
Abstract
A structured lipid (SL) with a substantial amount of palmitic acid at the sn-2 position and enriched with capric acid (C), was produced in two enzymatic interesterification stages by using immobilized lipase, Lipozyme® TL IM (Novozymes North America Inc., Franklinton, NC, USA). The substrates for the reactions were high melting point palm stearin, high oleic sunflower oil and tricaprin. The SL was characterized for total and positional fatty acid profiles, triacylglycerol (TAG) molecular species, free fatty acid content, melting and crystallization profiles. The final SL contained 20.13 mol% of total palmitic acid, of which nearly 40 % was located at the sn-2 position. The total capric acid content was 21.22 mol%, mostly at the sn-1 and sn-3 positions. The predominant TAGs in the SL were oleic–palmitic–oleic, POP and CLC. The melting completion and crystallization onset temperatures of the SL were 27.7 and 6.1 °C, respectively. The yield for the overall reaction was 90 wt%. This SL might be totally or partially used in commercial fat blends for infant formula.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The value of the composition of maternal breast milk is widely recognized. Human milk is a complex mixture of lipids, carbohydrates, proteins, vitamins, and minerals, as well as growth and hormonal factors that not only nurture, but also protect infants from infections and certain chronic diseases [1]. Lipids are the second largest component of breast milk by concentration (3–5 % in mature milk). They provide essential fatty acids (EFAs) required as structural cell components of membrane tissues and count for approximately 50 % of the total energy used by infants. Lipids are also necessary for physiological functions, such as fat-soluble nutrient absorption and eicosanoids syntheses [2]. Although breast milk is the preferred choice among mothers for the nutrition of their infants, breastfeeding cannot always be practiced and commercial infant formulas exist as reasonable substitutes. Over the years, infant formulas have become more sophisticated regarding nutrient additions that naturally appear in human breast milk, including prebiotic oligosaccharides, nucleotides, carotenoids and probiotic bacteria. However, lipids as present in human breast milk have not been fully matched because of their distinctive molecular structure, which differentiates them from other common sources of lipids found in nature [3]. A similar fatty acid (FA) profile to human milk fat (HMF) can be achieved in commercial infant formulas by combining edible oils from palm, rapeseed and corn with algal oils [4]. However, the specific placement of palmitic acid on the triacylglycerol (TAGs) in HMF differs from most commercial formulations and cannot be matched by mere blending of vegetable oils. While in HMF, palmitic acid largely exists (>60 %) on the sn-2 position of the TAGs and the sn-1,3 positions are commonly occupied by oleic and linoleic acids, in most infant formulas palmitic acid exists largely (about 90 %) in the sn-1,3 positions [5]. This difference in palmitic acid placement on the TAGs plays a crucial role in the uptake, digestion, and metabolism of milk fat for infants [6]. A higher palmitic acid absorption and utilization have been observed in human milk rich in sn-2 palmitic acid when compared to formulas in which this fatty acid was mainly esterified at sn-1,3 positions [7].
TAGs that have been chemically or enzymatically modified by incorporating new molecules or by rearranging the position of their original FAs are called structured lipids (SLs). For certain applications, these novel structures can be desirable, as they exhibit special properties that affect their melting behavior, digestion, absorption, and their nutritional value. Some applications of these SLs include the production of cocoa butter substitutes, partial acylglycerols as emulsifiers, modified fish oil products, margarines and HMF analogs [8]. The ideal SL for infant formula must contain palmitic acid mostly esterified at the sn-2 position and unsaturated FAs at the sn-1,3 positions to resemble HMF [9]. Betapol® (Loders Croklaan, The Netherlands) was the first HMF analog produced by using a 1,3-specific lipase and reacting tripalmitin with oleic acid. It contained 53.5 % of palmitic acid at the sn-2 position and 42.1 mol% of total oleic acid [10]. Following the Betapol® development, numerous studies have been conducted over the past years on the production of infant formula fat analogs by enzymatic esterification and/or acidolysis reactions. Recently, Zou et al . [11] reported the synthesis of a set of SLs with more than 60 % of palmitic acid at the sn-2 position via enzymatic acidolysis of palm stearin, using a commercially available 1,3 specific lipase from Rhizomucor miehei (RML). Wang et al. [12] reported the production of an infant formula fat analog synthesized by lipase-catalyzed reaction of interesterified palm stearin with a blend of free fatty acids (FFAs) from rapeseed, sunflower, and palm kernel oils in a continuous packed bed reactor. The final product contained 61.6 % of palmitic acid at the sn-2 position. Some other recent works include the use of either ethyl palmitate or tripalmitin with stearidonic acid soybean oil [13], hazelnut oil [14], extra virgin olive oil [15], and amaranth oil [16]. Although Betapol and the other products successfully achieved high sn-2 palmitic acid levels, all of them lacked high reaction yields because of the nature of the substrates used (FFAs and ethyl esters), and the type of reaction conducted that promotes an excessive formation of new FFAs or their ethyl esters. Common yields for acidolysis reactions in SL production for different applications can range from 55 to 70 wt% [15–17].
In the past decades, TAGs rich in medium-chain fatty acids (so called MCTs) have received considerable attention, because of their distinctive metabolic pathways for digestion and absorption when compared to long-chain triacylglycerols (LCTs). MCTs may not be assimilated via lymphatic transport like LCTs, but rather absorbed via portal transport to provide quick energy to the infants. MCTs are broken down almost immediately by enzymes in the saliva and gastric juices, so pancreatic enzymes for digestion are barely required [18]. Furthermore, it has been reported that medium-chain fatty acids (MCFAs) such as capric acid protect infants from harmful microorganisms, as they exhibit antiviral and antimicrobial properties [19]. Most commercial infant formulas are enriched with lauric acid (C12:0) as a source of MCFAs in concentrations up to 13 mol% [20]. Although lauric acid is frequently used as an MCFA for food applications, there is still disagreement within the scientific community, as some researchers consider it to be at the edge of the long-chain fatty acids (LCFAs) category, and therefore its functional properties have been questioned [21]. Instead, capric acid (C10:0) was recommended as a source of MCFAs [22].
