
Abstract
Thin Au-Fe bilayers (3–30 nm in total thickness) were deposited on sapphire substrates. Annealing in a temperature range of 600–1100°C resulted in solid-state dewetting and the subsequent formation of micro- and nano-particles. Electron microscopy, atomic force microscopy and in situ x-ray diffraction were employed to systematically study two phase transformations in the Au-Fe system: (1) precipitation of α-Fe from supersaturated Au-Fe solid-solution particles; and (2) α↔γ transformation in Fe and Au-Fe thin films and particles. In both cases, the transformations proceeded differently than in the bulk already for sub-micron (100 nm to 1 μm) particles. These results were explained by the low defect concentration in the particles, nucleation difficulties, slow diffusivity on facets, and Au segregation. A “reverse size effect” was observed in thin Fe films, and discussed in terms of nucleation model taking into account the small size of the parent phase. The main conclusion is that phase transformations in the particles and in the bulk proceed differently, not only for nano-sized particles as was customarily believed but also for particles of sub-micrometer size. We suggest that this size effect is governed by two different length scales: the inter-defect spacing (upper limit) and the bulk critical nucleus size (lower limit).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phase transformations in small solid particles of metals and metallic alloys have been the subject of many theoretical and experimental works in the last two decades. It has been established that phase transformations proceed differently at the nanoscale compared with bulk material. This “size effect” stems from a change in balance between the different thermodynamic driving forces, consequently altering the conditions for thermodynamic stability. The roles of surface energy, surface stress and elastic energy in the size effect in phase transformations have been demonstrated theoretically,1–3 computationally4,5 and experimentally.6,7
The aforementioned thermodynamic quantities are customarily used to treat the size effect in phase equilibria by incorporating their contribution to the relevant thermodynamic potential for the system.8 For example, for an isotropic spherical particle, the capillary term in the free energy scales with (γ/r), where γ is the surface energy and r is the particle radius. This contribution becomes significant at sufficiently small radii, typically r < 10 nm for isotropic metallic nanoparticles. Particles larger than 100 nm can then generally be considered as bulk-like since this contribution becomes negligible. This had been demonstrated experimentally for structural phase transformations in different materials.9–11 In the case of the α↔γ allotropic transformation in nanocrystalline Fe, the size effect was shown to favor γ-Fe at room temperature (RT) when the grain size was smaller than 8–40 nm, depending on the excess volume of the α/γ interface.12 It should be noted that in all of the previous examples9–12 the effect occurred only at the nanoscale (<100 nm), and the properties changed monotonically with decreasing length scale. For example, if a phase transformation is suppressed below a grain size of 50 nm, it is not expected to occur again with a further decrease in grain size.
There is, however, evidence in the literature that phase equilibria in sub-micrometer particles (i.e. 100 nm–1 μm) can be affected by additional factors beyond capillarity.13 This holds true especially in the case of single crystal particles obtained by high-temperature methods, since they often contain a very low density of structural defects. This results in a reduction in the available sites for heterogeneous nucleation and, consequently, increases the nucleation energy barrier and critical nucleus size for the transformation. If the barrier becomes prohibitively high, the transformation is suppressed. For example, in the classical experiments by Cech and Turnbull, no martensitic transformation was observed in Fe-Ni microparticles of 30 μm in size, and this was attributed to the scarcity of heterogeneous nucleation sites.14 Single crystal sub-micrometer particles, having even a weak surface energy anisotropy, exhibit a faceted equilibrium crystal shape.15 Since atomically-flat facets exhibit low surface energy and area-density of mobile atoms, nucleation on such surfaces is also associated with a high energy barrier, further limiting available nucleation sites. When attached to a rigid substrate, the contribution of the stored elastic energy, which scales with particle volume, is equally important in phase equilibria.2
The Au-Fe system shows great promise for examining size effects on phase transformations in sub-micrometer particles. This is due to a combination of high and strongly temperature-dependent Fe solubility in Au, an absence of intermetallic phases,16 and a relatively large difference in surface energies of Fe (2.4 J m−2) and Au (1.4 J m−2).15,17 A variety of phase transformations and morphologies were observed in the bulk system.18,19 The large solubility of Fe in Au allows ample “room” (in thermodynamic degrees of freedom) for exploring various phase transformations in the system. When using sapphire as a substrate for Au-Fe particles and thin films, the effect of thermal and misfit stress can be examined due to a large difference in thermal expansion coefficients and a relatively small lattice parameter misfit.20 Furthermore, due to the high energy of metal/ceramic interfaces, single crystal Au-Fe particles can be easily obtained by solid-state agglomeration (dewetting) of Au-Fe thin films.21,22
In this paper, we summarize the results of our investigations of two phase transformation in Au-Fe nano- and microparticles and thin films on a sapphire substrate: precipitation of α-Fe from Au(Fe) particles23 and the α↔γ Fe allotropic transformation.24 We discuss the relevant length scales which govern the size effect in these phase transformations, and propose a new nucleation model for nanoparticles.
