
Abstract
Although Agave angustifolia Haw is one of the most cultural and industrial promising species in Mexico, no extensive genomic resources were available. Herein, we explore the transcriptome data set of this species. Total RNA from stem, roots, young and mature leaves was purified and sequenced by NovaSeq6000 system. From the de novo assembly of A. angustifolia, a total of 358,170 transcripts with average length of 1117 bp, N50 of 858 bp, and GC mean content of 43.5% were obtained. About 48.7% of the transcripts had open reading frame (ORF). Hence, 63.4% and 42.7% of the ORFs were annotated using Blastp and Blastx against UniProtKB database, respectively, uncovering the presence of at least 29,273 genes in the A. angustifolia transcriptome. The six pairwise comparisons between plant tissues resulted in the identification of 123,181 differentially expressed transcripts (23,978 were upregulated and 79,203 were downregulated). Several carbohydrate metabolism pathways were significantly enriched from the comparisons between leaves and stems. Thus, genes regulated for the fructan, starch and sucrose biosynthesis were discussed. Furthermore, genes involved in cellulose synthesis were explored being the members of the cellulose synthase genes subfamily were highly expressed in young leaf, while the members of the cellulose synthase-like subfamily were preferentially expressed in mature leaf. Moreover, A. angustifolia transcriptome served for the identification of 30,766 SSR and 21,686 VNTR potential markers. Overall, the transcriptomic data generated in this study provide an invaluable resource to understand the biology of A. angustifolia, offering new insight to understand their physiology and adaptation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Agave is a diverse plant genus originated from the arid area of North America and account for approximately 200 species, where three quarters of Agave species are endemic to Mexico (García-Mendoza 2002). Their morphological and physiological characteristics allow them to survive and thrive in harsh environments (Borland et al. 2009; Simpson et al. 2011). Agaves contribute to Mexico’s economic income and have been a valuable cultural tradition since pre-columbian times. Agaves are primarily used for spirit beverages processing (Pérez-Zavala et al. 2020). Currently, they are used to produce tequila (A. tequilana Weber var. azul), mezcal (A. angustifolia Haw, A. potatorum Zucc, A. salmiana Otto and other species), and bacanora (A. angustifolia Haw) (Espinosa-Andrews et al. 2021). Moreover, Agave plants have been used to produce natural fibers, food, medicinal compounds, and other uses (López-Romero et al. 2018).
Agave angustifolia Haw, the most employed to produce mezcal, is the largest geographically distributed species (Gentry 1982). Taxonomically, A. angustifolia belongs to “Rigidae” section along with 12 other species (Gentry 1982; Simpson et al. 2011). In Mexico, the state of Oaxaca has the highest species richness with 39 species (García-Mendoza and Chiang 2003; Rivera‑Lugo et al. 2018). Thus, the cultivar “Espadín” (A. angustifolia) is the most domesticated and cultivated in Oaxaca.
For mezcal production, the main carbohydrate material is fructan. In addition, Agave fructans can be used to prevent or treat numerous human diseases (Ritsema and Smeekens 2003), and they are used as ingredients to improve color, flavor and other sensory attributes (Espinosa-Andrews et al. 2021). Four fructosyltransferase enzymes are implicated in the fructan biosynthesis: fructan:fructan 1-fructosyltransferase (1-FFT), sucrose:sucrose 1-fructosyl transferase (1-SST), fructan:fructan 6G-fructosyltransferase (6G-FFT) and sucrose:fructan 6-fructosyltransferase (6-SFT). Previously, some genes encoding fructan exohydrolases, fructosyltransferases and invertases were molecularly characterized from A. tequilana, A. deserti, A. victoriae–reginae and A. striata (Cortés-Romero et al. 2012; Suárez-González et al. 2014; Avila de Dios et al. 2015).
Compared to plant model species, countable studies have been carried out on Agave species at the genomic level despite their social and economic importance. This is mainly because of the absence of any reference genome sequence, limited basic genetic knowledge, high number of repetitive elements, large size of the genome (about 4 Gbp) and median-long-life cycle (5–12 years) (Simpson et al. 2011; McKain et al. 2012). Moreover, the ploidy level varies from 2 to 8n (n = 30), even within species, and hybridization could occur in wild cultivars, contributing to increase complexity of polyploid–hybrid genomes (McKain et al. 2012; Palomino et al. 2017).
In recent years, high-throughput transcriptome sequencing has been widely used to study gene expression to compare different biological and agricultural conditions (Conesa et al. 2016). The researchers were focused on bioenergy production in A. tequilana (Simpson et al. 2011), CAM photosynthesis in A. americana (Abraham et al. 2016; Yin et al. 2018) and response to drought stress in A. sisalana (Sarwar et al. 2019). Moreover, conservation of a genetic program for regeneration in A. salmiana (Cervantes-Pérez et al. 2018) and identification of cellulose synthase genes in Agave hybrid H11648 (Huang et al. 2019) were explored via RNA-seq analysis. Avila de Dios et al. (2019) used transcriptome analysis to study the regulatory mechanisms of the flowering initiation in A. tequilana. Moreover, RNA-seq analysis was used to survey the flavonoid production in A. lechuguilla (Morreeuw et al. 2021), cell wall metabolism and fiber lignification in Agave tequilana, hybrid 11,648, A. sisalana and A. fourcroydes (Raya et al. 2021; Maceda-López et al. 2022), and the expression of Expansin genes from Agave amanuensis (Wang et al. 2023). Likewise, RNA-seq studies were also used to determine the relationships between agave species. The global transcriptome profile similarity between A. deserti and A. tequilana was deciphered by Gross et al. (2013), and phylogenetic analysis between A. deserti, A. sisalana and A. tequilana was studied by Huang et al. (2018). Recently, a transcriptomic study from A. angustifolia leaves was focused on revealing conservation and diversification in the expression of cinnamyl alcohol dehydrogenase genes in Agaves (Huang et al. 2022).
To enhance genomics studies in Agave species, the objective of the present study was to present a comprehensive de novo transcriptome assembly of A. angustifolia based on different plant tissues to cover a broad variety of transcripts. In addition, tissue transcriptome profiles were compared through differential gene expression analysis. This could facilitate the identification of candidate genes involved in carbohydrates and cellulose biosynthesis, and to develop several types of molecular markers.
Materials and methods
Plant sampling and RNA sequencing
A healthy 3-year-old plant of A. angustifolia was collected from the “Paraje el Limón” farm in Santiago Yosondúa, Oaxaca, Mexico (16° 47′ 58.8″ N; 97° 35′ 15.6″ W). The plant was dissected into young leaves, mature leaves, roots and stems. Three individual samples were obtained from each tissue representing biological repeats. The samples were cut into small pieces of ~ 0.5 cm3 using sterile scalpel and forceps, and then, they were frozen in liquid nitrogen. Total RNA was purified using PureLink™ RNA Mini Kit (Invitrogen, USA) following the manufacturer's instructions. The quality and quantity of the RNAs were determined using RNA Pico Sensitivity Reagent Kit and LabChipGX Touch PE (PerkinElmer, USA). Twelve libraries were synthesized using the TruSeq Stranded mRNA LT Sample Prep Kit. To verify the size of the PCR-enriched fragments, the fragment size distribution was analyzed by an Agilent 2100 bioanalyzer using a DNA 1000 chip. Sequencing of 101 bp paired end reads was achieved using NovaSeq6000 (Illumina, USA) by Macrogen (Seoul, South Korea).
Data filtering and de novo assembly
Quality of raw reads was examined using FastQC v0.11.8 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Trimmomatic v0.36 (Bolger et al. 2014) was used to remove the low-quality reads (Phred quality score < 30) and discard short sequences (length < 60 bp). As no reference genome was available for the genus Agave, we assembled the transcriptome de novo to avoid the potential bias introduced using other species as base for genome-guided assembly (Conesa et al. 2016). The filtered reads were de novo assembled using Trinity v2.9.1 (Grabherr et al. 2011) applying the default parameters and contig length of 300 bp. The assembly was performed on Galaxy web platform (Cock et al. 2015). Subsequently, the potential duplicated transcripts for a similarity threshold of 95% were eliminated using CD–HIT–EST program (Fu et al. 2012). Assembly quality was assessed using Bowtie2 (Langmead and Salzberg 2012) and Trinity (Grabherr et al. 2011) mappers by aligning back the reads to the representative assembly. Moreover, Benchmarking Universal Single-Copy Orthologs (BUSCO; Simão et al. 2015) against eukaryota_odb10, Embryophyta, Eudicotyledons and viridiplantae_odb10 data sets were used to further ensure assembly quality. The TransDecoder v5.5 tool, implemented on the Galaxy web platform (Cock et al. 2015), was used to predict coding regions that were at least 100 amino acids length from the transcripts resulted from the representative assembly. Moreover, the transcriptome similarities between A. angustifolia (this study), A. tequilana, A. deserti (Gross et al. 2013) and A. lechuguilla (Morreeuw et al. 2021) were analyzed using reciprocal BLAST searches for percent identity cutoff of 90% and set expectation value cutoff of 10–5, using Megablast (NCBI BLAST + blastn) integrated in Galaxy (Camacho et al. 2009; Cock et al. 2015). Furthermore, Orthologs between the four Agave species were identified from OrthoVenn3 web server (Sun et al. 2023) using the translated protein sequences with an e value threshold of 1e−10. Likewise, A. angustifolia transcriptomic was compared against multiple species (Arabidopsis thaliana, Vitis vinifera, Oryza sativa and Zea mays), selected from the best hits of Blast analyses.