The objective of this study was to synthesize a SL at a high reaction yield that contained reasonable levels of palmitic acid at the sn-2 position and capric acid at the sn-1,3 positions. To achieve this, a two-step enzymatic interesterification approach was followed using immobilized lipase, Lipozyme® TL IM, as biocatalyst. In the first stage, high melting point palm stearin (HMPS) and high oleic sunflower oil (HOSO) were reacted to obtain a high sn-2 palmitic acid intermediate SL. In the second stage, tricaprin (TC) was interesterified with the intermediate SL to obtain the final product. With this SL, we propose an alternative to the fat blends currently used for the production of commercial infant formulas.
Materials and Methods
Materials
HMPS was provided by Team Foods S.A (Bogotá, Colombia) and HOSO was purchased under the commercial brand Frymax Sun Supreme from Stratas Foods® (Memphis, TN, USA). The immobilized lipase, Lipozyme® TL IM (sn-1,3 specific Thermomyces lanuginosus lipase), was obtained from Novozymes North America Inc. (Franklinton, NC, USA). The specific activity of Lipozyme® TL IM was 250 IUN/g at pH 6–8 and temperature of 50–75 °C, where IUN stands for interesterification units novo, as defined by the manufacturer [23]. The Lipid standards, Supelco 37 Component FAME mix, and 2-oleoylglycerol were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). TC and pentadecanoic acid (C15:0) were obtained from TCI America (Portland, OR, USA). The TAG standard mix (GLC-437) was provided by Nu-Chek Prep Inc. (Elysian, MN, USA). Other solvents and chemicals were purchased from Fischer Scientific (Norcross, GA, USA) and Sigma-Aldrich Chemical Co.
SL Synthesis
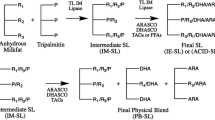
The SL was synthesized in two enzymatic interesterification stages, as shown in Fig. 1. In the first stage, HMPS and HOSO were blended and reacted in the presence of Lipozyme® TL IM. This enzyme, which is a sn-1,3 specific lipase, was used with the aim of increasing the amount of palmitic acid at the sn-2 position, predominantly in the form of oleic–palmitic–oleic (OPO) type of TAG. For the second stage, the intermediate SL obtained in stage 1 was blended and reacted with TC in a second interesterification reaction, with the aim of incorporating capric acid into the TAG structure while conserving the palmitic acid at the sn-2 position. In order to establish the optimal substrate molar ratio and reaction time for both the intermediate SL in stage 1 and the final product in stage 2, a set of experiments was conducted at small scale (mg), and samples were analyzed for total and positional fatty acid profiles. Once the reaction conditions were defined, scale-up reactions of the intermediate and final SLs were performed to determine the reproducibility of the results and to obtain enough product for further analysis of TAG molecular species, melting and crystallization profiles, FFA assay, and reaction yield.

Reaction scheme for SL synthesis. Stage 1: enzymatic interesterification of HMPS and HOSO by using sn-1,3 specific lipase, Lipozyme® TL IM. Stage 2: stage 1 product was filtered and excess FFAs were removed. Then, a second enzymatic interesterification with TC using Lipozyme® TL IM was conducted to obtain the final SL. P palmitic acid, O oleic acid, C capric acid
Reaction Procedure
All the enzymatic reactions at small scale (mg) for stage 1 and 2 were performed following a modified version of the Teichert and Akoh method [13]. Stage 1: 200 mg of blend (depending on substrate molar ratio tested) was weighed into a labeled Teflon-lined test tube and 3 mL of n-hexane was added to reach a concentration of 0.06 g/mL. Ten percent by weight of the lipase Lipozyme® TL IM was used as the biocatalyst. The reaction was carried out at 60 °C in a water bath while shaking at 200 rpm. After reaction, the product was filtered two times through a sodium sulfate column to remove both water and enzyme, and excess FFAs were removed by neutralization (as described below). The substrate molar ratios tested (HMPS:HOSO) were 2:3, 1:2, 1:3 and 1:4. The molar ratios were chosen taking into consideration that total palmitic acid in the SL should not exceed 30 mol% based on the average levels of this fatty acid in HMF (see Table 1 discussion section). The resulting SLs were named SL1–1, SL1–2, SL1–3 and SL1–4, respectively. Reactions were performed independently at 4, 8, 12 and 24 h to determine the time at which conversion rates (in terms of sn-2 palmitic acid content) were the highest for each substrate molar ratio tested. Finally, SLs were compared to each other and the best product was scaled-up for further characterization and selected as the substrate for the following stage. Stage 2: the same procedure as in stage 1, but in this case the substrate molar ratios tested (SL1:TC) were 2.8:0.5, 2:0.5, 1.5:0.5 and 1.2:0.5. The molar ratios were chosen taking into consideration that total capric acid in the SL should not exceed 30 mol%. The resulting SLs were named SL2–1, SL2–2, SL2–3, SL2–4, respectively. Reactions were performed independently at 4, 8, 12 and 24 h to determine the time at which conversion rates (in terms of capric acid incorporation and high sn-2 palmitic acid content) were the highest for each substrate molar ratio tested. SLs were compared to each other and the best product was scaled-up and selected as the final SL for further analysis and discussion.
The scale-up reaction for both stage 1 and 2 followed the Pande et al. method [15]. Four hundred grams of substrate blend (at the defined substrate molar ratio) was weighed into a stirred tank reactor without solvent. Ten percent by weight of the lipase Lipozyme® TL IM was used as the biocatalyst. For this procedure, the use of n-hexane was not recommended for safety and environmental reasons, but the tank stirrer at 200 rpm created the necessary turbulence to keep the mixture in continuous contact with the enzyme. The reaction was carried out at 60 °C. After reaction, the product was filtered two times through a sodium sulfate column to remove both water and enzyme, and excess FFAs were removed by short-path distillation (SPD).
