
Abstract
Lead zirconate (PZ) nanopowders were synthesized by the treatment of precursors with high-energy ball milling for a relatively short time. The effects of ball milling time and rotational speed on the produced nanomaterial were investigated. The calcination temperature of the ball-milled powder was determined from the thermogravimetric analysis (TGA) and the differential scanning calorimetry (DSC) results. The structural properties of the calcined nanoparticles were studied by Fourier transmission infrared (FT-IR), x-ray diffraction (XRD), and transmission electron microscope (TEM). Well-crystallized PZ nanopowders were obtained after heating at 800°C for 3 h. The diffraction data were refined by the Rietveld method to accurately determine the crystallographic information. Williamson-Hall, Halder-Wagner, and size-strain plot methods were employed to investigate the average crystallite size and lattice microstrain in the prepared samples. The XRD and TEM images confirmed the formation of nanoparticles with an average size in the range of 20–43 nm. The band gap of the nanopowders varied from 3.11 eV to 3.28 eV as established by diffuse reflectance measurement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Oxide ceramics are widely used in modern technologic applications such as micro-actuators, high-energy storage capacitors, and infrared detectors. PbZrO3 is a well-known antiferroelectric (AFE) material with a Curie temperature of 230°C. These materials lack piezoelectricity because of the presence of a centre of symmetry, which distinguishes this class of materials from ferroelectrics (FE) and affords them the name antiferroelectrics (AFE).1
When the AFE phase is subjected to an electric field or heating, phase transitions from AFE to FE or paraelectric (PE) can occur. These transitions from AFE to either FE or PE are always accompanied by significant growth in the volume (V). The maximum strain is always achieved near the AFE–FE transition temperature, where AFE and FE co-exist. Therefore, like the piezoelectric effect at the morphotropic phase boundary (MPB) in FEs, AFEs with a composition close to MPB would also enhance the generated strain. This conclusion has been supported by experimental data that are widely available in the open literature,2,3,–4 where a maximum longitudinal strain of 0.3–0.4% has been achieved.
The formation of nanocrystalline materials is now a practical approach of great interest in different fields of theoretical and industrial research because of their unusual properties. The production of nanopowders with precise stoichiometry depends on many factors such as the starting substances, temperature, and processes. PZ nanomaterials have been prepared by several chemical methods such as homogeneous precipitation, oxidant-peroxo, hydrothermal, and sol–gel techniques.5,6,7,–8 These methods are generally aimed at minimizing the crystallite size and preparation temperature with reliable homogeneity and stoichiometry.9 High-energy ball milling is one of the available techniques for the production of nanosized powders and enhancing the phase transformation in the material with the advantage of using simple equipment and cheap precursors.10,11,–12
X-ray diffraction patterns of individual phases of ball-milled samples are partially or completely overlapped as a result of several polymorphous transformations and the amount of contamination that might emerge from grinding media. Consequently, without a reliable profile fitting method, incorrect information could be extracted regarding full width at half maximum (FWHM), integrated intensity, etc., of individual profiles by any traditional x-ray method for particle size and strain analysis. Based on these considerations, the Rietveld method is one of the best methods for microstructural refinement as well as for quantitatively estimating multiphase materials.13
Although there are numerous publications on antiferroelectric PbZrO3, there are insufficient investigations available that focus on the microstructural aspects, which is critical to understand the AFE-FE transition. In the present work, nano-PbZrO3 was synthesized and characterized by different techniques such as x-ray powder diffraction (XRD), transmission electron microscope (TEM), thermal analysis, x-ray fluorescence spectroscopy (XRF), and UV–Vis absorption spectroscopy. The effect of ball milling rotational speed and time on the microstructure was studied by applying both conventional and advanced XRD line-broadening analysis methods.
