
Abstract
Alkaline esterase (carboxylic-ester hydrolases; EC 3.1.1.1) extracted from germinated soybean seeds (Glycine max) was purified approximately 3.6 times by chromatography in a DEAE-cellulose anion exchange column and filtration in Sephadex G100 gel. The molecular mass of the enzyme was estimated at 45 kDa by gel electrophoresis (SDS-PAGE). The purified enzyme showed a specific activity of 5.6 U mg−1 using p-nitrophenyl butyrate as substrate. The esterase showed optimal activity at 47 °C in moderately alkaline pH, low stability in temperatures higher than 50 °C, and high stability at pH values between 6 and 9.5. The Ca2+ and Co2+ ions proved to have a positive effect on enzyme activity; however, Hg2+ completely inhibited esterase activity. Using p-nitrophenyl butyrate as substrate, the enzyme showed a K m of 0.39 mM, V max of 31.5 mM mg−1 min−1 and k cat 7.60 × 106 s−1. Regarding substrate affinity, the enzyme showed greater activity for substrates containing short-chain fatty acids, especially p-nitrophenyl acetate. Such characteristics give the enzyme great potential for application in the production of low molecular weight esters, in the food industry, and in chemical products. This enzyme is another new member of the family of lipases and esterases from vegetable seeds with high activity and stability in alkaline pH.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Esterases (carboxyl ester hydrolases; EC. 3.1.1.1) and lipases (triacylglycerol acylhydrolase EC. 3.1.1.3), collectively known as ““lipolytic enzymes,″” are characterized by their potential to hydrolyze hydrophobic short- and long-chain carboxylic acid esters, respectively. Lipases catalyze the hydrolysis of ester bonds at the transition between an insoluble substrate phase and the aqueous phase, whereas esterases catalyze the hydrolysis of ester bonds of water-soluble substrates. However, medium-length fatty acid chains can be hydrolyzed by both enzymes [1–4].
Lipolytic enzymes present in vegetable seeds are responsible for the hydrolysis of triglycerides stored in the endosperm into free fatty acids and glycerol [4, 5]. Recently, lipolytic enzymes from seeds have been the focus of great attention as biocatalysts. In some cases, these enzymes present advantages over animal and microbial lipases due to some quite interesting features, such as specificity, low cost, availability, and ease of purification, representing a good alternative for potential commercial exploitation as industrial enzymes [4, 6–10].
The growing interest in esterases is due to the fact that these enzymes can be applied in many sectors, such as the food industry, pharmaceuticals, fine chemistry, etc. Various features of esterases have been reported, primarily wide distribution, quantification, production, synthesis target, purification, and molecular biology [11]. The esterases are applied in dairy products, wine production, fruit juices, beer and alcohol, synthesis of low molecular weight esters, etc. They are also applied in cosmetics, detergents, and transformation of low molecular weight fats into oils with greater commercial value and resolution of racemic compounds, among others [4, 11].
Esterases and lipases from seeds present some advantages in comparison to esterases from microorganisms, because, in some cases, they can be applied without being purified [3]. Due to their promising potential for commercial application, multiple studies have assessed the characteristics of lipases and esterases from seeds, including beans [8], sunflowers [13, 14], flaxseeds [15], peanuts [12], and cotton seeds [16].
Staubmann et al. [19] studied the physicochemical characteristics of two purified esterases and one purified lipase from Jatropha curcas. The authors observed that the enzymes showed optimum pH activity at a moderately alkaline pH level of about 8 and high stability at high temperatures (50 °C). Polizelli et al. [10] evaluated the physicochemical properties of lipase from Pachira aquatica and also observed that the lipolytic enzyme showed stability at 40 °C and optimum activity at alkaline pH 8. These are interesting characteristics that give this enzyme great potential for industrial application.
This study identified, purified, and evaluated the biochemical, biophysical, and biocatalytic characteristics of esterase from soybean (Glycine max) seeds after germination.