Functional annotation
The de novo transcriptome assembly, TransDecoder results and SwissProt protein database (563,972 sequences) (The UniProt Consortium 2021) were uploaded to and analyzed on the Galaxy web platform (usegalaxy.org, usegalaxy.eu and usegalaxy.org.au; Afgan et al. 2018). The assembled transcripts were aligned against the Diamond_makedb of SwissProt database, using Diamond Blastp and Diamond Blastx (Diamond v0.9.29.130) for best hits with an e value threshold of 1e−5. Moreover, EggNOG v5.0 (Huerta-Cepas et al. 2019) was used to annotate the assemblies. Furthermore, Clusters of Orthologous Groups (COG; Tatusov et al. 2000) and Mercator4 (Schwacke et al. 2019) were used to further annotate the assembled transcripts.
Differential expression transcripts (DETs), gene ontology (GO) and KEGG analyses from DETs
To quantify the relative expression abundance of each transcript, differential expression analyses were performed using Limma-voom program (Liu et al. 2015). Several pairwise comparisons between tissues were evaluated: young leaf (YL) vs mature leaf (ML), young leaf (YL) vs stem (P), mature leaf (ML) vs stem (P), young leaf (YL) vs roots (R), mature leaf (ML) vs roots (R), and stem (P) vs roots (R). Differentially expressed transcripts (DETs) with at least an absolute log (fold-change) > 2 and an adjusted P value threshold < 0.01 were extracted and queried against the Arabidopsis thaliana protein database using KOBAS-i (Bu et al. 2021) to identify enriched KEGG pathways and GO terms (e value of 1e −5).
Quantitative RT-qPCR validation
Selected transcripts for fructan and cellulose synthesis were selected to verify the RNA-seq expression data. Gene-specific primers were designed using Primer3 software (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). Gene quantification was carried out on the CFX96 system (Bio-Rad, USA) using the SYBR® Green PCR Master Mix. Thermal settings were: 95 °C for 1.5 min for initiation, followed by 40 cycles at 95 °C for 30 s for denaturation, then 60 °C for 45 s for annealing and extension. Data were calculated according to the 2−ΔΔCT method. The transcript levels were normalized relative to the ALDH gene. Three biological and three technical replicates were used for the RT-qPCR assays.
Molecular markers prediction form A. angustifolia
Krait v1.3.3 (Du et al. 2018) was used to mining the single sequence repeats (SSR) markers present in the A. angustifolia transcriptome assembly. The minimum repeat for each perfect SSR type was set to 20 for mono-, 10 for di-, 6 for tri, 5 for tetra-, 5 for penta-, 4 for hexanucleotide. For compound microsatellites (cSSRs), the maximum distance allowed between adjacent SSRs as to account as compounded (dMax) was specified to 10 bp. For variable number tandem repeats (VNTRs), the minimum motif length and maximum motif length were set to 7 and 30, respectively. The Primer3 tool, included in Krait 1.3.3, was used for primer designing. Furthermore, single-nucleotide polymorphism (SNPs) and insertion–deletion polymorphism (InDels) callings were carried out using the Samtools mpileup v2.1.4 (Li et al. 2009) and VarScan mpileup v2.4.3.1 (Koboldt et al. 2012), by aligning the cleaning reads against the representative assembled transcriptome. Minimum coverage and minimum supporting reads were set as 100 and minimum base quality as 30.
Identification of putative orthologs and phylotranscriptomic analysis
RNA-seq data for ten available agave species data were downloaded from public archives and analyzed as for A. angustifolia. Peptides sequences for the agave assemblies were clustered at a similarity threshold of 90% using CD–HIT–PROTEIN program. Ortholog genes among the ten species were identified using the OrthoVenn3 web server applying OrthoMCL algorithm with an e value threshold of e−10 (Sun et al. 2023). Based on 5863 single-copy genes, FastTree2 was used to construct a phylogenetic evolutionary tree by maximum likelihood method using the evolutionary model JTT + CAT (Sun et al. 2023).
Results
Transcriptome sequencing and assembling
A total of 610,057,174 raw 101 bp paired-end reads were generated from young leaf (YL), mature leaf (ML), stem (P) and roots (R) of A. angustifolia (Supplementary Table S1). The raw sequence files have been uploaded to the National Center for Biotechnology Information, Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra/) under the BioProject accession numbers SRX11362104-06, SRX11366017-19, SRX11377409-11 and SRX11369602-04.
After quality control, 582,033,692 (95.4%) high-quality reads were survived, ranging from 41,850,658 (P1 library) to 54,176,454 reads (ML1 library). The percentage of duplicate reads ranged from 54.4% (ML2 and ML3 libraries) to 74.4% (R3 library) with an average of 61.5% (Supplementary Table S1). For the initial de novo assembly, 487,028 transcripts were assembled. Furthermore, the use of CD–HIT–EST to remove duplicates and to cluster assembled transcripts into representative sequences resulted in 358,170 transcripts, representing a total of 185,092 loci (after clustering of isoforms in unigenes). The overall GC content was 43.5%, while the average length of transcripts and the N50 were 858 bp and 1,117 bp, respectively.
Assembly quality was assessed by mapping back the reads to the assembly. From Bowtie2, the average alignment rate was 96.2%, ranging from 94.2% (R2) to 97% (HM1). From Trinity mapper, about 90.4% of the reads were mapped back to the transcriptome. Moreover, BUSCO revealed the presence of 98.8% of complete BUSCOs out of the 255 orthologous genes searched from eukaryota_odb10, 97.2% of the 1,614 orthologues searched from Embryophyta data set, 91.8% of the 2,326 orthologues searched from Eudicotyledons data set and 99.1% of the 425 orthologues (57 species) searched from viridiplantae_odb10 data set. From the later, 421 orthologues were complete (53 were single copy and 368 were duplicated), four were fragmented and no missing orthologue was reported.
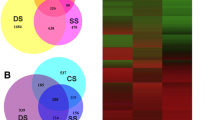
Based on the representative assembly, the four tissues shared 38.3% (137,102 transcripts) of the total transcripts. The leaves shared more transcripts (194,620) compared to those shares with stem (190,213 and 183,874 transcripts shared between mature leaf and stem, and between young leaf and stem, respectively) or roots (158,019, 156,827 and 152,644 transcripts shared between mature leaf and roots, between stem and roots, and between young leaf and roots, respectively). In addition, young leaf, mature leaf, roots, and stem presented 2904, 5590, 115,628 and 2341 “unique” transcripts, respectively (Fig. 1a).

a Shared and unique transcripts from the four tissue samples of Agave angustifolia. b Venn diagram of the annotated transcripts by Blastx and Blastp against UniProtKB/Swiss-Prot database
Structural and functional annotation A. angustifolia transcriptome
The representative transcriptome assembly of A. angustifolia was annotated using multiple methods and resources to provide comprehensive annotation. Predict coding regions from TransDecoder analysis resulted in 174,414 (48.7%) peptide sequences (95,752 loci, when only Trinity unigenes were considered). Blastp research against the UniProtKB/Swiss-Prot database uncovered 110,525 significant hits (60,879 Trinity unigenes, 25,966 genes) representing 63.4% of A. angustifolia assembly. Blastx research against the same protein database resulted in 153,007 significant hits (78,348 unigenes and 24,051 genes), representing 42.7% of the total 358,170 transcripts. Overall, 29,273 genes were identified from Blastp and Blastx analyses (Fig. 1b).
EggNOG-mapper analysis revealed that 65,024 transcripts were associated with at least one GO term and 41,371 transcripts with at least one KEGG Pathway. Overall, a total of 11,404 GO terms were identified (26% of total 43,850 in the GO database). From these, 7521 GO terms were assigned to biological process (BP) category (26.4% of the 28,503 GO terms in BP category), 2,702 GO terms were classified as molecular function (MF) category (24.2% of the 11,168 GO terms in MF category), and 1,181 GO terms were sorted in cellular component (CC) category (28.3% of the 4179 GO terms in CC category). Moreover, a total of 296 KEGG map pathways were identified (72.4% out of the total of 547 pathways in KEGG database).
From the COG annotation, A. angustifolia transcripts could be divided into 24 COG categories. The cluster for “Function Unknown” represented the largest group (36,878 transcripts), followed by “Signal Transduction” (14,178 transcripts), “Posttranslational modification, protein turnover, chaperone functions” (12,800 transcripts), “Transcription” (11,014 transcripts) and “Carbohydrate metabolism and transport” (8,253 transcripts).
Similarly, from the MapMan4 ontology annotation, out of the 358,170 submitted DNA sequences to Mercator4, 142,439 were annotated and 74,279 were classified. A total of 4694 (96.4%) transcripts occupied Bins, from the 4868 available Bins. The occupied Bins by category was greater than 88% for most of the categories except for the “Clade-specific metabolism” category (23%). Twenty of the 29 categories had occupied Bins greater than 96% and 7 of them had full occupancy. The average of occupied Bins (94%) was similar to that of Arabidopsis thaliana (96%) and higher than that of Solanum lycopersicum (91%) and Zea mays (90%).