Experimental
Electron beam deposition was employed to deposit Fe and Au thin films of varying total and relative thicknesses. The size of the particles obtained at the end of the dewetting process was larger by a factor of ~10 than the total film thickness, which varied between 3 nm and 30 nm.22 The global composition, which scales with the relative thickness, was varied between 32 at.% and 100 at.% Fe. The films were deposited on c-plane sapphire ((0001) single crystal α-Al2O3) substrates. Annealing of the samples was performed either in a quartz-tube furnace or in a rapid thermal annealing (RTA) furnace under forming gas flow (Ar-10%H2), at temperatures between 600°C and 1100°C for times between 1 min and 48 h. The α↔γ transformation was studied primarily by in situ x-ray diffraction (XRD). The main characterization methods were high-resolution electron microscopies (HRSEM and HRTEM), as well as atomic force microscopy (AFM) and XRD. Quantitative compositional analysis was performed by energy-dispersive x-ray spectroscopy (EDXS) employing standards. The complete experimental details may be found in Refs. 23 and 24, and only a brief description is provided here.
Results and Discussion
α-Fe Precipitation
Precipitation of α-Fe from supersaturated Au(Fe) solid–solution particles was studied by employing a two-step annealing regime. First, the as-deposited films were annealed at 900°C (corresponding to a single-phase region of the phase diagram), so that they dewet as a homogeneous Au(Fe) alloy, resulting in homogenous solid-solution particles. These particles were subsequently cooled down to 600°C at which both face-centered cubic (fcc) Au(Fe) and body-centered cubic (bcc) Fe(Au) phases are in equilibrium. The purpose of this two-step annealing was to prevent the precipitation of an Fe-rich phase during the dewetting process, so that the transformation could be studied in the homogeneous single crystal particles. Several compositions and particle sizes were investigated. According to conventional wisdom, the large difference in surface energies between Au and Fe should result in precipitation of Fe inside the parent Au particle, i.e. in a core–shell morphology.3 This is illustrated in Fig. 1.

Illustration of the dewetting of thin Au-Fe bilayers, resulting in core–shell particles. Here, the two-stage annealing is described, where the films are first annealed at a higher temperature corresponding to a single-phase region of the phase diagram until homogeneous solid solution particles are obtained. The annealing temperature is then reduced so that the precipitation of an Fe-rich phase takes place in the particles
Instead, however, precipitation was observed at the side of the Au particle (~1 μm in size), as shown in the HRSEM micrograph in Fig. 2a. This location of Fe precipitates—on the side facets of the parent Au particles—is related to both nucleation and growth aspects of the transformation. Due to the paucity of nucleation sites in the bulk of the single crystal Au particle and on its faceted top and side surfaces, nucleation of α-Fe is most likely to occur along the triple line with the substrate, at the vertices (intersections between facets), or on facets exhibiting the highest surface energy, as was indeed observed during the initial moments of precipitation.23 Subsequent growth of Fe precipitated at the side of the particle was explained by the presence of Au-rich segregation layers on all surfaces of the Fe precipitate and at its interface with the sapphire substrate. This is shown in the high-angle annular dark field scanning TEM (HAADF STEM) micrographs in Fig. 2b and c, taken from a cross-section of an Fe precipitate.