Materials and Methods
Materials
The soybean seeds (Glycine max) studied were donated by VITAO Brazilian industry LTDA, São Paulo-Brazil. The substrates for esterase p-nitrophenyl acetate (pNPA), p-nitrophenyl butyrate (pNPB), p-nitrophenyl caprylate (pNPC), p-nitrophenyl laurate (pNPL) and p-nitrophenyl palmitate (pNPP) were obtained from Sigma-Aldrich (St Louis, USA). All other chemical agents used were of analytical grade. The materials used in the purification process: Ultrafiltration tubes Millipore/UFC 903024, Sephadex G100 gel column, DEAE-Cellulose anion exchange column, were obtained from Sigma-Aldrich (São Paulo, Brazil).
Enzyme Extraction
The soybean esterase was extracted using the method described by Kermasha and van de Voort [17]. Samples of 150 g (dry weight) of seeds were triturated after 24 h of germination, and the flour obtained was resuspended in a solution containing 1 × 10−3M CaCl2 and 5 × 10−3M EDTA (1:3 w/v) and homogenized in a blender at 4 °C for 3 min. The sample was transferred to a 500-mL Erlenmeyer flask, stirred mechanically at 5 °C for 30 min, and then subjected to ultrasound (125–200 W) for 1 min to release the bound enzyme. The resulting suspension was centrifuged at 1,645g for 45 min to release the suspended residue. The supernatant containing the enzyme was lyophilized, and the enzymatic powder obtained was kept in a refrigerator at 4 °C.
Enzyme Purification
DEAE Cellulose Column Ion Exchange Chromatography
The crude enzymatic extract was eluted in a DEAE-Cellulose anion exchange column (44 cm diameter × 1 cm radius, particle size 100–200 µm). The buffer used for elution was 0.1 M Tris–HCl, pH 7.5, with a flow rate of 5 mL every 20 min. A total of 175 fractions were collected, and the absorbance was measured at 280 nm. An NaCl salt gradient was applied, 0–1 M, starting at fraction 59. The esterase activity in the fractions was determined using p-nitrophenyl butyrate (pNPB), as described below.
Sephadex G100 Gel Filtration Column Chromatography
The enzymatically active protein fractions collected after elution in DEAE cellulose were concentrated by ultrafiltration. The process of ultrafiltration was conducted in ultrafiltration tubes with a regenerated cellulose membrane, 30 kDa. Fifteen mL of protein fractions eluted from DEAE cellulose were added to each tube and centrifuged at 1,645×g for 25 min at 5 °C. After the ultrafiltration, 5 mL of concentrated enzymatic solution were obtained, and the sample was eluted in a Sephadex G100 gel column (1.1 cm × 95 cm) containing 80 cm3 of resin (particle size 40–120 µm) previously equilibrated with 0.05 M Tris–HCl buffer, pH 7.5, containing 0.1 M KCl. The proteins were eluted by applying 300 mL of the same buffer and 3-mL fractions were collected every 30 min using a fraction collector. The course of protein elution was followed by measuring the absorbance at 280 nm and observing the catalytic activity of the soybean esterase.
Gel Electrophoresis
The molar mass of the partially purified enzyme was determined by Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis (SDS-PAGE) in a 12 % polyacrylamide gel, according to the methodology described by Laemmli [18]. A mixture of low molecular mass standards (Bio-Rad 161-0304) was used, which contained phosphorylase B (97.4 kDa), serum albumin (66.2 kDa), ovalbumin (45 kDa), carbonic anhydrase (31 kDa), soybean trypsin inhibitor (21.5 kDa) and lysozyme (14.4 kDa).Coomassie blue was used to detect the protein bands.
Determination of Esterase Activity by Spectrophotometry
The esterase activity was determined by spectrophotometry at 405 nm in a DU 640, Beckman Coulter™, using p-nitrophenyl butyrate (pNPB) as substrate. 70 μL of the enzymatic solution was added to 3.43 mL of a reaction mixture containing the following compounds, added in the following order: 1.12 mM pNPB dissolved in a 50 mM phosphate buffer at pH 7.0, plus 0.2 % (N/P) Triton X-100, and 3 % tetrahydrofuran. The enzymatic reaction was carried out for a period of 5 min at 37 °C and monitored for 1 min against a blank solution. The enzyme activity was calculated by the molar extinction coefficient of 1.5 × 10−4 M−1 cm−1 and path-length of the cell 1 cm. One unit of activity was defined as the quantity of enzymes necessary to release 1 μmol micromole of p-nitrophenyl per minute under the specified conditions [18].