Experimental
Nano lead zirconate, PbZrO3, ceramics have been synthesized by processing raw materials with high-energy ball-milling (HEBM). The stoichiometry ratios of PbCO3 and ZrO2 fine powders with 99.8% purity (Loba Chemie, Mumbai, India) were weighed using a high-precision electronic balance. A small amount of stearic acid was added to the raw materials and then mixed thoroughly for approximately 5 min. The mixture was then ball milled for different times and at different rotational speeds using a high-energy ball mill (Resch EMax) under air atmosphere at room temperature. A set of two grouped powder samples was synthesized. The first group consists of three samples processed at different rotational speeds of 300 rpm, 350 rpm, and 400 rpm for a constant 2 h milling time, hereafter called (PZ1, PZ2, and PZ3). Similarly, the second group contains the PZ1 sample along with two samples processed at different milling times for 1 h and 3 h at a constant rotational speed of 300 rpm, hereafter called (PZ4 and PZ5). To cool down the system and maintain the temperature stability, the milling process was stopped for 5 min after every 20 min of milling. The ball-to-powder (BPR) weight ratio, total ball and powder weight were 5:1, 20 g, and 4 g, respectively.
The behaviour of the ball-milled powder was analysed by TGA and DSC (LINSEIS STA PT-1000), in which approximately 25 mg of the material was heated at a rate of 10°C/min to 900°C. The milled powder was then heated at 800°C under static air for 3 h. X-ray powder diffraction was recorded using Cu Kα (λ = 1.5406 Å) radiation for the synthesized samples with a Philips PW 1710 diffractometer system. The step-scan data with step size of 0.02° and a scan rate of 0.05° min−1 were recorded for the angular range of 20°–90°. A profile analysis (LeBail method) of the XRD patterns was performed. The extracted data were then used to evaluate the crystallite size and lattice microstrain. Rietveld structural refinement was employed to accurately determine the structural parameters of the obtained powders. The Fourier transmission infrared (FT-IR) spectra of the prepared samples were recorded using a JASCO FT-IR 4100 spectrometer in the wavelength range 400–4000 cm−1. The morphology of the powder was studied using a JEOL, JEM 1010 transmission electron microscope (TEM). The compositional analysis of the synthesized powders was carried out by x-ray fluorescence spectroscopy (XRF). Optical studies were performed using a (JASCO V-670, UV–VIS-NIR) spectrophotometer in the wavelength range of 200–1000 cm−1.
Results and discussion
TGA and DSC analysis
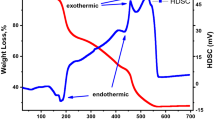
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were carried out to study the thermal decomposition process of the ball-milled powder. Figure 1 displays the TGA/DSC spectra of the ball-milled powder for sample PZ1 up to 900°C at a heating rate of 10°C/min in air. It is clearly visible that there are three main stages of weight loss in the temperature range up to 850°C. The first small notable loss in the temperature range up to 250°C may be attributed to the removal of residual water and decomposition of stearic acid. It is also observed that the weight becomes almost constant above 400°C up to 850°C with a slight change, which shows an enhancement in the crystallization of the powder. The main weight loss (about 20%) occurred in the range from 250°C to 400°C, which may correspond to the decomposition of the raw materials and formation of the pure phase. The changes in the DSC curve are observed as exothermic and endothermic peaks in the related temperature range. Based on the DSC, there are two exothermic peaks at 340°C and 400°C and one endothermic peak at 90°C. According to the TGA/DSC, the weight becomes almost constant above 400°C, which indicates the formation of the PbZrO3 compound above this temperature.

TGA and DSC spectra of ball-milled powder for sample PZ1.
Structural properties of PZ nano-powder
XRD spectral analysis
Supplementary Figure S1 (refer to online supplementary material) shows the XRD pattern of the calcined PbZrO3 nanopowders. The diffractogram showing the (1 1 2), (0 4 0), (2 4 0), (3 2 2), (0 0 4), (0 4 4), and (2 0 2) peaks indicates that the major phase in this composition had orthorhombic symmetry.4 The peaks were indexed in space group (SG) Pbam [QualX card number: 00-200-3149]. There are two small extra peaks of trace amounts of PbO at 28.34° and 29.16°, which could be eliminated by increasing the calcination temperature.14,15 To assess the crystallographic information associated with PbZrO3 nanocrystalline powders considering all permitted reflections from the XRD patterns of highly overlapping reflections, it is important to fit the experimental pattern with a profile fitting function based on the structural information.16 Thus, a profile refinement (LeBail fit)17 was carried out using the FuLLProf18 software package.