(a) HRSEM backscattered electron micrograph of a Au(Fe) particle with several Fe precipitates. (b, c) HAADF STEM micrographs of a cross-section taken from a precipitate similar to those in (a). Thin Au segregation layers (bright contrast) are marked. The contrast around the precipitate in (b, c), a thin C layer in dark contrast and a thick, grainy Pt layer, is due to sample preparation
An anisotropic, quantitative model based on the concept of weighted mean curvature25 was developed for describing the precipitation of Fe during dewetting. The static analysis of the model showed that, below some critical particle height, Fe precipitation occurs inside the parent particle when the surface energy of the Fe precipitate is lower than that of pure Fe and closer to that of Au. Above this height, the stable precipitate position is adjacent to the particle. This behavior was consistent with the experimental observations of the precipitation process,23 and demonstrated the effect of segregation, an interfacial phenomena, on a phase transformation in large, single-crystal particles.
The segregation layers’ composition, thickness and coverage determined quantitatively by EDXS agreed well with the results of first-principles calculations and classical thermodynamic models.26 The composition of the segregation layer at the Fe(110) surfaces was ~10 at.% Au, corresponding to a smaller decrease in surface energy than anticipated in the precipitation model. However, the elastic energy contribution, which was not included in the model, plays an important role in the transformation as well. While the Au-Fe interface energy is much lower than the respective surface energies, such an interface is also associated with a lattice misfit that increases the total elastic strain energy of the particle with increasing particle volume. So long as plastic relaxation (via misfit dislocations, for example) does not take place, meaning that the particle does not yield, the retained elastic energy may contribute to the driving force for segregation, in addition to surface/interface energy reduction.26,27 This may be the case for single crystal particles in the size ranges reported in this work. In addition, the substrate also affects the phase transformation since the lattice misfit between Fe and sapphire depends on the Au composition at or close to that interface. Thus, the associated composition-dependent elastic energy constitutes an additional driving force which affects the composition distribution in the system. Our finite element analysis demonstrated that this elastic effect overshadows the contribution of interfacial energies for particles larger than ~10 nm.27
In particles larger than 1 μm, lamellar Fe precipitates appeared inside the Au particle. An inter-lamellar spacing of ~200 nm was consistent with the results for bulk alloys19 (see Fig. 3a and b). The large Au sub-particle contains several defects, including a twin boundary running parallel to the substrate, and a complex interface is observed between Au and the Fe lamellae (Fig. 3c). This is an example of a size effect which affects the precipitation mechanism for relatively large particles: once the particle size is below the lamellar spacing typical for bulk transformations, precipitation occurs on the side facets of the Au particle. This lamellar spacing is generally a function of experimental conditions and of various material parameters and can vary significantly, yielding a size effect for particles larger than 1 μm.

(a) HRSEM secondary electron micrograph of a Au(Fe) particle with a side Fe precipitate and several lamellar Fe precipitates. (b) HAADF STEM micrograph of a cross-section taken from the particle in (a). (c) Magnified TEM micrograph of the lamellar precipitates, showing the Au–Fe interfaces. In (a, b) the Fe phase is in dark contrast, whereas in (c), it is in bright contrast
α↔γ-Fe Transformation
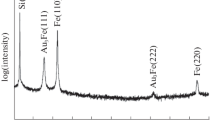
The α↔γ transformation was studied by in situ XRD of Fe and Au-Fe thin films and particles up to a temperature of 1100°C.24 Several Fe-rich compositions and film thicknesses/particle sizes were employed. The thin films were studied by in situ annealing of the as-deposited samples, while particles were studied by pre-annealing the respective films by RTA at 860°C for 1 h prior to in situ investigations. This temperature was selected since it is lower than the transformation temperature for bulk pure Fe (912°C) and Au-Fe alloys (868°C),16 and the annealing time was sufficient to obtain individual, single crystal particles. A bulk Fe sample was used as a reference. An example of a complete experiment for Fe-Au films of 25 nm in total thickness is shown in Fig. 4. The results from that sample are shown for an illustrative purpose, since they exhibit most of the possible observable effects in such experiments, namely the transformation and composition- and temperature-related peak shifts. The shifts in peak positions are a result of thermal expansion and temperature-dependent solubility which changes the lattice parameter of the alloy. The approximate angular range corresponding to each peak is marked. For each experiment, the temperatures at which a γ-Fe(111) peak appeared and disappeared (if at all) were determined.