The soluble protein content was quantified using the method described by Bradford [19] with a bovine serum albumin standard solution, and the specific esterase activity was determined as the relationship between the enzymatic activity (U) and the protein content, and expressed as U mg−1.
Determination of Esterase Activity by Titration
The esterase activity was determined using 1 mL of triacetin substrate, 7 mL of 0.1 M phosphate buffer solution at pH 7.0 and 1 mL of enzyme extract. The reaction mixture was incubated at 37 °C for 30 min in a thermostatic bath with stirring at 130 oscillations per minute. The reaction was stopped with the addition of 15 mL of acetone solution:ethanol (1:1 v:v), and the short acyl chain acids released were titrated with 0.05N KOH solution, using phenolphthalein as the indicator. One unit of esterase activity was defined as the quantity of enzymes necessary to release 1 μmol of fatty acids per minute, under the described conditions [9].
Determination of Lipase Activity by Titration
One milliliter of enzyme extract added to a reaction mixture containing 2 mL of 0.1 M phosphate buffer at pH 7.0 and 5 mL of an emulsion having the following composition (v/v): 75 % Arabic gum (7 %) and 25 % olive oil. The solution was incubated at in a thermostatic bath with stirring at 130 oscillations per minute. After incubation, the reaction was stopped by adding 15 mL ethanol:acetone 1:1 (v/v), the fatty acids being released and titrated with 0.05N KOH solution, using phenolphthalein as the indicator. One unit of lipase activity was defined as the amount of enzyme required to release 1 μmol of oleic acid in 1 min per mL under the specified conditions.
Experimental Design to Determine the Optimum Temperature and pH for Soybean Esterase Activity
When the problem involves data that are subject to experimental errors, statistical methods measure the effects of change in operating variables and their mutual interactions on the process performance through factorial experimental designs. The independent variables are usually referred as factors [20]. The data analysis were carried out on the basis of a central composite rotatable second-order design experiment (CCRD) which includes 11 experiments of 2 variables at 5 levels (−1.41, −1, 0, +1, +1.41).
The fractional factorial design consisted of 4 factorial points, 4 axial points (2 axial points on the axis of each design variable, at a distance of 1.41 from the design center) and 3 central points. The important factors consider for soybean esterase enzymatic activity were pH and temperature. Their corresponding coded levels are presented in Table 1.
The values for pH and temperature were established based on previous experiments [4].
The coefficients of the equation were determined by employing Statistica® 8.0 from Statsoft (Tulsa, OK, USA) software. The ANOVA analysis was done using the same software. The results of experimental design were fitted to a second-order model equation using the coded variables in Table 1.
Effect of Temperature and pH on Soybean Esterase Stability
To assess the stability pH, aliquots from purified soybean esterase were kept for 4 h at 30 °C in 0.1 M acetate buffer, pH 3.6, 4.0, 4.5, 5.0 and 5.6; 0.1 M phosphate buffer, pH 6.0, 6.5 and 7.0; 0.1 M Tris–HCl buffer, pH 7.0, 8.0, 8.5 and 9.0; and 0.1 M Borax-NaOH buffer, pH 9.5 and 10.0. The residual activity was determined as previously described. For the temperature and stability tests, aliquots of 70 μL of purified enzyme were kept for 1 h at 30, 40, 50, 60, 70 and 90 °C. The residual activity was determined at 37 °C, as previously described. The tests were conducted in triplicate, and the averages were subjected to the Tukey test with a confidence interval of ±95 % and a significance level of p ≤ 0.05.
Soybean Esterase Specificity Study
The specificity of the enzyme was studied by evaluating the hydrolysis activity indifferent synthetic substrates: pNPA (2:0), pNPB(4:0), pNPC (6:0), pNPL (12:0) and pNPP (16:0), at 37 °C for 5 min, as described above.
