing exposure temperature. Hence, the mechanism of oxidation did not change significantly between 550 and 650 °C for most steels exposed without SO2.

Parabolic oxidation rate constants of steels exposed at 550, 600, 650 °C and 1 atm to (a) 95% CO2, 4% H2O, 1% O2 and (b) the same gas containing 0.1% SO2.
Figure 6b shows two primary differences for exposures done with SO2. First, several additional steels/exposure conditions now fall within the zone of intermediate Cr-oxide and Fe-oxide growth, specifically all steels with Cr ≤ 18.7 wt%. Hence, exposure in SO2 required higher levels of Cr to facilitate Cr-rich oxide growth. Second, the high-Cr steels exposed at three temperatures clearly show non-Arrhenius behavior. Specifically, the oxidation rates decreased between 550 and 600 °C and increased between 600 and 650 °C. In short, SO2 generally showed a negative effect for high-Cr steels by making it more difficult for the steel to form and maintain a Cr-rich oxide scale and this effect had a complex dependence on the exposure temperature.
Possible reasons for the negative effect of SO2 on the high-Cr steels can be generally separated into two categories: (1) it may change the compounds that form by reaction of the alloy with the gas, such that protective (Cr-rich) oxides are no longer stable,16 and (2) SO2 (or another sulfur-containing species) may enter the growing oxide scale and prevent the establishment of the Cr-rich oxide, cause its failure and make it more difficult to reform a Cr-rich oxide in the event of failure.27,31,32,33,34,35,36 The first possible reason appears unlikely, considering no evidence of metal sulfates was found during characterization. Regarding the second explanation, significant previous work studying the oxidation of metals and alloys in mixed oxidant (e.g., CO2, H2O, O2) environments containing SO2 has highlighted the importance of competitive adsorption processes in controlling the oxidation behavior.37 Specifically, SO2 (and other sulfur-containing molecules) are very strong adsorbates and therefore the fraction of reacting SO2 (and SO3) molecules on the oxide surface can be expected to be much higher than that present in the bulk gas. Furthermore, the results show that some sulfur-containing species successfully diffused through the Cr-oxide oxide scales formed on the high-Cr steels, as evidenced by formation of sulfide particles in the underlying alloy (Figs. 3–5). This likely occurred by diffusion through defects (such as grain boundaries) in the oxide scale,27 and thus this diffusion process (and therefore the amount of sulfur ingress through the scale) is likely subject to the same competitive adsorption process as the initial adsorption of the reactive molecules on the oxide surface.28 Importantly, competitive adsorption processes generally become less prominent as temperature is increased and the adsorption rate decreases.30,38,39 Therefore, the effect of competitive adsorption of the sulfur-containing species was likely most pronounced during the 550 °C exposure. Thus, it appears likely that the increased participation of sulfur-containing species during oxidation at 550 °C contributed to the worse oxidation performance of the high-Cr steels at this temperature. The precise mechanism whereby SO2 prevented the formation of a Cr-rich oxide scale, or caused its failure (i.e., by affecting the growing oxide scale, the underlying alloy, or both), is currently unclear. It is also noteworthy that the equilibrium SO3/SO2 ratio is significantly higher at 550 °C (2.3) compared with 600 °C (1.0) and 650 °C (0.5), based on thermodynamic calculations. Therefore, if SO3 is the problematic sulfur species, this may further explain the worsened effect at the lowest exposure temperature. Finally, alloy diffusion was slowest at 550 °C. Because selective oxidation (of Cr in this case) is favored by faster diffusion, this likewise makes the alloy more susceptible to the disruption in establishment of a Cr-rich oxide scale caused by SO2.