A typical in situ XRD result, for a sample having a 25-nm initial total thickness and a composition of 91 at.% Fe. Several diffraction patterns are shown sequentially, where the first step corresponds to the front pattern. The last two patterns (labeled 1100 and RT) represent another heating–cooling cycle of the sample. The temperature- and composition-dependent positions of the relevant peaks are marked, and the color coding represents temperature (blue cold; red hot)
The transformation did not occur in pure Fe particles regardless of their size, which varied between 50 nm and 500 nm. In the films, it took place only for the thinnest (3 nm) and thickest (200 nm) films, and not in films of intermediate thicknesses (7 nm and 25 nm). A significant superheating of ~50–190°C was observed for the 3-nm films, and of 0–50°C for the 200-nm films, with hysteresis in the reverse transformation. These ranges correspond to the temperature increments of the annealing regime. The phenomenon observed for the pure Fe films can therefore be viewed as a “reverse size effect”: the transformation occurs in bulk Fe and in the thickest studied film (200 nm); in the intermediate range of film thicknesses (7 nm and 25 nm), the transformation is suppressed; and with further decrease in film thickness, the transformation occurs again. In the case of Au-Fe films and particles, the transformation was always observed and the γ-Fe phase persisted down to RT. The superheatings of the α-phase varied between 50°C and 230°C.
We now address the behavior of the pure Fe thin films which exhibited the “reverse size effect”. These results can be explained by a combination of a low concentration of structural defects in the pure Fe particles, their faceted morphology, and a reduced nucleation barrier in small systems. The immediate effect of the former is the lack of heterogeneous nucleation sites for γ-Fe, leading to a superheating exceeding 200°C. A similar explanation holds for the films of intermediate thickness, since even during the course of dewetting, faceted rims and singular (110) surfaces are present, significantly increasing the nucleation barrier. The 200-nm-thick film is substantially thicker than the rest of the employed samples, and exhibits a bulk-like behavior. This can be attributed to the presence of grain boundaries and other crystallographic defects (misfit dislocations, etc.) which are present in thicker films and cannot be completely annihilated prior to reaching the transformation temperature. The transformation was observed for the thinnest films, since at the onset of dewetting the average film thickness is still comparable with the critical nucleus size, calculated to be between 5 nm and 10 nm,28 whereas for the samples of intermediate thickness the average thickness had already increased beyond this value at the corresponding stage of in situ annealing. Therefore, the critical nucleus size serves as a lower length scale, below which the nucleation barrier is greatly decreased and facilitates the transformation, resulting in the reverse size effect. We have illustrated this in our recent work by a simple nucleation model of a lens-shaped nucleus in a spherical particle.24 In that model, we assumed the simplest case of an isotropic particle and equal surface energies between the α and γ phases. Here, we relax the latter assumption by including this contribution of a difference in surface energy to the energy balance, and studying its effect on the nucleation barrier.
Neglecting the contribution of elastic energy, the free energy of a γ-Fe nucleus is given by:
where ΔG v is the difference in free energies between γ-Fe and α-Fe per unit volume, V is the nucleus volume, σα / γ is the energy of the α/γ interface and Sα / γ is its area, Δσ = σγ–σα is the difference between the surface energies of γ-Fe and α-Fe, and Sγ is the surface area of the γ-Fe nucleus. The following analysis is identical to that of Ref. 24, with the exception of the third term in the RHS of Eq. 1, which accounts for the difference in surface energies between the two phases. All the geometric parameters of the model are shown in Fig. 5.

Geometrical construction (projection) employed in calculating the volume, surface and interface area of the γ-Fe nucleus [shown in gray]
Here, we assume that the surface energies of the two phases are different, and therefore the contact angle at the triple line, β eq, is given by:
The free energy of the lens-shape nucleus formed in a spherical parent particle of radius R and having a constant contact angle, is given by:
The angle λ is defined as \( \beta _{1} + \beta _{2} = \pi /2 + \lambda \), having the meaning of a deviation from a contact angle of 90°, and therefore satisfies:
To obtain the nucleation barrier, ΔG cr n , we seek a maximum of ΔG n (see Eq. 3) with respect to the angle β 1 at a given particle radius. This was performed numerically for different values of R and Δσ, employing the parameters from Ref. 29, for a superheating of ΔT = 5 K. The results are shown in Fig. 6 for several values of Δσ (positive, negative and zero). The green curve in Fig. 6 corresponds to Δσ = 0 which reproduces the result obtained in our previous work,24 along with the nucleation barrier and critical nucleus radius in the bulk as dashed lines. Positive values of Δσ increase the nucleation barrier compared to Δσ = 0, and negative values result in a lower barrier at a given particle radius, as expected. Shriniyan et al. reported a negative surface energy difference Δσ = −40 mJ m−2;29 however, the scatter of surface energy data in the literature does not lead to a clear conclusion on whether Δσ is positive or negative.30 Furthermore, this value may depend strongly on temperature and annealing ambient. Regardless, in all cases, the nucleation barrier decreases with decreasing parent particle size, with a strong decrease of up to two orders of magnitude when the particle size is below the bulk critical nucleus radius. It should be mentioned that the curves shown in Fig. 6 are not simply shifted by a constant with respect to each other, which means that the difference in surface energies has an intrinsic effect on the nucleation barrier.