Comparisons of A. angustifolia transcriptome against other plant species
The transcriptome of A. angustifolia (358,170 transcripts) was compared against the previously published transcriptomes of A. tequilana (204,530 transcripts; Gross et al. 2013), A. deserti (128,869 transcripts; Gross et al. 2013) and A. lechuguilla (636,383 transcripts; Morreeuw et al. 2021). A significant portion of each Agave transcriptome aligns to its counterpart. A total of 115,117, 83,075, 75,875 transcripts of A. angustifolia was aligned to A. tequilana, A. lechuguilla and A. deserti, respectively. Furthermore, the comparison of 174,414 peptide sequences of A. angustifolia using OrthoVenn3 (Sun et al. 2023) against the Agave species [A. tequilana (85,381 protein sequences), A. deserti (70,404 sequences) and A. lechuguilla (89,938 sequences)] revealed that 14,508 orthologous gene clusters were shared between the four compared Agave species. Moreover, 23,780, 22,793, and 22,791 orthologous were shared between A. angustifolia and A. tequilana, A. angustifolia and A. lechuguilla, A. angustifolia and A. deserti, respectively (Fig. 2a). In addition, A. angustifolia, A. lechuguilla, A. tequilana and A. deserti presented 13,442, 3525, 2617 and 1255 unique transcripts, respectively (Fig. 2a). Likewise, the comparison of 174,414 peptide sequences of A. angustifolia using OrthoVenn against various plant species, revealed that 11,806, 12,094, 12,669 and 12,408 orthologous gene clusters were shared between A. angustifolia and A. thaliana, A. angustifolia and V. vinifera, A. angustifolia and O. sativa, and A. angustifolia and Z. mays, respectively (Fig. 2b). Besides, 7672 orthologous were shared between the five compared species (Fig. 2b).

Venn diagram for unique and shared genes between a Agave angustifolia, A. tequilana, A. deserti and A. lechuguilla, and between b A. angustifolia, Arabidopsis thaliana, Vitis vinifera, Oryza sativa and Zea mays
Differentially expressed transcripts (DET) and functional enrichment of DET
Limma-voom method was used to extract differentially expressed transcripts with adjusted P value threshold < 0.01 and |LogFC|> 2. Multidimensional scaling plots showed that the first three dimensions accounted for approximately 73% of the data variability. The first dimension explained the most variation (43%), where a clear separation between root, stem and leaves was revealed. While the separation between the two leaf types was more apparent on the second and third dimensions. Several pairwise comparisons between plant tissues were evaluated, resulting in 123,181 DETs (23,978 were upregulated and 79,203 were downregulated) from the six pairwise comparisons. As expected, transcriptome profiles differ between Agave tissues according to their respective physiological functions. Clear variations were found on the composition of the DET between any two tissues comparison (Fig. 3). Out of the total 123,181 DETs, only 72 DETs were shared between all comparisons and 1,057 DETS were shared when comparisons that include roots were excluded, suggesting the specialized functions and differentiation between A. angustifolia tissues that might be involved in different sets of genes with distinct patterns of expression.

Differentially expressed transcripts (DETs) between A. angustifolia tissue pairwise comparisons [young leaf (YL), mature leaf (ML), stem (P) and roots (R)]. Limma-voom method was used to extracted DETs with adjusted P value threshold < 0.01 and |LogFC|> 2
To understand the molecular difference between these comparisons, functional annotation was determined from DETs using KOBAS tool via KEGG and GO enrichments.
For the YL vs ML comparison, 31 KEGG pathways were significantly enriched (P < 0.01) and the most enriched pathways were “Metabolic pathways”, “Biosynthesis of secondary metabolites” and “Carbon metabolism” (Supplementary Table S2). Moreover, 221 GO terms were significantly enriched (P < 0.01). These GO terms fell under BP category (BP, 126), followed by MF (62) and CC (33). The most enriched GO terms were “plasma membrane” (CC), “protein phosphorylation” (BP) and “protein binding” (MF). Moreover, “plant-type secondary cell wall biogenesis”, “photomorphogenesis”, “leaf senescence”, “cellulose biosynthetic process”, and “lignin biosynthetic process” were also enriched in the BP category (Supplementary Table S3).
For the ML vs P comparison, 32 KEGG pathways were significantly enriched (P < 0.01) and the most enriched pathways were “Biosynthesis of secondary metabolites” and “Metabolic pathways”. Furthermore, “Plant hormone signal transduction”, and “Starch and sucrose metabolism” pathways were also enriched (Supplementary Table S2). Moreover, 230 significantly enriched GO terms (P < 0.01) were recovered. These GO terms fell under BP category (133), followed by MF (63) and CC (34). The most enriched GO terms were “plasma membrane” (CC), “protein phosphorylation” (BP) and “kinase activity” (MF) (Supplementary Table S8).
For the YL vs P comparison, 40 KEGG pathways were significantly enriched (P < 0.01) and the most enriched pathways were “metabolic pathways” and “biosynthesis of secondary metabolites”. Likewise, “starch and sucrose metabolism”, “plant hormone signal transduction”, and “fructose and mannose metabolism” pathways were also enriched (Supplementary Table S2). Moreover, 227 GO terms were significantly enriched (P < 0.01). These GO terms fell under BP category (133), followed by MF (63) and CC (34). The most enriched GO terms were “plasma membrane” (CC), “protein phosphorylation” (BP) and “kinase activity” (MF). In the BP category, “leaf development”, “carbohydrate metabolic process” and “photomorphogenesis” were also enriched (Supplementary Table S3).
For the YL vs R comparison, no one of the KEGG pathways or GO terms was significantly enriched (P < 0.01). Moreover, for the ML vs R comparison, only one KEGG pathway (“Plant hormone signal transduction”) was enriched (Supplementary Table S2). However, 12 significant enriched GO terms (P < 0.01) were retrieved. These GO terms fell under BP category (5), MF (5) and CC (2) (Supplementary Table S3).
For the P vs R comparison, 5 KEGG pathways were significantly enriched (P < 0.01). “Plant hormone signal transduction” and “Ribosome” were the most enriched pathways (Supplementary Table S2). Moreover, 51 GO terms (P < 0.01) were significantly enriched. These GO terms fell under BP (21), followed by MF (21) and CC (9) (Supplementary Table S3).
Carbohydrate pathways from A. angustifolia transcriptome
From the 15 carbohydrate metabolism pathways reported in KEGG database, 8, 11 and 6 pathways were significantly enriched (P < 0.01) from the YL vs ML, YL vs P, and ML vs P comparisons, respectively. Any carbohydrate metabolism pathway was enriched in the comparisons involving root samples (YL vs R, ML vs R, and P vs R). Moreover, some carbohydrate metabolism pathways were enriched exclusively from YL vs P, namely, “citrate cycle (TCA cycle)”, “pentose and glucuronate interconversions”, “inositol phosphate metabolism” and “ascorbate and aldarate metabolism”, while “amino sugar and nucleotide sugar metabolism” pathway was uniquely enriched from the YL vs ML comparison (Table 1).
For the comparisons of young leaf vs stem and mature leaf vs stem, the annotated genes related to starch metabolism showed similar behavior: alpha-amylase 2 and alpha-amylase isozyme 3C (AMY2, AMY1.7; EC 3.2.1.1), beta-amylase 1, 3 and 7 (MBY1, BAM1, BAM3, BAM7; EC 3.2.1.2), Isoamylase 3 (ISA3; EC 3.2.1.68), glycogen phosphorylase 1 (glpV), alpha-glucan phosphorylase, H isozyme (EC 2.4.1.1), Hexokinase-3 (HXK3; EC 2.7.1.1), granule-bound starch synthase 1 and 2 (GBSSI1 and GBSSI2; EC 2.4.1.21), and glucose-1-phosphate adenylyltransferase large subunit 1 (ADG2; EC 2.7.7.27) were unregulated in leaves. Furthermore, the most annotated genes involved in the sucrose metabolism: sucrose synthase 2, 3 and 7 (SUS2, SUS3, SUS7; EC 2.4.1.13), probable sucrose-phosphate synthase (SPS1; EC 2.4.1.134), fructokinase-1 (FRK1; EC 2.7.1.4), and glucose-6-phosphate isomerase 1 (PGI1; EC 5.3.1.9) were upregulated in stem. Likewise, genes involved in fructan biosynthesis: 6(G)-fructosyltransferase (6G-FFT; EC 2.4.1.243), bifunctional 6(G)-fructosyltransferase/2,1-fructan:2,1-fructan 1-fructosyltransferase (EC 2.4.1.243) and sucrose:sucrose 1-fructosyltransferase (1-SST, EC 2.4.1.99) were upregulated in stem (Supplementary Table S4). For the comparisons of young leaf vs mature leaf, the genes involved in the metabolism of sucrose and starch [beta-amylase (BMY1; EC 3.2.1.1), beta-amylase 1 (BAM1; EC 3.2.1.2), alpha-amylase isozyme 3C (AMY1.7; EC 3.2.1.2), sucrose synthase 1, 2 and 3 (SUS1, SUS2, SUS3; EC 2.4.1.13), fructokinase-1 (FRK1; EC 2.7.1.4), and probable sucrose-phosphate synthase 2 (SPS2; EC 2.4.1.14)] tended to be upregulated in the mature leaf, whereas the genes involved in the biosynthesis of starch, i.e., granule-bound starch synthase 1 (GBSSI; EC 2.4.1.21), and putative fructokinase-8 (At1g50390; EC 2.7.1.4) tended to be upregulated in the young leaf. Furthermore, the genes involved in fructan biosynthesis [6(G)-fructosyltransferase (6G-FFT; EC 2.4.1.243), bifunctional 6(G)-fructosyltransferase/2,1-fructan:2,1-fructan 1-fructosyltransferase (EC 2.4.1.243), and beta-fructofuranosidase CWINV2; EC 3.2.1.26] were upregulated in the young leaf (Supplementary Table S4).