Almost all possible parameters were changed in the fitting process by means of the least square procedures.19 The pseudo-Voigt function was deployed as a peak profile function. The structures obtained by S. Teslic and T. Egami (COD entry ID 2003149)20 were used as initial parameters (a = 5.8808 Å, b = 11.7703 Å, c = 8.2539 Å, SG = Pbam, IN = 55). The background was adjusted as a linear interpolation between a set of manually chosen points. The refinement was continued until a reasonable matching between the observed and calculated patterns achieved with the best possible values for conventional R parameters [Rp (profile error), Rwp (weighted residual error), and Rexp (expected error)].
The conventional fitted profile parameters for the PZ nano-crystallite heat-treated at 800°C for 3 h are shown in supplementary Table SI. It is noticed that the goodness of fit (GOF) parameters are close to one and the χ2 is low, which indicates good agreement between the fitted and the calculated profile. The obtained cell constant parameters of the PZ powder samples are listed in supplementary Table SII. The average values of the cell parameters are a = 5.85869 ± 0.00029 Å, b = 11.74552 ± 0.00058 Å, and c = 8.21778 ± 0.00039 Å, which is in close agreement with the reported values for PZ ceramics21 and well matched with (JCPDS card no. 20-608 and 35-739). The occurrence of well-crystalline PbZrO3 as confirmed from the XRD patterns is fully consistent with the TGA/DSC analysis.
Crystallite Size and Lattice Strain
Accurate estimation of crystallite size is essential when the produced material crystallite size is < 100 nm. Although transmission electron microscopy is an effective method for estimating particle size, it has some restrictions. Since its images represent a local region, many samples and images are required to provide average information for the entire sample. Additionally, its sample preparation is time consuming. The x-ray powder diffraction technique is a suitable and accurate approach for the determination of crystallite size by precisely quantifying the peak broadening. The Scherrer equation, Williamson-Hall (WH), Halder and Wagner (HW), size-strain plot (SSP), and Rietveld methods are examples of methods that are based on that principle.22
Scherrer’s formula can be written as,23
where D is the average crystallite size (nm), K is related to the shape (here, K = 0.91), λ is the wavelength of the x-rays (λ = 1. 54056 Å for Cu Kα), θ is the diffraction angle, and β is the width of the diffraction peak determined at FWHM. The most intense peaks—(1 2 0), (0 4 0), (1 2 2), (2 0 2), (2 4 0), (0 0 4), (1 6 2), (3 2 2), (0 4 4), and (2 4 4)—were employed for the determination of crystallite size and lattice strain by different methods used in the study. The FWHM calculation was performed by considering the instrumental broadening by using the following equation:24
where βmeasured and βinstrumental are the observed FWHM and FWHM of a well-crystallized reference material. In the present study, Si (silicon) standard (supplied along with the XRD instrument) was used to determine the instrumental broadening.
The Williamson-Hall formula is a modification of Scherrer’s equation considering the contribution of the lattice strain in the peak broadening. The WH equation can be written as follows:25,26,–27
where B = 2 β cos Θ/λ and S = 2 sin Θ/λ. The WH plot is a very useful diagnostic tool for learning the type(s) of profile broadening and determining approximate values of crystallite size and strain.28 A plot of x = 2 sin Θ/λ versus y = 2 β cos Θ/λ is presented in Fig. 2. The size and strain values were extracted from the intercept and slope of the straight fitted line.

WH plots for a PZ1, b PZ2, c PZ3, d PZ4, and e PZ5 synthesized nanopowders.