Summary and Conclusion
Herein we evaluated the oxidation performance of several candidate steels in the gas compositions expected in direct-fired CO2 power cycle environments. Steels with Cr contents ranging from 2 to 27 wt% were exposed to 95% CO2, 4% H2O, 1% O2 without/with 0.1% SO2 at temperatures of 550–650 °C for up to 2500 h to simulate the compositions expected by the oxyfuel combustion of natural gas and coal syngas, respectively. The transition from Fe-rich oxide growth to (at least partial) Cr-rich oxide growth occurred for Cr levels between 10.5 and 11.5 wt% regardless of SO2, although the transition was less distinct in SO2. The expected temperature dependence on the oxidation rate was observed for both low-Cr steels (which formed Fe-rich oxide scales) and high-Cr steels (which formed Cr-rich oxide scales) in the absence of SO2. Furthermore, SO2 had little effect on the oxidation behavior of low-Cr steels. Alternatively, it had a strong effect on the behavior of high-Cr steels, particularly at the lowest exposure temperature of 550 °C. The most likely explanation appears to be due to strong preferential adsorption of sulfur-containing species on and within the growing oxide scale, preventing the formation/growth of a protective Cr-rich oxide.
From an application perspective, this work shows that many candidate steels may possess the required oxidation resistance for use in the intermediate-to-high temperature portions of a direct sCO2 power cycle fueled by natural gas, where sulfur levels are very low. For example, several 400 series steels showed good oxidation performance. Therefore, these low-Ni steels should be considered as a cost-effective alternative to austenitic stainless steels, up to their maximum use temperature (likely ≈ 450 °C in an sCO2 power cycle). Likewise, certain creep-strength-enhanced ferritic-martensitic steels (which can contain up to ≈ 12 wt% Cr) may also prove suitable for portions of the cycle. Caution is needed when sulfur-based impurities are expected, especially at relatively low temperatures. Additional research is needed (and ongoing) to establish the oxidation performance with and without SO2 at even lower temperatures, to further inform cost-effective materials selection for direct-fired sCO2 power cycles.
References
K. Brun, P. Friedman and R. Dennis, Fundamentals and applications of supercritical carbon dioxide (sCO2) based power cycles (Woodhead Publishing, Sawston, 2017).
R. Allam, S. Martin, B. Forrest, J. Fetvedt, X. Lu, D. Freed, G.W. Brown, T. Sasaki, M. Itoh and J. Manning, Energy Procedia 114, 5948. (2017).
R.P. Oleksak, J.H. Tylczak, C.S. Carney, G.R. Holcomb and Ö.N. Doğan, JOM 70, 1527. (2018).
B.A. Pint, R. Pillai, M.J. Lance and J.R. Keiser, Oxid. Met., 94, 505 (2020).
H.J. Lee, H. Kim, S.H. Kim and C. Jang, Corros. Sci. 99, 227. (2015).
V. Firouzdor, K. Sridharan, G. Cao, M. Anderson and T.R. Allen, Corros. Sci. 69, 281. (2013).
R.I. Olivares, D.J. Young, T.D. Nguyen and P. Marvig, Oxid. Met. 90, 1. (2018).
B. Adam, L. Teeter, J. Mahaffey, M. Anderson, L. Árnadóttir and J.D. Tucker, Oxid. Met. 90, 453. (2018).
Y. Gui, Z. Liang, H. Shao and Q. Zhao, Corros. Sci. 175, 108870. (2020).
M.H.S. Bidabadi, S. Chandra-ambhorn, A. Rehman, Y. Zheng, C. Zhang, H. Chen and Z.-G. Yang, Corros. Sci. 177, 108950. (2020).
F. Rouillard and T. Furukawa, Corros. Sci. 105, 120. (2016).
J.P. Shingledecker, S.C. Kung and I.G. Wright, Predicting the Oxidation/Corrosion Performance of Structural Alloys in Supercritical CO2, Report No. DE-FE0024120, Electric Power Research Institute (2017).
R.P. Oleksak and F. Rouillard, Materials performance in CO2 and supercritical CO2, in Comprehensive Nuclear Materials, 2nd edn. ed. by R.J.M. Konings and R.E. Stoller (Elsevier, Oxford, 2020), pp. 422–451. https://doi.org/10.1016/B978-0-12-803581-8.11622-4.
R.P. Oleksak, J.H. Tylczak, G.R. Holcomb and Ö.N. Doğan, Corros. Sci. 157, 20. (2019).
R.P. Oleksak, J.H. Tylczak, G.R. Holcomb and Ö.N. Doğan, Corros. Sci. 164, 108316. (2020).
R.P. Oleksak, J.H. Tylczak, G.R. Holcomb and O.N. Dogan, JOM 72, 1822. (2020).