Calculated nucleation barrier for γ-Fe in an α-Fe particle as a function of its radius, R, at a superheating of ΔT = 5° and for several values of the difference between the surface energies of the two phases, Δσ. The energy barrier and critical radius for nucleation in the bulk (for Δσ = 0) are marked by dashed lines. Both axes are presented in logarithmic scale to emphasize the differences between curves and the deviation from bulk behavior
While the model presented here demonstrates that for particles smaller than the bulk critical nucleus radius the nucleation barrier is greatly decreased, some major differences between it and the experimental results should be mentioned. First, the model does not include the presence of a substrate. For a hemispherical particle on a substrate, the model presented here yields a nucleation barrier which is one-half of the obtained results, assuming equal interface energies between α/γ-Fe and sapphire. Since data on the γ-Fe/sapphire interface energy are not available in the literature, we were not able to estimate the error associated with this assumption. The role of elastic energy was also not taken into account in the model. While there are several orientation relationships between both α and γ phases which results in a small amount of strain energy,31 the rigid sapphire substrate may impose a crystallographic orientation on each phase which results in a higher elastic energy in the α/γ particle (but lower for the whole system, including the substrate). Evidence for this may be found in the complex shape of the Au-Fe interface in Au-Fe particles.23,32 Finally, the reverse size effect was observed in the “thin film samples” rather than in the pre-annealed “particle samples”, since the particles obtained by pre-annealing are large, defect-free single crystals in which the nucleation barrier is prohibitively high. The thin films, however, agglomerate quickly during the in situ annealing. Once the transition temperature is reached, the film is still largely intact with only sporadic holes indicating the onset of dewetting, as shown in Fig. 7. Nucleation of γ-Fe is then likely to take place around the edges of the holes, where the geometry resembles that of a hemispherical particle attached to a continuous thin film,22 or in the nanoparticles present at the center of some of these holes. To simplify our analysis, we approximated the aforementioned nano-scale features of the thin films exhibiting the “reverse size effect” by spheres. The retracting edge of a thin film and the facetted nanoparticles present at this stage could be reasonably approximated as such by our model, since neither their anisotropic character nor the presence of the substrate should impact the trend of diminishing nucleation barrier with decreasing particle size. The presence of a substrate, for example, should simply reduce all nucleation barriers by a constant factor (2, for hemispherical particles). Therefore, the proposed model captures the essential features of our experiment.