Effect of Substrate Concentration
Soybean esterase was assayed in reaction buffer (pH 7) with different concentrations (0.5–2 mM) of pNPB (p-nitrophenyl butyrate) as a substrate. The values of V max (maximum velocity) and K m (Michaelis constant) were calculated from Lineweaver–Burk, Hofstee and Hanes–Woof plots. The value of k cat was determined using Eq. (1)
where V 0 is the initial reaction velocity (mM s−1), k cat is the turnover number, the number of times each enzyme site converts substrate to product per unit time (s−1), K m is the Michaelis–Menten Constant (mM), and Et is the concentration of enzyme at the catalytic enzyme sites (mM).
Influence of Metal Ions on Enzyme Activity
The measure the influence of metal ions (Ca2+, Mg2+, Li+, Co2+, Na+, K+, Zn2+, Mn2+, Hg2+) on enzymatic activity, salts were added to the reaction buffer at a concentration of 10 mM. The enzymatic activity of esterase with no addition of salts (CaCl2, MgCl2, CoCl2, NaCl2, KCl2, ZnSO4, MnCl2, HgCl2) was considered to be 100 %, and the values of enzymatic activity from the reaction mixtures containing salts were expressed as a percentage of relative activity.
Statistical Analysis
The data from the experiments are presented as the mean ± standard error mean (SEM), and were analyzed using a one-way analysis of variance (ANOVA), with the differences analyzed by the Tukey test at p < 0.05 (v.9.2; SAS Institute, Cary, NC, USA).
Results and Discussion
Identification of Alkaline Esterase from Soybean
The presence of esterase activity in crude soybean extract was observed using triacetin, triolein and p-nitrophenyl esters as substrate (Table 2). High activity in triacetin hydrolysis was observed; however, the enzyme did not act on triolein. The same happened with the esterases (JEA and JEB) from J. curcas L. studied by Staubmam et al. [21]. When p-nitrophenyl acetate, butyrate, caprylate and palmitate were used as substrate, higher activity was observed in p-nitrophenyl acetate. As the carbon chain of the substrate increased, enzymatic activity decreased.
Purification of Soybean Esterase
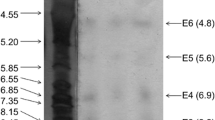
The crude soybean extract containing esterase was, firstly, eluted in a DEAE-cellulose ion exchange column equilibrated in a 0.1 M Tris–HCl buffer, pH 7.5. Esterase activity was observed in fractions 20–35, where there was also a higher concentration of protein (Fig. 1; Table 2). After fraction 60, the proteins were eluted with a gradient from 0.1 M Tris–HCl buffer, pH 7.5, containing NaCl 0–1 M. Analyses in polyacrylamide gel electrophoresis at 15 % showed the appearance of more than one band, demonstrating that the enzyme was not completely purified. The fractions containing enzymatic activity collected from the DEAE-Sepharose column were again eluted in a Sephadex G-100 gel column using a 0.05 M Tris buffer, pH 7.5, containing 0.1 M KCl. Two peaks of protein were observed during the process of elution in the gel column, showing that esterase was eluted in the first peak from fraction 32–50 (Fig. 2). SDS-PAGE polyacrylamide gel electrophoresis analyses showed that, after elution in the gel column, the enzyme was purified, with only one band being observed.

Elution profile of soybean esterase using chromatographic DEAE Cellulose anion exchange column. The buffer used for elution was 0.1 M Tris–HCl, pH 7.5. An NaCl salt gradient was applied, 0–1 M starting at fraction 59

Elution profile of soybean esterase in chromatographic filtration column in Sephadex G 100 gel using 0.05 M Tris–HCl buffer, pH 7.5, containing 0.1 M KCl
All phases involving the soybean esterase purification process are shown in Table 3. The overall purification was 3.6-fold, with an activity yield of 20 %. Esterase purification was achieved to an apparent homogeneity in two steps. The soybean esterase showed a molecular mass of 45 kDa estimated by Polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 3). Esterases EI and EIIfrom Jatropha curcas, studied by Staubmam et al. [21], showed molecular masses of 23 and 30.2 kDa, respectively.