Fructans are the main storage polysaccharides found in Agave species. Thus, the seven fructosyltransferases and invertases genes from A. tequilana, previously characterized by Cortés-Romero et al. (2012) and Mancilla-Margalli et al. (unpublished), were retrieved and blasted against our A. angustifolia assembly. Only 1-SST1 was found to be completely assembled (2896 bp). The remaining genes (1-SST2, 6G-FFT1, 6G-FFT2, 1-FFT, Vinv1 and Cwinv1) were found to be fragmented, composed of 2–4 transcripts. However, the expression behavior of the transcripts within the same gene was similar (Supplementary Fig. S1). Globally, the genes 1-SST1, 1-SST2 and 6G-FFT1 were upregulated in stem and roots. Moreover, 1-SST2 was also upregulated in young leaf and 1-FFT was only upregulated in stem (Fig. 4a). Likewise, 6G-FFT2 was upregulated in all tissues except in roots, and Vinv1 and Cwinv1 were upregulated in young leaf and roots. Furthermore, Cwinv1 was also upregulated in mature leaf (Fig. 4a).

In silico RNA-seq-based expression analysis of A. angustifolia Fructan (a) and cellulose synthesis (b) genes. Heatmap graph was constructed basted on Z-scores punctuation of standardized read counts from mature leaf (ML), young leaf (YL), stem (P) and roots (R)
Cellulose synthesis genes
The transcript expression profile of cellulose synthesis was explored and compared between the four tissues. Differentially expressed transcripts for each comparison was extracted and blasted against the Cellulose_synt (PF03552) family (10,927 sequences). A total of 24 members (from 366 transcripts of A. angustifolia) of the Cellulose Synthase Gene Superfamily [11 cellulose synthase genes (CesA) and 13 cellulose synthase-like (Csl)] were identified and 20 members (10 CesA and 10 Csl; 122 transcripts) were differentially expressed (P < 0.05 and (|LogFC|) > 1) in at least one of the tissues comparisons (Supplementary Table S5). Globally, the differentially expressed CesA and Csl were upregulated in roots. Moreover, the members of the CesA subfamily were preferentially upregulated in young leaf, whereas the members of the Csl subfamilies were preferentially upregulated in mature leaf (Fig. 4b; Supplementary Fig. S2).
Validation of the RNA-seq data
To validate the result of the RNA-seq analysis, 11 genes (Supplementary Table S6) were chosen for qRT-PCR quantification to compare their expression levels among the various tissues of A. angustifolia. The genes 1-SST1 and 6G-FFT2 had the highest expression in pineapple. Likewise, 1-SST2 and 6G-FFT1 had the higher expression in pineapple and roots. Similarly, Cwinv1 had the highest expression in the root. The gene Vinv1 had the highest expression in young leaf and lowest expression in mature leaf (Supplementary Fig. S3). For the cellulose genes and focusing on the comparison between the two types of leaves, the genes CesA5, CesA7 and CesA8 had higher expression in the young leaf, while CslD5 and CslG3 had higher expression in the mature leaf (Supplementary Fig. S3). The gene expression trends estimated by qRT-PCR were consistent with those found in the RNA-seq data, meaning that the in-silico transcriptomic analysis was reliable.
SSRs, VNTRs, InDels and SNPs prediction
Using Krait v1.3.3 tool (Du et al. 2018), we identified a total of 30,175 perfect SSR and 591 compound SSR markers, representing about 0.25% of the A. angustifolia transcriptome. The average relative density of microsatellites was one SSR per 2.5 kb. Based on the motif repetition, these SSRs were further categorized into di-nucleotide (12,747), followed by tri-(10,847), mono-(3353), hexa-(1756), tetra-(1099) and pentanucleotides motifs (373). Moreover, (A)n motif was the most frequent for mononucleotide, (AG/AC)n for di- while (AAG/AAA/ACG/AGG)n for trinucleotide microsatellites. Likewise, we identified a total of 21,686 VNTR markers, representing about 0.13% of the A. angustifolia transcriptome. The average relative density of VNTR was one VNTR per 1.3 kb. In addition, 18,897 pair primers for perfect SSR, 261 pair primers for cSSR, and 17,455 pair primers for VNTRs were verified for a unique amplification by in silico PCR for product size of 100–300 bp (Supplementary Table S7).
Furthermore, putative variants were called using VarScan by aligning the cleaning reads against the representative transcriptome. From 358,170 transcripts, 397,108 high confidence variant positions were identified including 382,589 SNPs and 14,519 InDels. The transition mutations were more frequent (64.93%) being the transition between A and G occurred most frequently (33.96%) and transition between C and T was the second most frequent (30.97%). The transversion frequency (35.07%) varied between 8.4% (transversion between A and C), 8.78% (transversion between A and T), 8.94% (transversion between G and T), and 8.95% (transversion between C and G).
Phylotranscriptomic analysis
The phylogenetic reconstruction of the ten Agave species (Supplementary Table S8) based on 5,863 single-copy orthologs showed the formation of two clusters (Fig. 5). The first cluster formed by A. lechuguilla, A. bracteosa and Manfreda scabra. The second cluster was formed by A. deserti, A. salmiana, A. sisalana, A. tequilana, Agave hybrid H11648, A. fourcroydes and A. angustifolia. These last four species formed a subgroup, although Agave hybrid H11648 was more closely related to A. tequilana than to A. angustifolia (Fig. 5).

Phylogenetic tree of Agave species based on single-copy orthologs retrieved from transcriptome assemblies and reconstructed by maximum likelihood method using the JTT + CAT evolutionary model. The branches frequency less than 90 is not shown in the figure
Discussion
Although A. angustifolia HAW is a plant of great economic importance for Mexico, few genomic resources are available. The knowledge about the molecular mechanisms of Agave to underlying its adaptability for xerophyte lands and tolerance to abiotic stresses, are still scarce due, in part, to the limited genomic and molecular information in public databases. Therefore, the transcriptomic data generated in the present research could provide an invaluable resource to comprehend the physiology of Agave, offering new resources that will facilitate the identification of candidate genes involved in development, physiology, and metabolism of this plant.
The de novo assembly of A. angustifolia transcriptome allowed the identification of 358,170 transcripts (185,092 loci). Depending on the RNA-seq technology, depth of sequencing, samples conditions, Agave species, and the bioinformatics programs and the specified parameters, the results of the assembly could vary. A total of 67,314 unigenes were assembled from A. angustifolia (Huang et al. 2022). Moreover, 204,530 transcripts (139,525 loci) and 128,869 transcripts (88,718 loci) were assembled from A. tequilana and A. deserti, respectively (Gross et al. 2013), 170,844 transcripts from A. salmiana (Cervantes-Pérez et al. 2018), 93,141 transcripts form A. sisalana (Sarwar et al. 2019), 148,046 transcripts from Agave hybrid H11648 (Huang et al. 2019), 136,692–170,474 transcripts from A. sisalana, A. fourcroydes and Agave hybrid 11,648 (Raya et al. 2021), and 991,511 transcripts from A. lechuguilla (Morreeuw et al. 2021). The N50 from A. angustifolia transcriptome showed an intermediate value (1117 bp) compared to the previous studies, where the N50 ranged from 658 to 1387 bp (Gross et al. 2013; Cervantes-Pérez et al. 2018; Morreeuw et al. 2021). Moreover, the transcripts average length (858 bp) from A. angustifolia was slightly higher than that reported in other agave species (ranging from 529 to 758 bp: Cervantes-Pérez et al. 2018; Huang et al. 2019, 2022; Sarwar et al. 2019; Morreeuw et al. 2021). The overall GC content was 43.5% in A. angustifolia, a value higher than that reported in A. tequilana (42.3%) (Gross et al. 2013) and lower than that reported in A. sisalana (45.3%) (Sarwar et al. 2019), A. deserti (45.1%) (Gross et al. 2013) and Agave hybrid H11648 (48.8%) (Huang et al. 2019).
The transcriptome completeness and the quality of the assemblies are critical for the accuracy of the downstream analyses, i.e., gene identification, differential gene expression analysis, and genetic molecular developments. Reads mapping back to transcriptome and BUSCO are the most widely used tools for the quality assessment of the de novo assembly. About 96% of the reads were mapped back to transcriptome by bowtie2, showing similar results reported by Sarwar et al. (2019). Moreover, BUSCO results (99.06% orthologues were complete (12.47% single copy and 86.59% duplicated), 0.94% fragmented and 0% missing) were higher compared to other studies (Raya et al. 2021; Sarwar et al. 2019). The high duplication level could be due to alternative splicing, low rate of RNA maturation or combination of both (Avila de Dios et al. 2019). Another possible explanation is the polyploidy level of the Agave genome; the studied variety “Espadín” was reported to have triploid genome (Rivera-Lugo et al. 2018). No partial or complete genome sequence has been published for A. angustifolia, although previous cytophotometry studies estimated that Agave species have genome size around 8 pg (2C DNA content), rating A. angustifolia as medium-sized genome specie (Palomino et al. 2017). All these indicators suggested that we have generated a high-quality transcriptome assembly that could be used for further downstream analyses.