HRSEM SE micrograph of a 3-nm-thin Fe film on sapphire, annealed in the RTA at 860°C for 1 min at a heating rate of 3°/s. This annealing simulates the first stages of the in situ annealing of the films and shows that they had not completely agglomerated into an array of equilibrium-shape particles
In contrast to the pure Fe samples, the Au-Fe samples (annealed in a two-phase region of the phase diagram) contain Au-Fe interfaces. This fcc/bcc interface may facilitate the nucleation of the fcc γ phase and may also (meta-)stablilize it down to RT. Indeed, many Fe particles attached to a Au sub-particle exhibited a characteristic fcc crystal shape, which has an hexagonal shape with a top {111} facet.15 In some cases, the Fe sub-particle appears to “complete” the shape of the fcc Au particle, as shown in Fig. 8a. In contrast, when no Au phase is present in a particle, or when its relative volume fraction is very small, the Fe particles adopt a bcc shape with a top {110} facet. Figure 8b compares two adjacent Fe particles. The particle on the left side exhibits traces of an attached Au sub-particle which may have evaporated due the prolonged annealing at high temperatures, since both the particle size and Au volume fraction are smaller compared with the particle in Fig. 8a. This particle exhibits a fcc shape, whereas the particle on the right side lacks any visible Au traces and exhibits a bcc shape. It should be noted that an Fe particle observed at RT with a fcc shape is not necessarily γ-Fe, and that similar particles were also observed in pure Fe samples where the γ phase was not retained at RT. In these particles, the (fast) transformation back to the α phase took place during cooling, whereas the associated change in crystal shape (which involves diffusion on singular surfaces) is significantly slower. Thus, a bcc particle may also exhibit a fcc-like crystal shape. Nevertheless, γ-Fe particles as large as 200 nm were observed at RT by HRTEM.24 In nearly all samples in which γ-Fe was formed, it coexisted with α-Fe. This is due to the wide size distribution associated with particles obtained by dewetting, especially of single crystalline thin films, and of alloy thin films.23

(a) HRSEM BSE micrograph showing a Au-Fe bi-particle. (b) HRSEM SE micrograph from a Au-Fe sample showing an α- and γ-shaped Fe particles on the right and left sides, respectively. The contrast adjacent to the left particle suggests the prior presence of a Au sub-particle
The Size Effect
Based on our observations of two phase transformations in Au-Fe particles and thin films, we were able to identify two length scales which characterize the size effect. The upper length scale, below which the transformation is significantly delayed, is the average distance between crystallographic defects, which serve as preferred sites for heterogeneous nucleation. A single crystal particle which does not contain such defects will generally not transform under reasonable superheatings, regardless of its size. In practice, such particles of up to 10 μm in size may be obtained by solid-state dewetting of thin films. Such particles are typically faceted, and nucleation on their atomically-flat surfaces is associated with a prohibitive nucleation barrier.33 However, if the particles exhibit a weak anisotropy of their surface energy, and therefore do not contain (many) facets, nucleation on their free surfaces becomes competitive with nucleation on bulk crystallographic defects due to the high concentration of surface defects. The same considerations can be applied to interphase boundaries: the complex Au-Fe interface observed in Au-Fe particles (e.g., Fig. 3) is not faceted and its presence facilitates nucleation of γ-Fe even in Fe particles with faceted surfaces. On the other hand, the planar interface between sapphire and Fe particle with a low-index orientation relationship23,34 does not facilitate nucleation of γ-Fe, whereas preliminary results indicate a different behavior for similar particles on an amorphous SiO2 substrate.24
The lower length scale, below which the phase transformation is observed again, is related to the bulk critical nucleus radius size. In nanoscale features (particles, films) which are smaller than this length scale the nucleation barrier at the surface can be reduced by up to 2–3 orders of magnitude, depending on the degree of superheating24 and the difference in surface energies between the two phases (Fig. 6). Since the nucleation rate is described by an Arrhenius dependence, such a reduction in the barrier yields a substantial increase in nucleation rate at a given temperature. Even though these predictions are based on our isotropic model, the nucleation barrier associated with atomically-flat facets is not expected to differ substantially.
Conclusion
In conclusion, we demonstrated a size effect in phase transformations in Au-Fe particles of up to 1 μm in size. For α-Fe precipitation, the effect of solute segregation, in combination with the anisotropy of surface energy and elastic strain energy, dictated the phase transformation kinetics and resulting morphology. The α↔γ transformation in pure Fe occurred only in the thinnest and thickest Fe films, resulting in a “reverse size effect”. In Au-Fe particles and films, the transformation occurred in all samples and γ-Fe particles persisted down to room temperature. These results were explained in terms of the lack of heterogeneous nucleation sites and the facilitating presence of an Au-Fe interface.
The size effect in large particles, and the reverse size effect in thin films reported in this work, subvert the existing paradigm that the size effect in phase transformations is mainly related to capillarity, and becomes significant when at least one of the characteristic dimensions of the system is smaller than 100 nm. The combination of both length scales described in this work presents a “window” in which phase transformations may be inhibited due to a size effect related to nucleation difficulties. In addition, these results could prove beneficial for nanostructures design. For example, Au segregation was utilized to form the shell in core(Fe)-shell(Au) micro- and nano-particles, by employing a sufficiently-high Fe concentration,35 or to alloy the grain boundaries of thin Au films.26
References
J.W. Cahn, Acta Metall. 28, 1333 (1980).