The modified WH method (Halder and Wagner29) was used to estimate the strain and crystallite sizes. Based on the assumptions that the Lorentzian and Gaussian components of β are solely due to the size and strain effects, respectively, the modified equation can be written as,28
In the Halder–Wagner (HW) plot, y = (β/tan θ)2 is plotted against x = β/(tan θsin θ). Then, the slope and y intercept of the resulting straight line afford Kλ/D and 16 ε2, respectively. Figure 3 shows the HW plot for the PZ samples. Despite the approximations and assumptions used in the derivation of Eq. 1, the HW method has the advantage that the reflection peaks at lower angles have more weight than those at higher angles, which are often less reliable. Furthermore, Eq. 4 does not contain a factor (2), unlike Eq. 3, which is another advantage of the HW plot over the WH plot.28

HW plots for a PZ1, b PZ2, c PZ3, d PZ4, and e PZ5 synthesized nanopowders.
The WH plot assumes that the strain-induced peak broadening is basically developing in a material with isotropic nature, wherein the strain is uniform in all crystallographic directions and the material properties are independent of the measuring direction. This indicates that the diffraction domains are isotropic and that there is also a uniform microstrain induced.30,31 It is also possible to estimate the size and strain by considering the size-strain plot method. In this SSP method, less statistical weight is assumed for the data of the high-angle reflection peaks, where more statistical fluctuations are generally observed. It is also assumed that the “crystallite size” profile is explained by a Lorentz function and the “strain” profile of a Gaussian.32,33 The equation used was as follows:34
The term (dhklβhkl cos θ)2 is plotted with respect to (d 2hkl βhkl cos θ) for the ten most intense orientation peaks. In this case, the crystallite size and strain were evaluated from the slopes of the straight fitted line and the intercepted line, respectively.34 The size strain plot for the PZ samples is shown in Fig. 4.

Size-strain plots for a PZ1, b PZ2, c PZ3, d PZ4, and e PZ5 synthesized nanopowders.
Table I summarizes the results of the estimated crystallite size and lattice microstrain estimated by the methods. It is observed that the crystallite size calculated from the Scherrer equation in most samples is smaller than those estimated by the HW method and larger than those obtained by the SSP method. It should be mentioned here that some available literature accepts the results of the Scherrer equation for strain-free materials.35 In addition, an acceptable agreement between the results of TEM and XRD analysis, with applications that consider strain broadening, has been reported for some materials. For example, the Scherrer and Rietveld approaches were compared to TEM observations for nanocrystalline TiO2.36 Likewise, an agreement was published among the Scherrer, WH, and TEM.37 Furthermore, a study of Scherrer, WH, and SSP methods was applied to CdS nanoparticles, wherein nearly similar values were observed.24 However, several publications reported significant differences in the estimated values from one method to another. For example, the crystallite size and microstrain evaluated by Williamson-Hall and Warren–Averbach are not the same as in a previous report.35 It is also clear that the obtained crystallite size calculated by various approaches is increased by increasing the ball milling time and rotational speed as well. On the other hand, the obtained strain values by different applied methods increase by increasing the ball milling process time and the rotational speed. A similar behaviour was observed in a previously reported work.38,39
Rietveld Refinement
Rietveld refinement was performed using the FULLPROF program to confirm the phase formation as well as to obtain crystallographic parameters. In the Rietveld method, a simulated diffraction pattern is established with the structural information, peak profile, background function, space group, lattice parameters, and any other related information. Consequently, the position and the intensity of all hkl reflections are calculated, and then they are refined by the least squares method until the matching occurs between the experimental and simulated ones. The simulated pattern was initially built up using the computer program based on the lattice parameters, space group, and atom coordinates of the well-matched pattern that was obtained from the crystallography open database (COD) (entry ID 2003149)20 listed in supplementary Table SIII. The reader is advised to refer to these studies40,41,–42 for more information about the atomic coordinates.