Removal of FFAs
For small scale reactions, the FFAs were removed through de-acidification by alkaline extraction method [24] with minor modifications. Once filtered over a sodium sulfate column, the SL (dissolved in n-hexane) was mixed with 3 mL of neutralized 95 % ethyl alcohol and 2 mL of 1 % phenolphthalein solution. Then, the sample was titrated with 0.5 N NaOH in 20 % ethanol. The aqueous layer was discarded, while the hexane layer was collected and once again passed through anhydrous sodium sulfate. The solvent was removed by nitrogen sparging. Finally, the SL was store at −20 °C until analysis.
For the scale-up reactions, the FFAs were removed through SPD, as reported previously [15]. Once filtered over a sodium sulfate column, the SL was fed into a KDL-4 (UIC Inc., Joliet, IL, USA) system under the following conditions: feed holding temperature of 65 °C, feeding rate of approximately 100 mL/h, evaporator heating temperature of 185 °C, condenser cooling temperature of 20–25 °C, roller speed of 200 rpm, and vacuum of <13.33 Pa. SL was passed through SPD twice to obtain FFAs of <0.1 %.
Determination of Fatty Acid Profiles
The substrates, namely HMPS, HOSO, TC, and the SLs, were converted to FA methyl esters (FAME) following AOAC Official Method 996.01, Section E [25] with minor modifications. Sample (0.1 g) was weighed into Teflon-lined test tubes and 0.20 mL internal standard (C15:0, 20 mg/mL in hexane) was added and dried under nitrogen to remove solvent. Two milliliters of 0.5 N NaOH in methanol was added and heated to 100 °C for 10 min for saponification of FFAs. The sample was cooled under tap water and 2 mL BF3 in methanol was added followed by vortexing for 1 min. Again, the sample was heated at 100 °C for 10 min for methylation and then cooled under tap water. To stop the reaction and extract the FAMEs, 2 mL of hexane and 2 mL of saturated NaCl solution were added. The sample was vortexed for 2 min and then centrifuged at 1,000 rpm for 5 min to separate the organic layer from the aqueous layer. The organic layer was passed through an anhydrous sodium sulfate column into a GC vial for analysis. The FAME external standard used was Supelco 37 component FAME mix and was run in parallel with the samples.
Positional Analysis
The pancreatic lipase-catalyzed sn-2 positional analysis of substrates and products was performed following a modified version of the Luddy et al. method [26]. One hundred milligrams of sample (extracted and dried TAG) was placed into Teflon-lined test tubes. Two milliliters of 1.0 M Tris–HCl buffer (pH 8), 0.5 mL of 0.05 % sodium cholate solution, and 2.2 % calcium chloride solution were added, and the mixture was vortexed for 2 min to emulsify. Then, 40 mg of pancreatic lipase was added, and the mixture was vortexed for 1 min and incubated in a water bath at 40 °C for 3 min while shaking at 200 rpm. The sample was vortexed again for 2 min. To stop the reaction and extract the hydrolyzed TAGs, 1 mL of 6 N HCl and 4 mL of diethyl ether were added. The sample was vortexed for 2 min and centrifuged at 1,000 rpm for 3 min. The upper layer, containing the lipid components, was filtered through an anhydrous sodium sulfate column. The sample was concentrated under nitrogen until approximately one-third of the original volume was left. A mixture of hexane, diethyl ether, and formic acid (60:40:1.6, v/v/v) was used as the mobile phase after equilibration in the TLC tanks for approximately 30 min. The concentrated sample was spotted onto an activated silica gel G dried TLC plate and placed into the tank. 2-Oleyglycerol was spotted as the standard and run in parallel with the sample for identification of the 2-monoacylglycerol (2-MAG) band. The plate was sprayed with 0.2 % 2,7-dichlorofluorescein in methanol and visualized under UV light. The 2-MAG band was scrapped off and converted to FAME (as described above). The determination of the fatty acid profile at the sn-2 position was quantified by GC analysis. The fraction of the FA that is esterified at the sn-2 position (out of the total content of that FA in the sample) was calculated using the following equation: FA sn-2 (%) = [100 × total FA at sn-2 (mol%)]/[3 × total FA in sample (mol%)].
GC Analysis
Fatty acid compositions of all samples were analyzed as FAME following the method of Pande et al. [24] with a Hewlett-Packard 6890 series II gas chromatograph (Agilent Technologies Inc., Palo Alto, CA, USA), and using a Supelco SP-2560, 100 m × 25 mm × 0.2 μm column. Helium was the carrier gas at a constant flow rate of 1.1 mL/min. The injection volume was 1 μL and a split ratio of 20:1 was used. Detection was with a flame ionization detector at 300 °C. The column was initially held at 140 °C for 5 min and then increased to 240 °C at 4 °C/min and held at 240 °C for 25 min. All samples were analyzed in triplicate and average values reported.
Triacylglycerol Molecular Species
TAG species were identified with high-performance liquid chromatography (HPLC) (Agilent Technologies 1260 Infinity, Santa Clara, CA, USA) equipped with an evaporative light scattering detector (ELSD) Sedex 85 (Richard Scientific, Novato, CA, USA). The reverse phase column was a Beckman Ultrasphere® C18, 5 μm, 4 mm × 250 mm with the temperature set at 30 °C. The sample was dissolved in chloroform with a final concentration of 5 mg/mL. The injection volume was 20 μL. The mobile phase at a flow rate of 1 mL/min consisted of solvent A, acetonitrile and solvent B, acetone. A gradient elution was used starting with 65–95 % solvent B for 55 min, followed by a 10 min post-run at 65 % B. The drift tube temperature was set at 70 °C, pressure at 3.0 bar and gain at 8. The TAG peaks were identified by comparison of retention times with those of the standards and also by equivalent carbon number (ECN). ECN is defined as CN-2n, where CN is the number of carbons in the TAG (excluding the three in the glycerol backbone) and n is the number of double bonds. Relative percent (%) of peak areas of the TAGs identified were reported as the average of three determinations.