S. Kung, J. Shingledecker, I. Wright, T. Lolla and A. Sabau. Proceedings of the 6th International Supercritical CO2 Power Cycles Symposium, Pittsburgh, Pennsylvania. (2018).
T. Lolla, J. Shingledecker, S. Kung, M. Gagliano, I. Wright and A. Sabau. Proceedings of the 6th International Supercritical CO2 Power Cycles Symposium. Pittsburgh, PA (2018).
B.A. Pint, R.G. Brese and J.R. Keiser. Proceedings of the 2018 NACE CORROSION Conference. Phoenix, AZ (2018).
B.A. Pint, J. Lehmusto, M.J. Lance and J.R. Keiser, Mater. Corros. 70, 1400. (2019).
J. Mahaffey, D. Adam, A. Brittan, M. Anderson and K. Sridharan, Oxid. Met. 86, 567. (2016).
J. Mahaffey, A. Schroeder, D. Adam, A. Brittan, M. Anderson, A. Couet and K. Sridharan, Metall. and Mater. Trans. A. 49, 3703. (2018).
J. Lehmusto, J. Kurley, M. Lance, J. Keiser and B.A. Pint, Oxid. Met. 94, 95. (2020).
B.A. Pint, R. Pillai and J.R. Keiser. Proceedings of the 2020 NACE CORROSION Conference. Virtual (2020).
K. Li, Y. Zeng and J.-L. Luo, Corros. Sci. 184, 109350. (2021).
S.R. Akanda, R.P. Oleksak, R. Repukaiti, K.A. Rozman and Ö.N. Doğan, Metall. Mater. Trans. A. 52, 82. (2021).
C. Yu, J. Zhang and D.J. Young, Oxid. Met. 90, 97. (2018).
P. Huczkowski, D.J. Young, T. Olszewski, A. Chyrkin and W.J. Quadakkers, Oxid. Met. 89, 651. (2018).
S.R. Akanda, R.P. Oleksak, R. Repukaiti, K.A. Rozman and Ö.N. Doğan, Corros. Sci. 192, 109795. (2021).
D.J. Young, High temperature oxidation and corrosion of metals, 2nd edn. (Elsevier, New York, 2016).
P. Singh and N. Birks, Oxid. Met. 19, 37. (1983).
S.-H. Choi and J. Stringer, Mater. Sci. Eng. 87, 237. (1987).
H. Xu, M. Hocking and P. Sidky, Oxid. Met. 41, 81. (1994).
R. Lobb and H. Evans, Corros. Sci. 25, 503. (1985).
C. Yu, J. Zhang and D.J. Young, Corros. Sci. 112, 214. (2016).
C. Yu, T.D. Nguyen, J. Zhang and D.J. Young, J. Electrochem. Soc. 163, C106. (2016).
D. Young and S. Watson, Oxid. Met. 44, 239. (1995).
Z.-S. Liu, W.-K. Li and M.-J. Hung, J. Air Waste Manag. Assoc. 64, 1038. (2014).
B. Li and C. Ma, Energy Procedia 153, 471. (2018).
Acknowledgements
This work was performed in support of the U.S. Department of Energy’s Fossil Energy Crosscutting Technology Research Program. We thank Jeffrey Hawk (NETL), Paul Jablonski (NETL) and Martin Detrois (NETL) for providing the NETL developed steels (JMP3, JMP4, CPJ7). We further thank Christopher McKaig (NETL) and Matthew Fortner (NETL) for preparing the sample cross-sections and Keith Collins (NETL) for performing the electron microprobe analysis.
Funding
This work was funded by the Department of Energy, National Energy Technology Laboratory, an agency of the United States Government, through an NETL Support Contractor. Neither the United States Government nor any agency thereof, nor any of their employees, nor the contractor, nor any of their employees, makes any warranty, expressed or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Oleksak, R.P., Tylczak, J.H. & Doğan, Ö.N. High-Temperature Oxidation of Steels in Direct-Fired CO2 Power Cycle Environments. JOM 73, 3965–3973 (2021). https://doi.org/10.1007/s11837-021-04960-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11837-021-04960-z