From the reciprocal Blast and orthologous clustering results, A. angustifolia shared more transcripts with A. tequilana compared to A. lechuguilla and A. deserti. These findings support the hypostasis that A. tequilana and A. angustifolia belong to the same genetic pool (Rivera‑Lugo et al. 2018; Trejo et al. 2018). Taxonomically, A. angustifolia and A. tequilana belong to the Rigidae section (Gentry 1982; Simpson et al. 2011). Moreover, orthologous analysis uncovered the tendency that A. angustifolia share more orthologs with other monocotyledonous species (O. sativa and Z. mays) than with dicotyledonous species (A. thaliana and V. vinifera). This was expected as agaves are evergreen monocots, although A. thaliana was the species with the highest occurrence in the annotation results (Blastx and Blastp). Furthermore, the results from the sharing and unique transcripts between young leaf, mature leaf, roots, and stem were similar to those reported for Croton tiglium (Haak et al. 2018) and could reflect the biological, physiological and functional differences of each plant tissues.
Predict coding regions analysis for the A. angustifolia transcriptome resulted in 174,414 (48.7%) potential protein sequences (95,752 unigenes). An amount higher than that reported by Cervantes-Pérez et al. (2018) for Agave salmiana (24%). About 63.4% and 42.7% of the longest ORFs were annotated using Blastp and Blastx analyses against UniProtKB/Swiss-Prot database, respectively, uncovering the presence of at least 29,273 genes in the agave transcriptome. This number is lower than that reported by Gross et al. (2013) for A. tequilana (34,870) and A. deserti (35,086). Given that the potential number of genes in plant species ranges from 20,000 to 94,000 (Michael and Jackson 2013), the assembled transcripts could represent a significant portion of the genes present in A. angustifolia. Moreover, genome size variation may cause changes in the number of genes in plants (Soltis et al. 2003). The proportion of annotated transcripts for agave species varied between 30 and 55% (Avila de Dios et al. 2019; Morreeuw et al. 2021; Raya et al. 2021). The absence of any reference genome or transcriptome of any Agave species or their related ones, and the scarce information in public databases could arguably be the reasons underlying the high number of un-assigned sequence in A. angustifolia transcriptome (Sarwar et al. 2019). Moreover, possible artifacts of the assembly process could cause non-identification of some transcripts (Conesa et al. 2016).
Overall, a total of 396 pathways (72.39% out of the total pathways in KEGG database) were identified. Likewise, for the COG annotation, A. angustifolia transcripts could be divided into 24 COG categories. The cluster for “Function Unknown” represented the largest group indicating that a significant number of agave genes are still not studied yet. Moreover, one of the most represented clusters was “Carbohydrate metabolism and transport” which could explain the active growth of the plant (3 years) and the formation and growth of stem (“pineapple”), an organ for carbohydrates storage. Similarly, from MapMan4 annotation, twenty of the 29 categories had occupied Bins greater than 96% and seven of them had full occupancy. The average of occupied Bins (94%) was similar to that of Arabidopsis thaliana (96%) and higher than that of Solanum lycopersicum (91%) and Zea mays (90%). Altogether, the annotations results suggest the suitability of the data for subsequent analyses viz. identification of genes involved in some important physiological features in A. angustifolia.
The studied agave plant was 3-year-old, corresponding to a very actively vegetative growing stage, and as expected, several GO terms (“leaf development”, “carbohydrate metabolic process”, “photomorphogenesis”, “glycolytic process”, “plant-type secondary cell wall biogenesis”, “cellulose biosynthetic process”, and “lignin biosynthetic process”) and pathways (“Metabolic pathways”, “Biosynthesis of secondary metabolites”, “Photosynthesis”, “Carbon metabolism”, “Biosynthesis of amino acids”, “Phenylpropanoid biosynthesis”, “Starch and sucrose metabolism”, “Plant hormone signal transduction”, “Fructose and mannose metabolism”) related to this phenology stage were enriched.
The “amino sugar and nucleotide sugar metabolism” pathway was uniquely enriched from the Agave leaves comparison. Figueroa et al. (2021) reviewed that nucleotide–sugars are critical intermediates in the biosynthetic pathways of complex carbohydrates. Moreover, the biosynthetic pathways of sucrose and starch represent ones of the major fluxes involving nucleotide–sugars in plants (Figueroa et al. 2021). This result suggests that carbohydrates biosynthesis precursors, including fructan, are located in the leaves of agave. The difference in the number of carbohydrate metabolism pathways suggests that the young leaves (11 pathways) are more active than the mature leaves (8 pathways). Moreover, no carbohydrate metabolism pathway was enriched in the comparisons involving root samples indicating that agave roots do not actively contribute to carbohydrates biosynthesis and reserve.
Starch has the function of a transient carbohydrate storage for actively growing tissues in Agave (Zavala-García et al. 2018). In this research, the genes related to starch metabolism (AMY2, AMY1.7, MBY1, BAM1, BAM3, BAM7, ISA3, glpV, PHS1, HXK3, GBSSI1, GBSSI2, and ADG) were more expressed in leaves than in stem. Similar results were reported by Zavala-García et al. (2018), where some genes of starch metabolism showed the highest levels of expression in apical meristem, followed by leaves. Moreover, low levels of starch were reported in the center of the stem (Mancilla-Margalli and López 2006; Zavala-García et al. 2018). Comparing leaf types, the genes involved in the biosynthesis of starch (GBSSI1 and At1g50390) tended to be more expressed in the young leaf, whereas the genes involved in the metabolism of sucrose and starch (BMY1, BAM1, AMY1.7, SUS1, SUS2, SUS3, FRK1 and SPS2) were predominately more expressed in the mature leaf. Accordingly, Avila de Dios et al. (2019) reported decreasing levels of sucrose in mature leaves in comparison with young leaves in A. tequilana. Moreover, the genes involved in fructan biosynthesis (FFT1 and CWINV2) were upregulated in the young leaf compared to mature leaf. In general, genes involved in fructan biosynthesis (1-FFT and 1-SST), and genes involved in the sucrose metabolism (SUS2, SUS3, SUS7, SPS1, FRK1 and PGI1) were upregulated in stem. Based on these results, we can generate a potential relationship for starch, sucrose and fructan biosynthesis: in photosynthetically active young leaf, starch biosynthesis is very active, and it is transiently accumulated before being metabolized in the young leaf for active growing and/or transited to mature leaf and/or stem. In addition, in mature leaves, starch metabolism is active ensuring the carbohydrate needs of the leaves for its maintenance. Likewise, the energetic sucrose metabolism in stem serves as carbohydrate source for active stem growing or to synthesize starch for short term storage or fructan for long term reserve. Moreover, biosynthesis of fructan was highly active in stems. Likewise, strong expression of genes involved in fructan biosynthesis in young leaf (compared to mature leaf) suggests that fructans are also synthesized de novo in leaves; however, these polymers probably act as osmotic regulators or could be transited to stem (Arrizon et al. 2010; Zavala-García et al. 2018). Moreover, levels of fructan accumulation in stem depend on the plant ages. Whereas 2-year-old plants have around 69% fructans, 4- and 7-year-old plants contain around 97% (Arrizon et al. 2010). Thus, 2–5-year-old plants mainly stored fructooligosaccharides, while 6–7-year-old plants mainly contained long-chain fructans (Mellado-Mojica and López 2012).
Fructans are water-soluble fructose-based polymers synthesized from sucrose (Van den Ende et al. 2009). Four fructosyltransferase (FT) and hydrolytic invertases genes (1-SST, 1-FFT, 6-SFT, and 6G-FFT) are needed to synthesize fructans, whereas exohydrolases (FEH) ensures its degradation. In-silico RNA-seq and RT-qPCR quantifications of genes involved in fructan metabolism uncovered very clear patterns associated with tissue type. The three fructosyltransferase detected genes were highly expressed in the stem contrary to the invertase genes. Although the main storage organ is the stem, fructans are found in almost all Agave organs (Mellado-Mojica and López 2012; Pérez-López and Simpson 2020). Previously, isoforms of 1-SST and 6G-FFT were retrieved from other agave species (Cortés-Romero et al. 2012; Suárez-González et al. 2014, 2016; Avila de Dios et al. 2015, 2019). However, gene encoding 6-SFT enzyme has not yet been reported in agaves (Pérez-López and Simpson 2020; Gomez-Vargas et al. 2022). Globally, the three detected FT genes were highly expressed in the stem contrary to the invertase genes. 1-SST gene showed distinct patterns of expression across A. angustifolia tissues, contrary to patterns previously reported for A. tequilana and similar to patterns found in A. victoriae–reginae in other tissues (Avila de Dios et al. 2015). 1-SST isoforms were highly expressed in stem and 1-SST2 isoform was upregulated in young leaf. Functionally, FTs are enzymes that catalyze the transfer of the fructosyl residue between sucrose molecules or a growing fructose chain (Van den Ende et al. 2009). Thus, 1-SST initiates fructan synthesis by forming the trisaccharide 1-kestose, which, in turn, serves as substrate for the neokestose synthesis by the involvement of 6G-FFT gene (Livingston et al. 2009). 6G-FFT2 isoform was highly expressed in all tissues except in roots. As well, 6G-FFT1 was upregulated in stem and roots. Distinct expression patterns of 6G-FFT isoforms were reported for other tissues form other agave species (Avila de Dios et al. 2015). The highly and distinct expression of 6G-FFT (1 and 2) in stem tissues than in other tissues could be explained by its role in the synthesis of more complex fructan molecules (Pérez-López et al. 2021). Therefore, the 6G-FFT enzymes produce “neo” type fructans (Van den Ende et al. 2009) and could serve as signals at specific developmental stages (Avila de Dios et al. 2019). Moreover, the presence of FE genes in leaves was correlated with the synthesis of low degrees of polymerization (DP) fructans, which suggested to be transported to the stem, where higher DP fructans were produced and stored (Cortés-Romero et al. 2012). The expression of 1-FFT was only upregulated in stem. As function, the 1-FFT enzyme is responsible for high DP fructan formation in plants (Van den Ende et al. 2006). It is worth mentioning that the genetic structure and molecular function of the 1-FFT gene (GenBank accession No. EU026119.2) has not yet been explored and enzyme activity has not been reported for this gene in Agave species. Moreover, cell wall and vacuolar invertases were also included in the analysis, Vinv1 and Cwinv1 were upregulated in young leaf and roots. Furthermore, Cwinv1 was also upregulated in mature leaf. Likewise, high levels of expression for invertase were observed in floral tissues (Avila de Dios et al. 2015). Invertases contribute to the hydrolysis of sucrose to fructose and glucose. These enzymes were associated with tissue growth and carbohydrates storage, and they are present in all plant species (Sturm 1999; Van den Ende et al. 2009).