Melting and Crystallization Profiles
The melting and crystallization profiles were determined using a differential scanning calorimeter DSC 204 F1 Phoenix (NETZSCH Instruments North America, Burlington, MA, USA) following AOCS Official Method Cj 1-94 [27]. First, 8–12 mg of the sample was weighed into an aluminum pan and hermetically sealed. Sample was rapidly heated to 80 °C at 20 °C/min, and held for 10 min to destroy any previous crystalline structure. The samples were then cooled to −80 °C at 10 °C/min (for crystallization profile), and held for 30 min and finally heated to 80 °C at 10 °C/min (for melting profile). Nitrogen was used as protective gas (purge). All samples were analyzed in triplicate and average values are reported.
Free Fatty Acid Assay and Reaction Yield
Free fatty acid content (FFA%) was determined following a modified version of the AOCS Official Method Ca 5a-40 [27] for refined and crude vegetable oils. Oil (0.3–6.0 g) was weighed in an Erlenmeyer flask according to the range of FFAs expected in the sample by using the pre-established values in the method. 10 milliliters of neutralized 95 % ethyl alcohol and 2 mL of 1 % phenolphthalein solution were added, and the mixture was heated to 70 °C for 3 min to keep the oil in solution. The sample was then titrated with a standardized 0.25 N NaOH solution until the appearance of the first permanent pink color indicating the end of the reaction. The volume of standardized sodium hydroxide was recorded for further calculations. All samples were analyzed in triplicate and average values were reported as percent of oleic acid.
The reaction yield of the final SL obtained in stage 2 was calculated using the following equation: Yield = [1 − (FFA%final SL/100)], where FFA%final SL corresponds to the combined FFA% of SLs in stage 1 and stage 2.
Statistical Analysis
All analyses were performed in triplicate. Statistical analysis was performed with SAS software package (SAS Institute, Cary, NC, USA). Duncan’s multiple-range test was performed to determine the significant difference (P ≤ 0.05) among SLs.
Results and Discussion
Total and Positional Fatty Acid Profiles
Table 1 shows the common fatty acid composition in HMF and some commercial fat blends used for infant formula, as reported by López-López et al. [20]. The major fatty acids in HMF are oleic (28.30–43.83 wt%), palmitic (15.43–24.46 wt%), linoleic (10.61–25.30 wt%), and stearic (4.60–8.13 wt%) acids. Commercial infant formulas achieve similar fatty acid compositions to those found in human milk by combining edible oils from vegetable origin. However, in these formulas, the fraction of the total palmitic acid that is esterified at the sn-2 position (11.64–13.43 %) is considerably lower than in HMF (57.11–67.15 %). The total and positional fatty acid profiles of the substrates used for stage 1 and stage 2 are shown in Table 2. HMPS is an edible fat rich in tripalmitin (PPP > 30 %) obtained by the fractionation process of palm stearin, in which it is separated from a softer oleic fraction rich in POP type of TAGs (also known as PMF) [28]. The major FA in HMPS is palmitic acid (70.98 mol%), from which 28.77 % is esterified at the sn-2 position. Other important FAs in HMPS include oleic (17.77 mol%), and stearic (5.61 mol%) acids. HOSO contains 74.08 mol% oleic, 9.59 mol% linoleic, and 7.12 mol% palmitic acids. TC, a pure substance, contains more than 98 mol% capric acid.
Table 3 compares the SLs obtained in stage 1 for each substrate molar ratio tested. The SLs correspond to the reaction time at which sn-2 palmitic acid was the highest among independent experiments performed at 4, 8, 12 and 24 h. The major FA in all the SLs was oleic acid (59.01–71.73 mol%). This high content can be directly attributed to the high molar ratio of HOSO used as a substrate for all the SLs (>60 mol%). SL1–1, SL1–2, SL1–3, SL1–4 had 26.40, 25.14, 18.37, and 18.51 mol% total palmitic acid, respectively. Compared to the total palmitic composition of both HMF and infant formulas (see Table 1), all the SLs were reasonably within the range. However, it was SL1–2 that showed the highest fraction of its palmitic acid esterified at the sn-2 position (35.21 %), when compared to the values obtained in SL1–1 (31.42 %), SL1–3 (32.56 %), and SL1–4 (33.16 %). TAGs containing sn-2 palmitic acid are preferred in HMF analogues, as it helps in fat digestion and absorption. A number of digestive enzymes are involved in the breakdown of the human milk TAGs. As a result of the pancreatic lipase activity, all FAs in the sn-1,3 positions of TAGs are hydrolyzed during digestion, in contrast to only around 20 % of fatty acids in the sn-2 position [29]. If palmitic acid is predominantly esterified at sn-1,3 positions, it is released as free palmitic acid. At the alkaline pH of the intestine, free palmitic acid readily forms insoluble soaps with divalent cations such as calcium that cannot be absorbed in the small intestine and are excreted as hard stools. This results in not just unavailability of both palmitic acid and calcium for the infants, but also in constipation problems [30]. Although Lipozyme® TL IM lipase is a sn-1,3 specific enzyme, a considerable amount of palmitic acid was esterified at the sn-2 position of TAGs, which influenced the final levels found in all the SLs. This may be attributed to acyl migration. Acyl migration is considered a side reaction involving migration of acyl groups from sn-1,3 to sn-2 positions and vice versa [31]. It mainly occurs due to the presence of diacylglycerols (DAGs) and monoacylglycerols (MAGs), which are intermediate products of enzymatic syntheses of lipids [32]. Neither DAGs nor MAGs were analyzed in the current study. Interesterification reaction comprises two steps. First, the original TAGs are hydrolyzed into DAGs and MAGs, and then fatty acid are re-esterified in other positions to form novel TAGs. Acyl migration can occur between these two steps. Furthermore, it has been found that acyl migration can be also influenced by a number of factors, including temperature, degree of unsaturation of FAs, reaction time, FFA%, presence of water, and carrier of the enzyme [33]. Acyl migration is mostly considered an undesirable phenomenon that affects the conversion rates and the product yield in enzymatic reactions of lipids, but in this particular case it was desirable since it contributed to the sn-2 palmitic acid increase in the final SLs. As SL1–2 was synthesized using a molar ratio 1:2 (HMPS:HOSO), which is intermediate between the ratios used for SL1–1 and SL1–4, it is plausible that there exists a maximum value where the substrate ratio affected acyl migration the most. Because of the highest content of palmitic acid at the sn-2 position, and its relative lower levels of total oleic acid (66.85 mol%), SL1–2 was selected as the substrate for the stage 2 reaction.