F.D. Fischer, T. Waitz, D. Vollath, and N.K. Simha, Prog. Mater Sci. 53, 481 (2008).
A.S. Shirinyan and M. Wautelet, Nanotechnology 15, 1720 (2004).
J. Diao, K. Gall, and M.L. Dunn, Nat. Mater. 2, 656 (2003).
Y. Mishin, M. Asta, and J. Li, Acta Mater. 58, 1117 (2010).
N. Braidy, G.R. Purdy, and G.A. Botton, Acta Mater. 56, 5972 (2008).
B. Kim, J. Tersoff, C.Y. Wen, M. Reuter, E. Stach, and F. Ross, Phys. Rev. Lett. 103, 155701 (2009).
W. Jesser, R. Shneck, and W. Gile, Phys. Rev. B 69, 144121 (2004).
G. Baldinozzi, D. Simeone, D. Gosset, and M. Dutheil, Phys. Rev. Lett. 90, 216103 (2003).
O. Kitakami, H. Sato, Y. Shimada, F. Sato, and M. Tanaka, Phys. Rev. B 56, 13849 (1997).
S.H. Tolbert and A.P. Alivisatos, Science 265, 373 (1994).
Q. Meng, N. Zhou, Y. Rong, S. Chen, T.Y. Hsu, and Xu Zuyao, Acta Mater. 50, 4563 (2002).
L. Pasquini, E. Callini, E. Piscopiello, A. Montone, M.V. Antisari, and E. Bonetti, Appl. Phys. Lett. 94, 041918 (2009).
R.E. Cech and D. Turnbull, Trans. AIME 206, 124 (1956).
H. Sadan and W.D. Kaplan, J. Mater. Sci. 41, 5099 (2006).
H. Okamoto, T.B. Massalski, L.J. Swartzendruber, and P.A. Beck, Bull. Alloy Phase Diagr 5, 592 (1984).
L. Vitos, A.V. Ruban, H.L. Skriver, and J. Kollár, Surf. Sci. 411, 186 (1998).
D. Favez, J.D. Wagnière, and M. Rappaz, Acta Mater. 58, 1016 (2010).
C.C. Chou, H. Chen, and C.M. Wayman, Mater. Charact. 26, 93 (1991).
D. Amram and E. Rabkin, Acta Mater. 61, 4113 (2013).
D. Amram, L. Klinger, and E. Rabkin, Acta Mater. 60, 3047 (2012).
C.V. Thompson, Annu. Rev. Mater. Res. 42, 399 (2012).
D. Amram, L. Klinger, and E. Rabkin, Acta Mater. 61, 5130 (2013).
D. Amram, O. Kovalenko, and E. Rabkin, Acta Mater. 98, 343 (2015).
W.C. Carter, A.R. Roosen, J.W. Cahn, and J.E. Taylor, Acta Metall. Mater. 43, 4309 (1995).
D. Amram, Y. Amouyal, and E. Rabkin, Acta Mater. 102, 342 (2016).
D. Amram, D. Barlam, E. Rabkin, and R.Z. Shneck, in preparation (2016).
Q. Meng, Y. Rong, and T. Hsu, Phys. Rev. B 65, 174118 (2002).
A.S. Shirinyan, Y.S. Bilogorodskyy, G. Wilde, and J.W.P. Schmelzer, J. Phys.: Condens. Matter 23, 245301 (2011).
R.G. Aspden, J.A. Berger, and H.E. Trout, Acta Metall. 16, 1027 (1968).
C. Cayron, Acta Mater. 96, 189 (2015).
D. Amram, O. Kovalenko, and E. Rabkin, in preparation (2016).
W.W. Mullins and G.S. Rohrer, J. Am. Ceram. Soc. 83, 214 (2000).
T. Epicier, C. Esnouf, M.A. Smith, and D. Pope, Philos. Mag. Lett. 65, 299 (1992).
D. Amram and E. Rabkin, ACS Nano 8, 10687 (2014).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Amram, D., Rabkin, E. Phase Transformations in Au-Fe Particles and Thin Films: Size Effects at the Micro- and Nano-scales. JOM 68, 1335–1342 (2016). https://doi.org/10.1007/s11837-016-1847-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11837-016-1847-8