A. angustifolia has been used as parental for hybridization and one derived hybrid, H11648, is the major cultivated Agave for fiber production in large tropical lands (Huang et al. 2019; Raya et al. 2021). For this reason, we take advantage of the A. angustifolia transcriptome to mine genes involved in the synthesis of cellulose. Cellulose is the dominant structural polysaccharide in the plant cell wall (Carroll et al. 2012; Little et al. 2018). The catalytic subunits of cellulose synthase encoded by CesA genes are the main catalysts that generate the plant cell wall cellulose (Little et al. 2018; Zhu et al. 2019). Moreover, the non-cellulosic cell wall polysaccharides (hemicellulosic polysaccharide backbones) are produced by the cellulose synthase-like (Csl) enzymes, members of the glycosyltransferase 2 (GT2) superfamily, which they were initially categorized into eight subfamilies: CslA, CslB, CslC, CslD, CslE, CslG, CslF and CslH (Speicher et al. 2018). In addition to the five CesA genes (CesA1/3/4/5/7) earlier analyzed from some Agave species (Huang et al. 2019; Raya et al. 2021), other six CesA genes (CesA2/6/8/9/10/11) could be retrieved from the A. angustifolia transcriptome. Moreover, only two Csl genes (CslD and CslG: 12 transcripts) were previously reported in agave species (Raya et al. 2021). Here, 13 Csl genes (CslD (5 genes), CslE (3 genes), CslG (3 genes), CslH (1 gene) were retrieved from the A. angustifolia transcriptome.
Transcription expression profile of cellulose synthesis genes was explored and compared between the four tissues. A total of 24 members of the cellulose synthase gene superfamily (11 CesA and 13 Csl) were identified and 20 members (10 CesA and 10 Csl) were differentially expressed in at least one of the pairwise tissue comparisons. As expected, and due to the anatomical and physiological nature of the roots, the expression of CesA and Csl was higher in roots. Moreover, the members of the CesA subfamily were preferentially upregulated in young leaf, whereas the members of the Csl subfamilies were preferentially upregulated in mature leaf. The generally accepted view is that the CesA genes encode enzymes that synthesize cellulose, whereas the Csl genes mediate in the biosynthesis of hemicellulose polysaccharides (Speicher et al. 2018).
Genetic markers-based studies in populations of A. angustifolia are limited. Hence, molecular marker types such as randomly amplified polymorphic DNA (RAPD) (Rodríguez-Garay et al. 2009), inter-simple sequence repeats (ISSR) (Vargas-Ponce et al. 2009), inverse sequence tagged repeat (ISTR) (Torres-Morán et al. 2008), amplified fragment length polymorphism (AFLP) (Rivera-Lugo et al. 2018) and simple sequence repeats (SSR) (Trejo et al. 2018) have been amplified in A. angustifolia germplasm for characterization and differentiation purposes. No SSR marker was specifically designed from A. angustifolia. In fact, only 8 SSR pair primers, designed for A. parryi (Lindsay et al. 2012), were amplified in A. angustifolia (Trejo et al. 2018).
The de novo transcriptome assemblies are useful to develop molecular markers for further efforts in Agave molecular studies and breeding. Given the polyploidy and large genome of A. angustifolia var “Espadín”, we expected to find a variety of molecular markers within the transcriptome. Thus, a large number of A. angustifolia transcripts displayed SSR, VNTR, SNPs and InDels, similar to those reported from the A. deserti and A. tequilana (Gross et al. 2013). Sarwar et al. (2019) identified 13,375 SSR markers within A. sisalana transcriptome, a smaller amount than found here in A. angustifolia transcriptome (30,766 SSR markers). The average relative density of microsatellites was one SSR per 2.5 kb, similar to that reported for A. deserti (one SSR per 2.1 kb; Gross et al. 2013) and for A. sisalana (one SSR per 2.9 kb; Sarwar et al. 2019) and lower than found in A. tequilana (one SSR per 4.39 kb; Gross et al. 2013). The genome size, ploidy level, sequencing depth, bioinformatics tools, and software and parameters used for the prediction of the molecular marker could be some of the causes of the differences found between the Agave species.
Furthermore, SNPs endure the ability to produce high-density genetic maps, association mapping and molecular markers with the promise of lower cost and error rate. In this study, 397,108 high confidence variant positions were identified including 382,589 SNPs and 14,519 InDels. Smaller amounts (35,059 SNPs and 1466 InDels) were reported in A. sisalana (Sarwar et al. 2019). The polyploid nature of Agave species may increase the possibility of high counts of SNPs due to the identical paralogous loci in the genome. Despite the differences in the number of SNPs and Indels mentioned above, transitions (64.9% in A. angustifolia vs in 57.3% in A. sisalana) were more frequent than transversions (35.1% in A. angustifolia vs in 42.6% in A. sisalana). However, validation of these SNPs is still required but their annotation could indicate potential polymorphism in biology and adaptative characteristics of A. angustifolia.
Transcriptomes hold a large number of genes that provide a potentially cost-effective resource to generate a considerable sequence data for phylogenetic analyses. Largely, the phylogenetic tree based on 5863 single-copy genes was similar to that obtained by Jiménez-Barron et al. (2020) using ITS region. Moreover, A. angustifolia and A. fourcroydes were clustered together confirming hypothesis of ancestry between these two species (Colunga-GarcíaMarín et al. 1999).
Conclusion
In the present study, the transcriptome of Agave angustifolia was assembled based on paired-end Illumina high-throughput RNA-seq. A total of 358,170 transcripts were assembled; from which, 174,414 transcripts had potential coding protein revealing the presence of at least 29,273 genes. Gene expression analysis and functional annotation revealed the identification and quantification of genes involved in the carbohydrate pathways metabolism and cellulose synthesis. Moreover, development of molecular markers was possible from the A. angustifolia transcriptome. The presented findings offer a comprehensive transcriptome resource for A. angustifolia that could facilitate improvements in genetic and molecular studies for this important plant species and suggest new insights to understand its physiology and adaptation.
Data availability
The data sets (raw sequence files) generated during the current study are available in the National Center for Biotechnology Information, Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra/) under the BioProject accession numbers PRJNA744214 (SRX11362104-06; https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA744214), PRJNA744267 (SRX11366017-19: https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA744267), PRJNA744577 (SRX11377409-11: https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA744577), and PRJNA744498 (SRX11369602-04; https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA744498). All data analyzed during this study are included in this published article and its supplementary information files.