Table 4 compares the SLs obtained in stage 2 for each substrate molar ratio tested. The SLs correspond to the reaction time at which capric acid incorporation was the highest (while conserving the sn-2 palmitic acid content achieved in stage 1) among independent experiments performed at 4, 8, 12 and 24 h. The major FA in all the SLs was oleic acid (52.98–61.85 mol%). SL2–1, SL2–2, SL2–3, and SL2–4 had 23.37, 25.12, 21.34, and 20.13 mol% total palmitic acid, respectively. The fraction of palmitic acid esterified at the sn-2 position in SL2–1 (38.33 %), SL2–2 (38.87 %), SL2–3 (38.93 %), and SL2–4 (37.57 %) did not show significant difference at p ≤ 0.05. On the other hand, the amount of capric acid incorporated in SL2–1, SL2–2, SL2–3, and SL2–4 was 8.23, 11.15, 18.68, and 21.22 mol%, respectively. The levels of major FAs such as capric and oleic acid at the sn-2 position in all the SLs tended to remain stable during the enzymatic synthesis when compared to the values found in the physical blends of substrates before the reaction. However, in the case of palmitic acid, there was a slight increase in the fraction esterified at the sn-2 position, as it went from 35.21 % (level in SL1–2 from stage 1, used as substrate) to nearly 38 % in the SLs of stage 2. Again, this may be attributed to acyl migration where palmitic acid displaced other fatty acids from the sn-2 position. TAGs rich in MCFAs, such as capric acid, have received considerable attention because of their distinctive metabolic pathways for digestion and absorption when compared to LCTs. MCTs oils are not assimilated via lymphatic transport like LCTs, but rather are absorbed via portal transport to provide quick energy to the infants [18]. Among the SLs synthesized, SL2–4 was considered the best option for a possible infant formula fat analog because of its high fraction of palmitic acid esterified at the sn-2 position (~38 %), its relatively low content of oleic acid (52.98 mol%), and the total amount of capric acid incorporated (21.22 mol%). Whereas sn-2 palmitic acid level of SL2–4 appeared to be below that found in HMF (see Table 1), it still seems to be a better alternative for a human fat analog than the fats currently used in infant formulas (37.57 % against 13.43 %) and other edible oil-based SLs recently reported by Teichert and Akoh [13], Turan et al. [14], and Pina-Rodriguez and Akoh [16], who achieved 32.85, 35.17, and 29.09 %, respectively (described as relative percentage of total palmitic acid). Although SL2–4 contained somewhat higher levels of total oleic acid compared to HMF (28.30–43.83 mol%), commercial infant formulas (34.34–44.69 mol%), and the SLs reported by Pande et al. [15] (37.10–43.22 mol%), Zou et al. [11] (32.57–34.15 mol%), and Wang et al. [12] (34.10 mol%), it can be physically blended with other vegetable oils in adequate proportions in order to adjust the oleic acid content, while still maintaining the desired amount of sn-2 palmitic acid and total capric acid in the final product. Neither HMF nor commercial formulas contain relevant amounts of capric acid as a source of MCFAs. Instead, they provided around 17 wt% of combined lauric and myristic acids, but none of them will provide the metabolic and physiologic effects of capric acid as MCFA. SL2–4 could be an alternative to provide these important fatty acids to the infants.
TAG Molecular Species
The TAG molecular species and their relative content in substrates and SLs of stages 1 and 2 are shown in Table 5. The fatty acids in the TAG species are not in any specific order on the glycerol backbone. HMPS showed a mixture of eight different types of TAGs. These species correspond to those concentrated originally from palm oil by fractionation process [28]. The main TAGs present in HMPS were PPP (43.01 %), POP (30.81), OPO (7.20 %) and PPSt (7.28 %). HOSO, which is a liquid oil, contained mostly OOO (70.73 %), OPO (11.94 %), and StOO (7.53 %) types of TAGs. TC is a pure substance containing above 99 % of tricaprin (CCC). For stage 1, PB1–2 represents the physical blend of substrates to produce SL1–2 (intermediate SL) before the enzymatic reaction occurs. This blend exhibited a more complex set of TAGs, since the species found in HMPS and HOSO were combined at the established ratio. Major TAGs found in PB1–2 included OOO (48.02 %), PPP (13.31 %), OPO (10.67 %) and POP (9.83 %). As shown in SL1–2, it was found that the amount of OOO and PPP after the reaction decreased to 15.93 and 1.84 %, respectively. On the other hand, the amount of OPO (a combination of sn-OPO and sn-POO) increased to 34.63 % and POP increased to 22.97 %. These values can be attributed to the sn-1,3 specific-type of interaction between OOO (from HOSO) with PPP and PPSt (from HMPS) to produce OPO and POP. In this case, OPO formation was favored over POP by using a molar ratio higher in OOO (1:2, HMPS:HOSO). Once SL1–2 was blended with TC in stage 2 (see PB2–4, Table 5), the TAG species found were CCC (20.57 %), OOO (12.40 %), OPO (27.75 %), and POP (18.49 %). After the reaction, a novel set of TAGs was synthesized as a result of the interesterification of CCC with all the pre-existing TAGs in the intermediate SL. As shown in SL2–4, the new species included CLC (11.13 %), COC (7.63 %), CPC (4.50 %), OCO (3.46 %), and OPC (2.00 %), whereas CCC was almost totally depleted. The levels of OOO (10.42 %), OPO (28.06 %) and POP (18.79 %) remained reasonably the same before and after the enzymatic incorporation of capric acid. Other minor species present included OLO (3.58 %), and PLO (2.63 %). There was no evidence of unreacted PPP.