The background points were chosen manually and adjusted using a linear interpolation between the points during the refinement. The used peak shape was a pseudo-Voigt (pV) function. A number of consecutive refinements to the zero shift and structural and microstructural parameters were applied. After a reasonable pattern was obtained, the refinement continued with background points, instrumental parameters, and atomic coordinates to advance the matching between the calculated and experimental values. Thereafter, a total of about 20 parameters, including the scale factor, lattice constants, structure factor, zero shift, occupancy, instrumental parameters, background points, etc., were refined. To achieve a good refinement model, the peak positions and intensities were generated and monitored from time to time. Figure 5 clearly shows that the experimental and simulated patterns are in good agreement. The red solid line represents the best Rietveld fit, Icalc, the squares are the observed data, Iobs, the vertical bars are the calculated angular Bragg positions of the reflections, and the green line is the difference curve between the calculated and the observed intensities Icalc − Iobs. The fit converges to residual values χ2 ≈ 1 and Rwp ≈ 15% with the independent variation of 20 parameters. The goodness of fit (GOF) of the refinement patterns was measured in terms of the profile R-factors. The relatively lower values of Rp and Rwp are usually used as a criterion for good Rietveld profile fit. Structural parameters and conventional R-factors are listed in supplementary Table SIV. The retrieved values of conventional R-factors are generally < 15%, and the agreement between the experimental and simulated patterns confirmed the goodness of refinement43

XRD Rietveld refinement patterns for PZ powders.
The obtained cell parameters of the PZ nanopowder samples are summarized in supplementary Table SV. The obtained Rietveld lattice constants are almost the same as those obtained by the Lebail profile fit. The average values for the cell parameters are a = 5.86113 ± 0.00041 Å, b = 11.74352 ± 0.00081 Å, and c = 8.22000 ± 0.00034 Å, which agrees with the reported values for PZ powders.21 The parameters a, b, c, and cell volume remained almost the same for all PZ samples.
FT-IR Study
Supplementary Fig. S2 illustrates the vibrational spectra of the calcined powder (FT-IR) obtained in the wavenumber range from 4000 cm−1 to 400 cm−1, which is most useful for chemical characterization of organic and inorganic materials.44,45 The spectrum shows three main broad vibrational bands. The first broad band was observed in the range of 500–580 cm−1, which is evidence for the development of the perovskite structure of the PZ compound at the calcination temperature (800°C), which is due to metal oxide binding.8,46 The second characteristic absorption band was in the range of 1380–1430 cm−1, which was assigned to the symmetric stretching vibration of C-O, which means that the stearic acid reacted with the raw materials during the ball-milling process.47 Moreover, the broad vibrational band centred at 3450 cm−1 could be assigned to the hydrogen O-H stretching band, while that at 1640 cm−1 is related to –COO– stretching modes. The FT-IR observations confirms the development of perovskite type structure, which corroborates our XRD and TGA/DSC studies.
Elemental Analysis and TEM
A representative XRF pattern in Fig. 6a for the calcined powder PZ1 brings about a homogeneous composition of the expected stoichiometry. Furthermore, there is no evidence of impurities due to wear debris that might be advanced into the specimens from the milling process. The morphologic evolution of the powders was revealed, as shown in Fig. 6b. In general, the grains were agglomerated and showed an irregular shape distribution, with a large variation in size. The powders consist of primary particles of nanometres in size, where the smallest particle size observed in the micrographs using an imaging computer software was approximately 20 nm.

(a) XRF and (b) TEM for PZ1 calcined powder.
Optical Properties
Diffuse reflectance spectra were measured (supplementary Fig. S3) in the wavelength range of 300–850 nm for the calcined nano PZ samples. The Kubelka–Munk function48 given below was used to evaluate the band gap of the prepared nanopowder samples by means of reflectance.
where \( R_{\infty } \) is the diffuse reflectance emanated by an infinitely thick sample and is related to the absorption coefficient (K) and scattering coefficient (S). Supplementary Fig. S4 shows the plot of [F(R)hν]1/2 versus hν, from which the band gaps of the PZ nanocrystalline powders were evaluated by extrapolating the linear portions of the curves. PZ nano-powders with crystallites in the range of 50–90 nm possess a gap of 3.11 eV, 3.18 eV, 3.28 eV, 3.13 eV, and 3.27 eV for PZ1, PZ2, PZ3, PZ4, and PZ5, respectively. Figure 7 shows a plot of the gap energy with respect to the rotational speed. It is clear that the band gap increased linearly with increasing rotational speed. On the other hand, the optical gap values were almost the same for the 1 h and 2 h samples, while it increased as the time increased to 3 h. It is reported that the exponential absorption edge and the gap energy are controlled by the degree of structural disorder.49 Therefore, the change in the estimated values of the optical band gap energy could be attributed to structural disorder.50,51

a, b Band gap energy versus time of ball milling and rotational speed, respectively.