References
Abraham P, Yin H, Borland A, Weighill D, Lim S, Cestari De Paoli H, Engle N, Jones PC, Agh R, Weston DJ, Wullschleger SD, Tschaplinski T, Jacobson D, Cushman JC, Hettich RL, Tuskan GA, Yang X (2016) Transcript, protein, and metabolite temporal dynamics in the CAM plant Agave. Nat Plants 2:1–10. https://doi.org/10.1038/nplants.2016.178
Afgan A, Baker D, Batut B, van den Beek M, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Grüning BA, Guerler A, Hillman-Jackson J, Hiltemann S, Jalili V, Rasche H, Soranzo N, Goecks J, Taylor J, Nekrutenko A, Blankenberg D (2018) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018update. Nucl Acids Res 46:W537–W544. https://doi.org/10.1093/nar/gky379
Arrizon J, Morel S, Gschaedler A, Monsan P (2010) Comparison of the water-soluble carbohydrate composition and fructan structures of Agave tequilana plants of different ages. Food Chem 122:123–130. https://doi.org/10.1016/j.foodchem.2010.02.028
Avila De Dios E, Gomez Vargas AD, Damian Santos ML, Simpson J (2015) New insights into plant glycoside hydrolase family 32 in Agave species. Front Plant Sci 6:594. https://doi.org/10.3389/fpls.2015.00594
Avilade Dios E, Delaye L, Simpson J (2019) Transcriptome analysis of bolting in A. tequilana reveals roles for florigen, MADS, fructans and gibberellins. BMC Genom 20:473. https://doi.org/10.1186/s12864-019-5808-9
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Borland AM, Griffiths H, Hartwell J, Smith JA (2009) Exploiting the potential of plants with crassulacean acid metabolism for bioenergy production on marginal lands. J Exp Bot 60:2879–2896. https://doi.org/10.1093/jxb/erp118
Bu D, Luo H, Huo P, Wang Z, Zhang S, He Z, Wu Y, Zhao L, Liu J, Guo J, Fang S, Cao W, Yi L, Zhao Y, Kong L (2021) KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucl Acids Res 49:W317–W325. https://doi.org/10.1093/nar/gkab447
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: architecture and applications. BMC Bioinform 15:421. https://doi.org/10.1186/1471-2105-10-421
Carroll A, Mansoori N, Li S, Lei L, Vernhettes S, Visser RG, Somerville C, Gu Y, Trindade LM (2012) Complexes with mixed primary and secondary cellulose synthases are functional in Arabidopsis plants. Plant Physiol 160:726–737. https://doi.org/10.1104/pp.112.199208
Cervantes-Pérez SA, Espinal-Centeno A, Oropeza-Aburto A, Caballero-Perez J, Falcon F, Aragon-Raygoza A, Sanchez-Segura L, Herrera-Estrella L, Cruz-Hernandez A, Cruz-Ramirez A (2018) Transcriptional profiling of the CAM plant Agave salmiana reveals conservation of a genetic program for regeneration. Dev Biol 442:28–39. https://doi.org/10.1016/j.ydbio.2018.04.018
Cock PJA, Chilton JM, Grüning B, Johnson JE, Soranzo N (2015) NCBI BLAST+ integrated into Galaxy. GigaSci 4:39. https://doi.org/10.1186/s13742-015-0080-7
Colunga-GarcíaMarín P, Coello-Coello JN, Eguiarte LE, Piñero D (1999) Isozymatic variation and phylogenetic relationships between henequen (Agave fourcroydes) and its wild ancestor A. angustifolia (Agavaceae). Am J Bot 86:115–123. https://doi.org/10.2307/2656960
Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL, Zhang X, Mortazavi A (2016) A survey of best practices for RNA-seq data analysis. Genome Biol 17:13. https://doi.org/10.1186/s13059-016-0881-8
Cortés-Romero C, Martinez-Hernandez A, Mellado-Mojica E, López MG, Simpson J (2012) Molecular and functional characterization of novel fructosyltransferases and invertases from Agave tequilana. PLoS ONE 7:e35878. https://doi.org/10.1371/journal.pone.0035878
Du L, Zhang C, Liu Q, Zhang X, Yue B (2018) Krait: an ultrafast tool for genome-wide survey of microsatellites and primer design. Bioinformatics 34:681–683. https://doi.org/10.1093/bioinformatics/btx665
Espinosa-Andrews H, Urías-Silvas JE, Morales-Hernández N (2021) The role of Agave fructans in health and food applications: a review. Trends Food Sci Technol 114:585–598. https://doi.org/10.1016/j.tifs.2021.06.022
Figueroa CM, Lunn JE, Iglesias AA (2021) Nucleotide-sugar metabolism in plants: the legacy of Luis F. Leloir. J Exp Bot 72:4053–4067. https://doi.org/10.1093/jxb/erab109
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152. https://doi.org/10.1093/bioinformatics/bts565
García-Mendoza A (2002) Distribution of Agave (Agavaceae) in Mexico. Cact Succ J 74:177–187
García-Mendoza AJ, Chiang F (2003) The confusion of Agave vivipara L. and A. angustifolia Haw., two distinct taxa. Brittonia 55:82–87
Gentry HS (1982) Agaves of continental North America. The University of Arizona Press, Tucson
Grabherr M, Haas B, Yassour M et al (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652. https://doi.org/10.1038/nbt.1883
Gomez-Vargas AD, Hernández-Martínez KM, López-Rosas ME, Alejo-Jacuinde G, Simpson J (2022) Evidence for light and tissue specific regulation of genes involved in Fructan metabolism in Agave tequilana. Plants 11:2153. https://doi.org/10.3390/plants11162153
Gross S, Martin J, Simpson J, Abraham-Juarez M, Wang Z, Visel A (2013) De novo transcriptome assembly of drought tolerant CAM plants, Agave deserti and Agave tequilana. BMC Genom 14:1–14. https://doi.org/10.1186/1471-2164-14-563
Haak M, Vinke S, Keller W, Droste J, Rückert C, Kalinowski J, Pucker B (2018) High quality de novo transcriptome assembly of Croton tiglium. Front Mol Biosci 5:62. https://doi.org/10.3389/fmolb.2018.00062
Huang X, Wang B, Xi J, Zhang Y, He C, Zheng J, Gao J, Chen H, Zhang S, Wu W, Liang Y, Yi K (2018) Transcriptome comparison reveals distinct selection patterns in domesticated and wild Agave species, the important CAM plants. Int J Genom 2018:5716518. https://doi.org/10.1155/2018/5716518
Huang X, Xiao M, Xi J, He C, Zheng J, Chen H, Gao J, Zhang S, Wu W, Liang Y, Xie L, Yi X (2019) De Novo transcriptome assembly of Agave H11648 by Illumina sequencing and identification of cellulose synthase genes in Agave species. Genes 10:103. https://doi.org/10.3390/genes10020103
Huang X, Xu B, Tan S, Huang Y, Xi J, Qin X, Chen T, Chen H, Yang X, Yi K (2022) Transcriptome sequencing of Agave angustifolia reveals conservation and diversification in the expression of cinnamyl alcohol dehydrogenase genes in Agave species. Agriculture 12:1003. https://doi.org/10.3390/agriculture12071003
Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, von Mering C, Bork P (2019) eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucl Acids Res 47:D309–D314. https://doi.org/10.1093/nar/gky1085
Jiménez-Barron O, García-Sandoval R, Magallón S, García-Mendoza A, Nieto-Sotelo J, Aguirre-Planter E, Eguiarte LE (2020) Phylogeny, diversification rate, and divergence time of Agave sensu lato (Asparagaceae), a group of recent origin in the process of diversification. Front Plant Sci 11:536135. https://doi.org/10.3389/fpls.2020.536135
Koboldt D, Zhang Q, Larson D, Shen D, McLellan M, Lin L, Miller C, Mardis E, Ding L, Wilson R (2012) VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22:568–576. https://doi.org/10.1101/gr.129684.111
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. https://doi.org/10.1038/nmeth.192
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) 1000 genome project data processing subgroup (2009) the sequence alignment/map (SAM) format and SAM tools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Lindsay DL, Edwards CE, Jung MG, Bailey P, Lance RF (2012) Novel microsatellite loci for Agave parryi and cross-amplification in Agave palmeri (Agavaceae). Am J Bot 99:e295–e297. https://doi.org/10.3732/ajb.1200033
Little A, Schwerdt JG, Shirley NJ, Khor SF, Neumann K, O’Donovan LA, Lahnstein J, Collins HM, Henderson M, Fincher GB, Burton RA (2018) Revised phylogeny of the cellulose synthase gene superfamily: insights into cell wall evolution. Plant Physiol 177:1124–1141. https://doi.org/10.1104/pp.17.01718
Liu R, Holik AZ, Su S, Jansz N, Chen K, Leong HS, Blewitt ME, Asselin-Labat ML, Smyth GK, Ritchie ME (2015) Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucl Acids Res 43:e97–e97. https://doi.org/10.1093/nar/gkv412
Livingston DP, Hincha D, Heyer A (2009) Fructan and its relationship to abiotic stress tolerance in plants. Cell Mol Life Sci 66:2007–2023. https://doi.org/10.1007/s00018-009-0002-x
López-Romero JC, Ayala-Zavala JF, González-Aguilar GA, Peña-Ramos EA, González-Ríos H (2018) Biological activities of Agave by-products and their possible applications in food and pharmaceuticals. J Sci Food Agric 98:2461–2474. https://doi.org/10.1002/jsfa.8738
Maceda-López LF, Góngora-Castillo EB, Ibarra-Laclette E, Morán-Velázquez DC, Girón Ramírez A, Bourdon M, Villalpando-Aguilar JL, Toomer G, Tang JZ, Azadi P, Santamaria JM, López-Rosas I, López MG, Simpson J, Alatorre-Cobos F (2022) Transcriptome mining provides insights into cell wall metabolism and fiber lignification in Agave tequilana Weber. Plants 11:1496. https://doi.org/10.3390/plants11111496
Mancilla-Margalli NA, López MG (2006) Water-soluble carbohydrates and fructan structure patterns from Agave and Dasylirion species. J Agric Food Chem 54:7832–7839. https://doi.org/10.1021/jf060354v
McKain MR, Wickett N, Zhang Y, Ayyampalayam S, McCombie WR, Chase MW, Pires JC, Depamphilis CW, Leebens-Mack J (2012) Phylogenomic analysis of transcriptome data elucidates co-occurrence of a paleopolyploid event and the origin of bimodal karyotypes in Agavoideae (Asparagaceae). Am J Bot 99:397–406. https://doi.org/10.3732/ajb.1100537