The major TAG molecular species found in HMF were OPO (17.56–42.44 %), POL (9.24–38.15 %), OOO (1.61–11.96 %) and OLO (1.64–10.18 %) [34]. SL2–4 contained all the TAGs of HMF within its levels except for POL, which fell below the range. Similar results were found by Pande et al. [15], who reported the enzymatic synthesis of extra virgin olive oil-based SLs with POL levels between 2.13 and 6.03 %. This can be explained by the poor levels of linoleic acid present in both substrates used for the production of SL1–2, which predominantly contained oleic and palmitic acids. The metabolic fate of TAGs from dietary fat during digestion and absorption is determined by the position of the fatty acids on the glycerol backbone and their chain length [15]. Compared to OOP, OPO is better metabolized and absorbed in infants [35]. Since the fraction of total palmitic acid esterified at the sn-2 position in SL2–4 increased up to nearly 40 %, and OOO decreased from an initial value of 48.02 % in PB1–2 to 10.42 %, it is plausible to assume that SL2–4 might contain higher amounts of OPO than OOP. TAG profile greatly influences the physical properties of the SLs. There are four major categories for TAG species depending on the degree of unsaturation, namely, SSS (trisaturated), UUU (triunsaturated), SUU (monosaturated–diunsaturated), and SUS (disaturated–monounsaturated). When the values of final SL2–4 were compared with PB1–2 before the reactions, it was found that SSS decreased from 15.68 to 5.33 %, and UUU decreased from 54.07 to 14.00 %. On the other hand SUU increased from 16.49 to 38.75 %, and SUS increased from 13.76 to 41.92 % as a result of the formation of the symmetric species of OPO, POP, and COC.
Melting and Crystallization Profiles
In the crystal state, TAG molecules adopt different arrangements in relation to their neighbors to optimize intramolecular and intermolecular interactions and accomplish an efficient close-packing. These arrangements, so-called polymorphs, determine the melting properties of fats and oils. There are three main types of polymorphs depending on their physical stability: α (unstable, lowest melting point), β′ (metastable, intermediate melting point), and β (stable, highest melting point) [36]. The occurrence of one or more polymorphs in a single fat or oil is highly influenced by the chain length and the degree of unsaturation of the FAs, which define the symmetry of the TAGs structures. In general, fats with high content of long-chain saturated fatty acids have higher melting points than those containing short and medium chains, and/or unsaturated fatty acids [37].
The melting profiles of substrates and SLs of stages 1 and 2 are shown in Fig. 2. HMPS, which contained 50.28 % of SSS type of TAGs (mostly PPP and PPSt), had a melting completion temperature (T mc) of 61.6 °C. On the other hand HOSO, which is liquid oil with 79.39 % of UUU (mostly OOO), was completely melted at 0.6 °C. Although TC contained nearly 99 % of SSS (tricaprin), its T mc of 41.3 °C is considerably lower than that found for HMPS because of the chain length of the FAs. In both stages 1 and 2, the melting temperature of the SLs decreased when compared to the physical blend of substrates before the reactions. For SL1–2, T mc went from 54.8 to 34.2 °C, and for SL2–4 went from 57.6 to 27.7 °C. This behavior is highly correlated with the loss of SSS and the formation of SUS and SUU type of TAGs during the enzymatic interesterifications. Both, HMF and oil blends used for commercial formulas are completely melted at normal body temperature (about 37 °C) [7]. SL2–4 had a T mc under this value, which may be helpful in the formulation of products with a proper consistency and texture for the infants. On the contrary, HMF analogs with high T mc are difficult to handle, to transport and to combine with other non-lipidic ingredients in a formulation. Such is the case of the final SLs synthesize by Pande et al. [15], which had high levels of PPP type of TAGs (up to 10.32 %) and consequently, high T mc values (up to 42 °C). Additionally, the crystallization onset temperature (T co) for HMPS, HOSO and TC were 36.3, −12.4, and 3.3 °C, respectively (see Fig. 3). T co for SL1–2 decreased from 26.6 to 15.0 °C, while SL2–4 decreased from 12.9 to 6.1 °C (before and after the reactions). The complexity and wide range of TAG species in SLs resulted in multiple crystallization peaks rather than a sharp event as in TC for example, which is made of a simple homogeneous TAG.