Polyacrylamide gel electrophoresis (SDS-PAGE) of the crude and purified preparations of soybean esterase in DEAE-cellulose and Sephadex G-100 columns. P standard protein, A crude esterase extract, B esterase purified in DEAE-cellulose column, C esterase purified in Sephadex G 100 column
Biochemical and Physicochemical Characterization of Purified Soybean Esterase
An experimental design was carried out to verify the pH and optimum temperature of the esterase activity from purified soybean. Table 4 illustrates the results of the experimental design for the study of the effect of temperature and pH and the interaction between them in the soybean esterase activity.
For soybean esterase, it was observed that the temperature and pH showed a linear positive effect on the enzyme activity. That is, an increase in temperature and pH had a positive influence on soybean esterase activity. The p value calculated for each variable, and for the interaction among them, indicates that the interaction between pH and temperature did not show an effect on enzyme activity. The model containing the significant coded terms can be seen in Eq. (2):
The ANOVA indicates that the regression was significant at a 95 % confidence interval, insofar as, in the F test, calculated F was greater than tabulated F. The square correlation coefficient was 0.92, meaning that the model is able to explain 92 %.
Figure 4 shows the effect of pH and temperature on the esterase activity from purified soybean.

Response surface plot showing the effect of pH and temperature on the purified esterase activity from soybean seed
The results show that, as temperature and pH rose, enzymatic activity also rose, indicating maximum activity at around pH 8 and 47 °C with 17.50 U mg−1 of activity. Barros and Macedo [4] studied the effect of pH on the activity of esterase from crude soybean extract. The authors observed higher activity around pH 8–8.5. At pH 9, the enzyme showed 60 % activity. Almond seed lipase, studied by Yesiloglu and Baskurt [23] presented optimum activity at a pH value of about 8.5. The laurel seed lipase was isolated and characterized by Isbilir et al. [22] and presented optimum activity at pH 8.
Figure 5a, b illustrate the effect of temperature and pH on the stability of the purified esterase.

Effect of temperature (a) and pH (b) on the stability of the purified esterase from soybean seed. The assays were carried out using pNPB as the substrate. Each point is the average of three replicates ±SD
Purified esterase from soybean showed stability after 1 h of incubation at 45 °C and pH 7.0 and was completely inactivated at temperatures higher than 50 °C. According to Barros and Macedo [4], the crude preparation of esterase from soybean proved to be stable after 1 h at 70 °C and pH 7.0 and was inactivated at temperatures higher than 80 °C, indicating that the crude preparation of esterase from soybean is more stable than the purified enzyme. Based on the literature, esterase from purified soybean can be considered a stable enzyme at medium temperatures, as it was approximately 100 % when incubated at 45 °C. Yesiloglu and Baskurt [23] studied lipase from peanuts (Antygadalus communis) and observed that the enzyme was stable in a temperature range from 10 to 90 °C. Lipase from Laurus nobilis seeds, studied by Isbilir et al. [22], showed 80 % relative activity after being kept for 1 h at 50 °C.
Regarding pH, esterase from purified soybean showed stability in a wide range of pH values (5–10), and the enzyme showed greatest stability at pH 7.5. At pH 9, the enzyme retained 55 % activity. The esterase from crude preparation showed greater stability in the pH range from 6.5 to 10, the greatest stability being at pH 7.5 [4]. Lipase from peanuts, studied by Yesioglu and Baskurt [23], showed a profile of stability at pH levels from 6 to 9.5, similar to that of soybean esterase.
Esterases (JEA and JEB) from J. curcas L. seed, studied by Staubmam et al. [21], showed optimum activity and stability at pH 8 and high activity and stability at higher temperatures (50 °C). The authors reported that such characteristics make application of these enzymes possible in various industrial sectors.
Influence of Metal Ions and Kinetic Parameters
Figure 6 illustrates the effect of some ions on the activity of purified esterase from soybean.