Mellado-Mojica E, de la Vara LEG, López MG (2017) Fructan active enzymes (FAZY) activities and biosynthesis of fructooligosaccharides in the vacuoles of Agave tequilana Weber blue variety plants of different age. Planta 245:265–281. https://doi.org/10.1007/s00425-016-2602-7
Mellado-Mojica E, López MG (2012) Fructan metabolism in A. tequilana Weber Blue variety along its developmental cycle in the field. J Agric Food Chem 60:11704–11713. https://doi.org/10.1021/jf303332n
Michael TP, Jackson S (2013) The first 50 plant genomes. Plant Genome 6:1–7. https://doi.org/10.3835/plantgenome2013.03.0001in
Morreeuw ZP, Escobedo-Fregoso C, Ríos-González LJ, Castillo-Quiroz D, Reyes AG (2021) Transcriptome-based metabolic profiling of flavonoids in Agave lechuguilla waste biomass. Plant Sci 305:110748. https://doi.org/10.1016/j.plantsci.2020.110748
Palomino G, Martínez J, Romero P, Barba-González R, Rodríguez-Garay B (2017) Nuclear genome size and karyotype analysis of Agave angustifolia Haw. “Cimarron” and “Lineño” (Asparagales, Asparagaceae). Caryologia 70:93–101. https://doi.org/10.1080/00087114.2017.1279931
Pérez-López AV, Simpson J (2020) The sweet taste of adapting to the desert: fructan metabolism in Agave species. Front Plant Sci 11:324. https://doi.org/10.3389/fpls.2020.00324
Pérez-López AV, Simpson J, Clench MR, Gomez-Vargas AD, Ordaz-Ortiz JJ (2021) Localization and composition of fructans in stem and rhizome of Agave tequilana Weber var. azul. Front Plant Sci 11:608850. https://doi.org/10.3389/fpls.2020.608850
Pérez-Zavala ML, Hernández-Arzaba JC, Bideshi DK, Barboza-Corona JE (2020) Agave: a natural renewable resource with multiple applications. J Sci Food Agric 100:5324–5333. https://doi.org/10.1002/jsfa.10586
Raya FT, Marone MP, Carvalho LM, Rabelo SC, de Paula MS, Campanari MFZ, Freschi L, Mayer JLS, Ferreira Silva ORR, Mieczkowski P, Carazzolle MF, Pereira GAG (2021) Extreme physiology: Biomass and transcriptional profiling of three abandoned Agave cultivars. Ind Crops Prod 172:114043. https://doi.org/10.1016/j.indcrop.2021.114043
Ritsema T, Smeekens S (2003) Fructans: beneficial for plants and humans. Curr Opin Plant Biol 6:223. https://doi.org/10.1016/S1369-5266(03)00034-7
Rivera-Lugo M, García-Mendoza A, Simpson J, Solano E, Gil-Vega K (2018) Taxonomic implications of the morphological and genetic variation of cultivated and domesticated populations of the Agave angustifolia complex (Agavoideae, Asparagaceae) in Oaxaca, Mexico. Plant Syst Evol 304:969–979. https://doi.org/10.1007/s00606-018-1525-0
Rodríguez-Garay B, Lomelí-Sención J, Tapia-Campos E, García-Galindo J, Rodríguez-Domínguez J, Urbina-López D, Vicente-Ramírez I (2009) Morphological and molecular diversity of Agave tequilana Weber var. Azul and Agave angustifolia Haw. var Lineño. Ind Crops Prod 29:220–228. https://doi.org/10.1016/j.indcrop.2008.05.007
Sánchez-Teyer F, Moreno-Salazar S, Esqueda M, Barraza A, Robert ML (2009) Genetic variability of wild Agave angustifolia populations based on AFLP: a basic study for conservation. J Arid Environ 73:611–616. https://doi.org/10.1016/j.jaridenv.2009.01.008
Sarwar MB, Ahmad Z, Rashid B, Hassan S, Gregersen PL, de la Leyva MO, NagyAspHusnain OTT (2019) De novo assembly of Agave sisalana transcriptome in response to drought stress provides insight into the tolerance mechanisms. Sci Rep 9:396–410. https://doi.org/10.1038/s41598-018-35891-6
Schwacke R, Ponce-Soto GY, Krause K, Bolger AM, Arsova B, Hallab A, Gruden K, Stitt M, Bolger ME, Usadel B (2019) MapMan4: a refined protein classification and annotation framework applicable to multi-omics data analysis. Mol Plant 12:879–892. https://doi.org/10.1016/j.molp.2019.01.003
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. https://doi.org/10.1093/bioinformatics/btv351
Simpson J, Martínez Hernández A, Abraham Juárez JM, Delgado Sandoval S, Sánchez Villarreal A, Cortés Romero C (2011) Genomic resources and transcriptome mining in Agave tequilana. GCB Bioenergy 3:25–36. https://doi.org/10.1111/j.1757-1707.2010.01079.x
Soltis DE, Soltis PS, Bennett MD, Leitch IJ (2003) Evolution of genome size in the angiosperms. Am J Bot 90:1596–1603. https://doi.org/10.3732/ajb.90.11.1596
Speicher TL, Li PZ, Wallace IS (2018) Phosphoregulation of the plant cellulose synthase complex and cellulose synthase-Like proteins. Plants 7:52. https://doi.org/10.3390/plants7030052
Sturm A (1999) Invertases. Primary structures, functions, and roles in plant development and sucrose partitioning. Plant Physiol 121:1–7. https://doi.org/10.1104/pp.121.1.1
Suárez-González EM, López MG, Délano-Frier JP, Gómez-Leyva JF (2014) Expression of the 1-SST and 1-FFT genes and consequent fructan accumulation in Agave tequilana and A. inaequidens is differentially induced by diverse (a)biotic-stress related elicitors. J Plant Physiol 171:359–372. https://doi.org/10.1016/j.jplph.2013.08.002
Suárez-González EM, Suárez PAP, Cruz-Rubio JM, Martínez-Gallardo NA, Hernández IC, Délano-Frier JP, Gómez-Leyva JF (2016) Differential fructan accumulation and expression of fructan biosynthesis, invertase and defense genes is induced in Agave tequilana plantlets by sucrose or stress-related elicitors. Agric Gene 2:17–28. https://doi.org/10.1016/j.aggene.2016.09.003
Sun J, Lu F, Luo Y, Bie L, Xu L, Wang Y (2023) OrthoVenn3: an integrated platform for exploring and visualizing orthologous data across genomes. Nucl Acids Res 51:W397–W403. https://doi.org/10.1093/nar/gkad313
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COGdatabase: a tool for genome-scale analysis of protein functions and evolution. Nucl Acids Res 28:33–36. https://doi.org/10.1093/nar/28.1.33
The UniProt Consortium (2021) UniProt: the universal protein knowledgebase in 2021. Nucl Acids Res 49:D480–D489. https://doi.org/10.1093/nar/gkaa1100
Torres-Morán MI, Almaraz-Abarca N, Velasco-Ramírez AP, Hernández-Vargas V, Orea-Lara G, Cifuentes-Díaz de León A, Oliver-Salvador C (2008) Taxonomic significance of ISTR to discriminate species in Agavaceae. Am J Agric Biol Sci 3:661–665. https://doi.org/10.3844/ajabssp.2008.661.665
Trejo L, Limones V, Peña G, Scheinvar E, Vargas-Ponce O, Zizumbo-Villarreal D, Colunga-GarcíaMarín P (2018) Genetic variation and relationships among agaves related to the production of Tequila and Mezcal in Jalisco. Ind Crops Prod 125:140–149. https://doi.org/10.1016/j.indcrop.2018.08.072
Van den Ende W, Clerens S, Vergauwen R, Boogaerts D, Le Roy L, Arckens L, Van Laere A (2006) Cloning and functional analysis of a high DP fructan:fructan 1-fructosyl transferase from Echinops ritro (Asteraceae): comparison of the native and recombinant enzymes. J Exp Bot 57:775–789. https://doi.org/10.1093/jxb/erj065
Van den Ende W, Lammens W, Van Laere A, Schroeven L, Le Roy K (2009) Donor and acceptor substrate selectivity among plant glycoside hydrolase family 32 enzymes. FEBS J 276:5788–5798. https://doi.org/10.1111/j.1742-4658.2009.07316.x
Vargas-Ponce O, Zizumbo-Villarreal D, Martínez-Castillo J, Coello-Coello J, Colunga-GarcíaMarín P (2009) Diversity and structure of landraces of Agave grown for spirits under traditional agriculture: a comparison with wild populations of A. angustifolia (Agavaceae) and commercial plantations of A. tequilana. Am J Bot 96:448–457. https://doi.org/10.3732/ajb.0800176
Wang X, Huang X, Chen L, Xie Z, Tan S, Qin X, Chen T, Huang Y, Xi J, Chen H, Yi K (2023) Transcriptome sequencing of Agave amaniensis reveals shoot-related expression patterns of Expansin A genes in Agave. Plants 12:2020. https://doi.org/10.3390/plants12102020
Yin H, Guo HB, Weston DJ, Borland AM, Ranjan P, Abraham PE, Jawdy SS, Wachira J, Tuskan GA, Tschaplinski TJ, Wullschleger SD, Guo H, Hettich RL, Gross SM, Wang Z, Visel A, Yang X (2018) Diel rewiring and positive selection of ancient plant proteins enabled evolution of CAM photosynthesis in Agave. BMC Genom 19:588. https://doi.org/10.1186/s12864-018-4964-7
Zavala-García LE, Sánchez-Segura L, Dios EA, Pérez-López A, Simpson J (2018) Starch accumulation is associated with active growth in A. tequilana. Plant Physiol Biochem 130:623–632. https://doi.org/10.1016/j.plaphy.2018.08.011
Zhu X, Xin X, Gu Y (2019) Cellulose and hemicellulose synthesis and their regulation in plant cells. In: Cohen E, Merzendorfer H (eds) Extracellular sugar-based biopolymers matrices. Springer, Cham, pp 303–353. https://doi.org/10.1007/978-3-030-12919-4_7
Acknowledgements
This work was supported by Institutional Strategic Project Code: 21004-EI from Chapingo Autonomous University.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Sample acquisition was performed by EVM and SS. Data analyses were performed by SS. The first draft of the manuscript was written by EVM and SS. In addition, all authors commented on previous versions of the manuscript. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors report no declarations of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Valadez-Moctezuma, E., Samah, S., Mascorro-Gallardo, J.O. et al. The de novo transcriptome assembly of Agave angustifolia Haw, mining for carbohydrates and cellulose synthesis genes profiling, and molecular markers development. Plant Biotechnol Rep 17, 889–904 (2023). https://doi.org/10.1007/s11816-023-00861-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-023-00861-6