Melting thermograms of substrates and structured lipids. The values shown correspond to melting completion temperatures. HMPS high melting point palm stearin, HOSO high oleic sunflower oil, TC tricaprin, PB1–2 physical blend of substrates before reaction using a molar ratio 1:2 (HMPS:HOSO), SL1–2 structured lipid synthesized in stage 1 (intermediate SL), PB2–4 physical blend of substrates before reaction using a molar ratio 1.2:0.5 (SL1–2:TC), SL2–4 structured lipid synthesized in stage 2 (final SL)

Crystallization thermograms of substrates and structured lipids. The values shown correspond to crystallization onset temperatures. HMPS high melting point palm stearin, HOSO high oleic sunflower oil, TC tricaprin, PB1–2 physical blend of substrates before reaction using molar ratio 1:2 (HMPS:HOSO), SL1–2 structured lipid synthesized in stage 1 (intermediate SL), PB2–4 physical blend of substrates before reaction using molar ratio 1.2:0.5 (SL1–2:TC), SL2–4 structured lipid synthesized in stage 2 (final SL)
Upscaling, Free Fatty Acid Assay, and Reaction Yield
In order to evaluate the reaction yield of the final SL synthesized, scaled-up reactions of SL1–2 (stage 1) and SL2–4 (stage 2) were performed in a stirred tank reactor without solvent. The objective of this test was to determine the FFA development in the SLs as the enzymatic reactions took place. Figure 4 shows the change in FFA% (expressed as oleic acid) in SLs as a function of time. SL1–2 exhibited a high rate of FFA formation in the first 8 h of reaction as it rose from 0.16 to 5.08 wt%. After this, it continued to increase gradually until it approached equilibrium at 24 h (6.10 wt%). The reaction time established for SL1–2 in small scale was also 24 h, as the conversion of the SL at this point proved to be the highest among the range tested. In stage 2, SL2–4 also showed a high rate of FFA formation until it reached a peak at 8 h (4.00 wt%), and then it dropped slightly to find equilibrium at 24 h (3.04 wt%). The reaction time established for SL1–2 in small scale was 8 h (higher incorporation of capric acid). In this case, the SL synthesis was finished before the system approached equilibrium (24 h). The environment for lipase-catalyzed interesterification involves water. The water content in a reaction system is the determining factor as to whether the reaction equilibrium will be towards hydrolysis or ester synthesis. If the water content increases in the system, the FFA content will increase (along with DAGs), and as a consequence, the yield of the new TAG products will decrease [38, 39]. Although most reactions require water content of <1 % for effective interesterification, too low water activity prevents all reactions from occurring because lipases need a certain amount of water to remain hydrated, which is essential for enzymatic activity [40, 41]. Both SL1–2 and SL2–4 produced in the stirred tank reactor were consistent with the physical and chemical properties obtained on small scale. Excess FFAs were removed from SL1–2 and SL2–4 by SPD to <0.1 wt%. The combined loss (expressed as %FFAs in SL) was 10 wt%, and therefore, the overall reaction yield was nearly 0.9 (90 wt%). This result is considerable higher compared to the common reaction yields observed for acidolysis reactions in SL production for different applications, such as 70 wt% [15], 60 wt% [16], and 55 wt% [17]. A higher product yield implies less operating expenses and higher return, making the SL somewhat more economically feasible for industrial purposes.

Free fatty acid development in SLs reaction. SL1–2 structured lipid synthesized in stage 1 using a substrate molar ratio 1:2 (HMPS:HOSO), SL2–4 structured lipid synthesized in stage 2 using a substrate molar ratio 1.2:0.5 (SL1–2:TC)
Conclusion
For those health conditions where infants cannot be breastfed, commercial formulas are the most convenient alternative. However, the differences in stereospecific structure of TAGs in vegetable oils used in infant formula relative to those in HMF lead to lower energy use and less calcium absorption by formula-fed infants. SLs that contain palmitic acid predominantly at the sn-2 position, and which are also enriched with capric acid as a source of MCFAs, can be used in the formulation of a new generation of commercial formulas that may closer fit the nutritional needs of infants and enhance their physiological and metabolic functions. Therefore, SL2–4 may be suitable for use in commercial infant formulas as an HMF analog, as it was synthesized at a relatively high product yield (90 wt%), and it contained substantial levels of palmitic acid at the sn-2 position and capric acid as a source of MCFAs for the proper growth and development of infants.
References
Jensen RG (1989) The lipids of human milk. CRC Press, Boca Raton
Wells JCK (1996) Nutritional considerations in infant formula design. Semin Neonatal 1:19–26
Scott CE (2010) Structured lipid: a functional and fundamental ingredient to more closely matches infant formula to breast milk. Nutrafoods 9:7–13
Innis SM (1991) Essential fatty acids in growth and development. Prog Lipid Res 30:39–103
Bracco U (1994) Effect of triglyceride structure on fat absorption. Am J Clin Nutr 60:1002S–1009S
Lucas A, Quinlan P, Abrams S, Ryan S, Meah S, Lucas PJ (1997) Randomised controlled trial of a synthetic triglyceride milk formula for preterm infants. Arch Dis Child Fetal Neonatal 77:F178–F184
Tomarelli RM, Meyer BJ, Weaber JR, Bernhart FW (1968) Effect of positional distribution on the absorption of the fatty acids of human milk and infant formulas. J Nutr 95:583–590
Høy CE, Xu X (2001) Structured triacylglycerols. In: Gunstone FD (ed) Structured and modified lipids. Marcel Dekker, New York, pp 209–239
Akoh CC, Kim BH (2008) Structured Lipids. In: Akoh CC, Min DB (eds) Food lipids. CRC Press, Boca Raton, pp 841–872
