
Abstract
We present a case of primary synovial sarcoma arising from the left heart, an extremely rare occurrence, with a large amount of necrotic tissue, which suggested a poor prognosis. After incomplete tumor resection, chemotherapy and radiation therapy were performed; however, PET/CT findings at 26 months after the operation revealed local recurrence. Although we performed two additional operations following chemotherapy, the patient died from local recurrence at 36 months after the initial operation. In this case of synovial sarcoma arising from the left heart, even though aggressive multimodality therapy was performed, the prognosis was still poor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Case
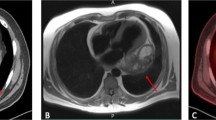
A 19-year-old male was referred to our medical center with dyspnea and orthopnea. Computed tomography (CT) of the chest showed a heterogeneously enhanced mass in the left side of the heart greater than 15 cm in diameter and severe compression of the left atrium (Fig. 1a, b). Echocardiogram findings revealed a large mass compressed left atrium and this was considered to be the cause of his symptoms.

Computed tomography (CT) of the chest showing a heterogeneously enhanced mass in the left heart and b severe compression of the left atrium. At 7 months after the operation, CT showed no recurrence (c, d). Ao aorta, RA right atrium, RV right ventricle, LA left atrium, LV left ventricle
At a week after diagnosis of cardiac tumor, surgical resection was performed via a median sternotomy. The tumor was found to be severely adhered to the anterior and lateral walls of the left ventricle, and extended to the right ventricle and aorta, with an unclear border between it and the heart, making complete surgical resection difficult. To achieve adequate exposure, we added a fifth intercostal incision on the left side (Fig. 2a). We speculated that the tumor had arisen from the left ventricle, though the primary site was not determined. Intraoperative direct echocardiography revealed a solid tumor containing a hematoma-like portion and the intraoperative histological diagnosis was high-grade sarcoma. The tumor wall and hematoma-like tissue were resected as much as possible, which relieved compression of the left atrium and ventricle. The patient recovered uneventfully, and preoperative dyspnea and orthopnea disappeared.

a Gross appearance of tumor from intercostal incision. The border between tumor (yellow arrow) and left ventricle was not clear. b Resected tumor contained necrotic and hemorrhagic portions. c Spindle cells with sparse cytoplasm and ovoid hyperchromatic nuclei with inconspicuous nucleoli were arranged in dense cellular sheets with staghorn-shaped vasculature. Mitotic figures were numerous (hematoxylin and eosin, ×200). LV left ventricle
Grossly, the tumor was gray–white with marked hemorrhage and necrosis. Microscopically, spindle cells with sparse cytoplasm and ovoid hyperchromatic nuclei with inconspicuous nucleoli were arranged in dense cellular sheets with staghorn-shaped vasculature. Mitotic rate was high (20–30/10 HPF) (Fig. 2b, c). Immunohistochemical staining showed diffusely positive for Bcl-2, partially positive for CK (AE1/AE3) and CD99, and negative for EMA, S-100 protein, myogenin, SMA, desmin, and CD34. These findings suggested that the tumor was a monophasic synovial sarcoma. Molecular genetic study showed transcription of the t(X;18) SYT–SSX1 fusion transcript, which led to a final definitive diagnosis of monophasic synovial sarcoma.
The patient underwent two courses of postoperative chemotherapy with adriamycin (45 mg/day on day 1–2) and ifosfamide (3.5 g/day on day 1–5) every 1 month, followed by radiation therapy at a dose of 5000 Gy over 5 weeks. At 7 months after the operation, PET/CT showed no recurrence (Fig. 1c, d). However, at postoperative 26 months, a PET/CT revealed local tumor recurrence without signs of metastasis. The recurrent tumor had spread around the left pulmonary vein, left atrium, and posterior wall of the left ventricle. After two courses of chemotherapy with adriamycin and ifosfamide, tumor reduction was observed.
A second operation was performed at 28 months after the initial surgery for prognostic improvement. We attempted resection of the tumor as much as possible, which showed expansion between the posterior left atrial wall and posterior left ventricular wall. The resected specimen consisted of white friable tissue and brown fluid. Prognosis was considered to be very poor without surgical resection, so we performed the third surgery at 36 months after the initial operation. Unfortunately, his heart function deteriorated after the operation and he died. The cause of heart failure was suspected to be invasion of tumor to coronary artery. Autopsy was rejected by his family.
Discussion
The incidence of primary cardiac sarcoma is extremely low. While angiosarcomas account for 31% of all sarcomas and rhabdomyosarcomas for 21%, synovial sarcomas comprise only 5% [1]. Most primary cardiac synovial sarcomas arise from the pericardium (41.4%) or right atrium (24%), while that from the left ventricle has only been reported in three cases prior to this one, with complete resection found to be anatomically difficult in each [2]. In the present case, we considered that the tumor arose from the left ventricle or left atrium. Complete resection was anatomically difficult because of invasion to the left ventricle, which was also a factor related to the poor prognosis of our patient.
A primary synovial sarcoma with a large amount of necrotic tissue is extremely rare, though gross pathological findings of a synovial sarcoma vary [2]. Some previous reports have noted that the tumor mass showed friable appearance with some necrotic regions or cystic regions. In the present case, the tumor was severely adhered on the left ventricle, and contained large amounts of necrotic tissue and hematoma. Burke et al. showed that the presence of necrosis in cases with other types of primary cardiac sarcoma was a risk factor for early mortality [3]. The poor prognosis of our patient was consistent with that report, though occurrence in the left ventricle was also a factor in early death.
The prognosis of patients with a cardiac synovial sarcoma is poor and no effective strategy has been established. Despite possible anatomic difficulties, a wide surgical excision should be attempted, because the main cause of death is local recurrence. Although surgical resection is indispensable for treatment, the survival rate at 1 and 5 years after surgery has been reported to be only 59.9 and 29.9%, respectively [2]. While it has not been established whether chemotherapy improves survival, some authors have reported that adjuvant chemotherapy improved prognosis [1, 2]. On the other hand, the efficacy of radiation therapy remains uncertain, though some have recommended that in cases with a positive resection margin [4].
Conclusion
We treated a case of primary synovial sarcoma of the left heart with a large amount of necrotic tissue, which suggested a poor prognosis. Although aggressive multimodal therapy including three surgical procedures was performed, the patient died at 36 months after the first operation.
References
Zhang L, Qian J, Li Z, Jing H. Primary synovial sarcoma of the heart. Cardiol J. 2011;18(2):128–33.
Wang J, Li N. Primary cardiac synovial sarcoma. Ann Thorac Surg. 2013;95:2202–9.
Burke AP, Cowan D, Virmani R. Primary sarcoma of the heart. Cancer. 1992;69:387–95.
Al-rajhi N, Husain S, Coupland R, et al. Primary pericardial synovial sarcoma: a case report and literature review. J Surg Oncol. 1999;70:194–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared that no conflict of interest existed.
Rights and permissions
About this article

Cite this article
Maeda, S., Takano, H., Yamauchi, T. et al. Primary synovial sarcoma of the left heart with large amount of necrotic tissue. Gen Thorac Cardiovasc Surg 66, 365–367 (2018). https://doi.org/10.1007/s11748-017-0828-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11748-017-0828-3