Conclusion
The PbZrO3 nanoparticles were successfully synthesized using high-energy ball milling and characterized by TGA/DSC, XRD, TEM, and XRF. Conventional and advanced techniques of XRD peak profile analysis, namely the Williamson-Hall (WH), Halder and Wagner (HW), size-strain plot (SSP), Lebail, and Rietveld methods, were applied to calcined ball-milled nano-lead zirconium oxide. A notable variation in the resulting crystallite sizes and micro-strain was clearly observed in the obtained results of various methods. The sizes calculated by the SSP technique are considerably smaller than those of the other applied approaches. Crystallite sizes obtained from the Scherrer, WH, and HW procedures were nearly the same. Moreover, the values of microstrain obtained from the WH, SSP, and HW methods are of the same order of magnitude. The estimated crystallite size and microstrain slightly increased with the increase of the rotational speed and ball milling time process. Moreover, the band gap energy increased significantly with the rotational ball milling speed. The TEM and XRF results showed the nanosize and purity of the prepared powder. The FT-IR analysis confirms the development of the perovskite structure. Furthermore, the nano-powder could be obtained efficiently by 1-h ball milling at a rotational speed of 300 rpm by utilizing high-energy ball milling.
References
J. Zhai, X. Li, Y. Yao, and H. Chen, Mater. Sci. Eng. B 99, 230 (2003).
N.H. Fletcher, A.D. Hilton, and B.W. Ricketts, J. Phys. D Appl. Phys. 29, 253 (1996).
I. Burn and D.M. Smyth, J. Mater. Sci. 7, 339 (1972).
U. Sukkha, R. Muanghlua, S. Niemcharoen, B. Boonchom, and N. Vittayakorn, J. Am. Ceram. Soc. 94, 3397 (2011).
M. M. Calzada, in edited by L. Pardo and J. Ricote (Springer Netherlands, Dordrecht, 2011), pp. 93–144.
M.P. Moret, M.A.C. Devillers, K. Wörhoff, and P.K. Larsen, J. Appl. Phys. 92, 468 (2002).
W. Wang, X. Chen, Q. Sun, T. Xin, and M. Ye, J. Alloys Compd. 830, 154581 (2020).
S. Li, R.A. Condrate, S.D. Jang, and R.M. Spriggs, J. Mater. Sci. 24, 3873 (1989).
O. Khamman, W. Chaisan, R. Yimnirun, and S. Ananta, Mater. Lett. 61, 2822 (2007).
S. Begin-Colin, G. Le Caër, M. Zandona, E. Bouzy, and B. Malaman, J. Alloys Compd. 227, 157 (1995).
I.J. Lin and S. Nadiv, Mater. Sci. Eng. 39, 193 (1979).
I.J. Lin, Miner. Sci. Eng. 7, 313 (1975).
B. E. Warren, X-Ray Diffraction (Courier Corporation, 1990).
C. Puchmark, S. Jiansirisomboon, G. Rujijanagul, and T. Tunkasiri, Curr. Appl. Phys. 4, 179 (2004).
K.H. Omran, M.S.A. El-sadek, M. Mostafa, and O.M. Hemeda, Appl. Nanosci. 10, 2315 (2020).
P. Scardi and M. Leoni, Acta Crystallogr. Sect. A Found. Crystallogr. 57, 604 (2001).
A. Le Bail, H. Duroy, and J.L. Fourquet, Mater. Res. Bull. 23, 447 (1988).
J. Rodríguez-Carvajal, CEA/Saclay, Fr. (2001).
G.S. Pawley, J. Appl. Crystallogr. 14, 357 (1981).
S. Teslic and T. Egami, Acta Crystallogr. Sect. B Struct. Sci. 54, 750 (1998).
L.B. Kong, J. Ma, W. Zhu, and O.K. Tan, Mater. Lett. 49, 96 (2001).