Zock PL, Gerritsen J, Katan MB (1996) Partial conservation of the sn-2 position of dietary triglycerides in fasting plasma lipids in humans. Eur J Clin Investig 26:141–150
Zou X, Huang J, Jin Q, Guo Z, Liu Y, Cheong L, Xu X, Wang X (2013) Characterization and oxidative stability of human milk fat substitutes enzymatically produced from palm stearin. J Am Oil Chem Soc 91:481–495
Wang X, Huang J, Jin Q, Song Z, Liu Y, Zou X (2012) Lipase-catalyzed synthesis of human milk fat substitutes from palm stearin in a continuous packed bed reactor. J Am Oil Chem Soc 89:1463–1472
Teichert SA, Akoh CC (2011) Modifications of stearidonic acid soybean oil by enzymatic acidolysis for the production of human milk fat analogues. J Agric Food Chem 59:13300–13310
Turan D, Yeşilçubuk NS, Akoh CC (2012) Production of human milk fat analogue containing docosahexaenoic and arachidonic acids. J Agric Food Chem 60:4402–4407
Pande G, Sabir JSM, Baeshen NA, Akoh CC (2013) Enzymatic synthesis of extra virgin olive oil based infant formula fat analogues containing ARA and DHA: one-stage and two-stage syntheses. J Agric Food Chem 61:10590–10598
Pina-Rodriguez AM, Akoh CC (2009) Synthesis and characterization of a structured lipid from amaranth oil as a partial fat substitute in milk-based infant formula. J Agric Food Chem 57:6748–6756
Ifeduba EA, Akoh CC (2013) Chemoenzymatic method for producing stearidonic acid concentrates from stearidonic acid soybean oil. J Am Oil Chem Soc 90:1011–1022
Babayan VK (1988) Medium chain triglycerides. In: Babayan VK, Beare-Rogers J (eds) Dietary fat requirements in health and development. American Oil Chemists’ Society Press, Champaign, pp 73–86
Francois CA, Connor SL, Wander RS, Connor WE (1998) Acute effects of dietary fatty acids on the fatty acids of human milk. Am J Clin Nutr 67:301–308
López-López A, López-Sabater MC, Campoy-Folgoso C, Rivero-Urgell M, Castellote-Bargalló AI (2002) Fatty acid and sn-2 fatty acid composition in human milk from Granada (Spain) and in infant formulas. Eur J Clin Nutr 56:1242–1254
St-Onge MP, Mayrsohn B, O’Keeffe M, Kissileff HR, Choudhury AR, Laferrère B (2014) Impact of medium and long chain triglycerides consumption on appetite and food intake in overweight men. Eur J Clin Nutr 68:1134–1140
St-Onge MP, Ross R, Parsons WD, Jones PJ (2003) Medium-chain triglycerides increase energy expenditure and decrease adiposity in overweight men. Obes Res 11:395–402
Novozymes Biopharma DK A/S (2014) Enzymes for Biocatalysis: for smarter chemical synthesis. http://www.novozymes.com/en/solutions/biopharma/Brochures/Documents/2014-12576-01_Biocatalysis-Product-Sheet-Immobilised-Lipases-2.pdf. Accessed 20 Apr 2015
Pande G, Sabir JSM, Baeshen NA, Akoh CC (2013) Synthesis of infant formula fat analogs enriched with DHA from extra virgin olive oil and tripalmitin. J Am Oil Chem Soc 90:1311–1318
Official Methods of Analysis of AOAC International (2012) Method 996.01. AOAC Int, Gaitherburg
Luddy FE, Bardford RA, Herb SF, Magidman P, Riemenschneider RW (1964) Pancreatic lipase hydrolysis of triacylglycerides as a semi-micro technique. J Am Oil Chem Soc 41:639–696
Official Methods and Recommended Practices of the American Oil Chemists’ Society (2011) Method Cj 1–94. AOCS, Champaign
Son JM, Lee KT, Akoh CC, Kim MR, Kim MJ, Lee JH (2010) Optimization of tripalmitin-rich fractionation from palm stearin by response surface methodology. J Sci Food Agric 90:1520–1526
Karupaiah T, Sundram K (2007) Effects of stereospecific positioning of fatty acids in triacylglycerol structures in native and randomized fats: a review of their nutritional implications. Nutr Metab 4:1–17
Kennedy K, Fewtrell MS, Morely R, Abbott R, Quinlan PT, Wells JC, Bindels JG, Lucas A (1999) Double-blind, randomized trial of a synthetic triacylglycerol in formula-fed term infants: effects on stool biochemistry, stool characteristics, and bone mineralization. Am J Clin Nutr 70:920–927
Yang T, Fruekilde MB, Xu X (2005) Suppression of acyl migration in enzymatic production of structured lipids through temperature programming. Food Chem 92:101–107
Xu X (2000) Production of specific-structured triacylglycerols by lipase-catalyzed reactions: a review. Eur J Lipid Sci Technol 102:287–303
Xu X, Skands ARH, Høy CE, Mu H, Balchen S, Adler-Nissen J (1998) Production of specific-structured lipids by enzymatic interesterification: elucidation of acyl migration by response surface design. J Am Oil Chem Soc 75:1179–1186
Morera S, Castellote AI, Jauregui O, Casals I, López-Sabater MC (2003) Triacylglycerol markers of mature human milk. Eur J Clin Nutr 57:1621–1626
Mu H, Høy CE (2004) The digestion of dietary triacyglycerols: review. Prog Lipid Res 43:105–133
Sato K (2005) Polymorphism in fats and oils. In: Bailey’s industrial oil and fat products. Wiley, Hiroshima
Strayer D (2006) Food fats and oils. Institute of Shortening and Edible Oils, Washington DC
Bornaz S, Fanni J, Parmentier M (1994) Limit of the solid fat content modification of butter. J Am Oil Chem Soc 71:1373–1380
Malcata FX, Reyes HR, García HS, Hill CG Jr, Amundson CH (1992) Kinetics and mechanisms catalyzed by immobilized lipases. Enzyme Microb Technol 14:426–446
Briand D, Dubreucq E, Galzy P (1994) Enzymatic fatty esters synthesis in aqueous medium with lipase from Candida parapsilosis (Ashford) Langeron and Talice. Biotechnol Lett 16(813):818
Svesson I, Wehtje E, Adlercreutz P, Mattiasson B (1994) Effects of water activity on reaction rates and equilibrium positions in enzymatic esterifications. Biotechnol Bioeng 44:549–556
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Álvarez, C.A., Akoh, C.C. Enzymatic Synthesis of Infant Formula Fat Analog Enriched with Capric Acid. J Am Oil Chem Soc 92, 1003–1014 (2015). https://doi.org/10.1007/s11746-015-2662-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11746-015-2662-z