N. Sahu and S. Panigrahi, Ceram. Int. 38, 1085 (2011).
A. Taylor (1961).
M.S. Abd El-Sadek, H.S. Wasly, and K.M. Batoo, Appl. Phys. A 125, 283 (2019).
G.K. Williamson and W.H. Hall, Acta Metall. 1, 22 (1953).
H.P. Klug and L.E. Alexander, X-Ray Diffraction Procedures: For Polycrystalline and Amorphous Materials (1974).
S. Murugesan, R. Thirumurugesan, E. Mohandas, and P. Parameswaran, Mater. Chem. Phys. 225, 320 (2019).
F. Izumi and T. Ikeda, Implementation of the Williamson-Hall and Halder-Wagner Methods into RIETAN-FP (2014).
N.C. Halder and C.N.J. Wagner, Acta Crystallogr. 20, 312 (1966).
K. Venkateswarlu, A. Chandra Bose, and N. Rameshbabu, Phys. B Condens. Matter 405, 4256 (2010).
P. Muhammed Shafi and A. Chandra Bose, AIP Adv. 5, 057137 (2015).
M.A. Tagliente and M. Massaro, Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 266, 1055 (2008).
D. Nath, F. Singh, and R. Das, Mater. Chem. Phys. 239, 122021 (2020).
H. Irfan, K. Mohamed Racik, and S. Anand, J. Asian Ceram. Soc. 6, 54 (2018).
V. Soleimanian and M. Mojtahedi, Appl. Phys. A Mater. Sci. Process. 119, 977 (2015).
V. Uvarov and I. Popov, Mater. Charact. 58, 883 (2007).
A. Weibel, R. Bouchet, F. Boulc, and P. Knauth, Chem. Mater. 17, 2378 (2005).
A. Pragatheeswaran, R. Ravi, and S.R. Bakshi, Adv. Powder Technol. 30, 2759 (2019).
H. Dutta, P. Sahu, S.K. Pradhan, and M. De, Mater. Chem. Phys. 77, 153 (2003).
H. Fujishita, Y. Ishikawa, S. Tanaka, A. Ogawaguchi, and S. Katano, J. Phys. Soc. Jpn. 72, 1426 (2003).
F. Jona, G. Shirane, F. Mazzi, and R. Pepinsky, Phys. Rev. 105, 849 (1957).
S. Piskunov, A. Gopeyenko, E.A. Kotomin, Y.F. Zhukovskii, and D.E. Ellis, Comput. Mater. Sci. 41, 195 (2007).
S. Verma, S. Rani, S. Kumar, and M.A.M. Khan, Ceram. Int. 44, 1653 (2018).
R.M. Silverstein and G.C. Bassler, J. Chem. Educ. 39, 546 (1962).
J.M. Hollas, Modern Spectroscopy (Wiley, 2004).
D. Sun, X. Jin, H. Liu, J. Zhu, Y. Zhu, and Y. Zhu, Ferroelectrics 355, 145 (2007).
A. Tawfik, O.M. Hemeda, A.M.A. Henaish, and A.M. Dorgham, Mater. Chem. Phys. 211, 1 (2018).
P. Kubelka, JOSA 38, 448 (1948).
G.F. Teixeira, M.A. Zaghete, G. Gasparotto, M.G.S. Costa, J.W.M. Espinosa, E. Longo, and J.A. Varela, J. Alloys Compd. 512, 124 (2012).
K. Suzuki, M. Terauchi, Y. Uemichi, and K. Kijima, Jpn. J. Appl. Phys. 44, 7593 (2005).
P. Bharathi, P. Thomas, and K.B.R. Varma, J. Mater. Chem. C 3, 4762 (2015).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Omran, K.H., Abd El-sadek, M.S., Mostafa, M. et al. Microstructural Analysis and Optical Properties of Lead Zirconate Nanoparticles. JOM 73, 630–639 (2021). https://doi.org/10.1007/s11837-020-04296-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11837-020-04296-0