Effect of some ions on the enzymatic activity of purified soybean esterase. The assays were carried out using pNPB as the substrate at 37 °C and pH 7.0. Each point is the average of three replicates ±SD
The addition of the salts CaCl2 and CoCl2 to the reaction mixture at a concentration of 10 mM activated the purified esterase around 4 times, compared to the control. Peanut lipase, studied by Yesioglu and Baskurt [23], showed an increase in relative activity of 3.5 and 1.5 times when ions Co2+ and Ca2+, respectively, were present in the medium reaction.
Isbilir et al. [22] studied the Laurus nobilis L. lipase and observed a significant increase in enzymatic activity when ions Ca2+, Co2+, Cu2+, Fe2+ and Mg2+ were added to the enzyme medium reaction. Lipase from oilseed (P. aquatica), studied by Polizelli et al. [10], also showed an increase in relative activity when Ca2+ and Mg2+ were present in the reaction medium.
The ion Hg2+, at a concentration of 10 mM, completely inhibited soybean esterase activity. The same was observed for esterase from Jatropha curcas L., studied by Staubmam et al. [21]. Almost all enzymes are inhibited by Hg2+. To test SH in the active site, p-chloro, mercury, benzoate, iodoacetamide, etc. have been used.
The values of K m, Vmax and k cat of the purified enzyme for substrate p-NPB were estimated at 0.481 mM, 22.8 U mg−1 and 7.60 × 106 s−1, respectively. Also calculated was the ratio between k cat/K m; the value was 15.83 × 106 M−1 s−1 Fig. 7.

Determination of K m and V max values of purified esterase from soybean seed, using substrate p-nitrophenyl butyrate, according to the Lineweaver–Burk method
Specificity of Esterase from Purified Soybean
The esterase from purified soybean showed the following order of activity on substrates: pNPA > pNPB > pNPC > pNPL > pNPP (data not shown), similar behavior to that exhibited by the enzyme in the crude extract (Table 2). The same profile was observed for esterases from J. crucas. The esterase from barley, studied by Kubicka et al. [24], showed great affinity for short-chain fatty acids, especially acetic acid esters.
Liaquat and Apeten [25] studied the synthesis of different aromatic esters using corn lipase and observed that this lipase presented greater affinity for short-chain substrates in the following order: acetic acid (2C) > butyric acid (4C) > caprylic acid (6C), in an organic medium using isoamyl alcohol, after 72 h of reaction. The authors reported that lipolytic enzymes that showed greater affinity for short-chain fatty acids have great potential for application in the synthesis of aromatic esters.
Conclusion
The esterase from soybean Glycine max showed some interesting characteristics, such as stability and elevated activity in moderately alkaline pH, stability and high activity at temperatures around 45 °C and elevated affinity for short-chain fatty acids. In addition to being an enzyme that is easy to extract, alkaline enzymes like esterase from soybean have great potential for application in various industrial sectors, such as in the synthesis of chiral molecules, wood pulping, detergents, food, production of esters of low molecular mass, and so on.
References
Lin H-Y, Wimer LT, Huang AHC (1993) Lipase in the lipid bodies of corn scutella during seedling growth. Plant Physiol 73:460–463
BhardwajK Raju A, Rajasekharan R (2001) Identification, purification, and characterization of a thermally stable lipase from rice bran. A new member of the (Phospho) lipase family. Plant Physiol 127:1728–1738
BarrosM Fleuri LF, Macedo GA (2010) Seed lipases: sources, applications and properties—a review. Braz J Chem Eng 27:15–29
Barros M, Macedo GA (2011) Biochemical characterization and biocatalytic potential of Esterase from Brazilian Glycine Max. J Food Sci Biotechnol 20:1195–1201
QuettierAL Eastmond PJ (2009) Storage oil hydrolysis during early seedling growth. Plant Physiol Biochem 47:485–490
Hellyer SA, Chandler IC, Bosley JA (1999) Can the fatty acid selectivity of plant lipases be predicted from the composition of the seed triglyceride? BBA Mol Cell Biol Lipids 1440(2–3):215–224
Villeneuve P (2003) Plant lipases and their applications in oils and fats modification. Eur J Lipid Sci Tech 105:308–317
EnujiughaVN Thani FA, Sanni TM, Abigor RD (2004) Lipase activity in dormant seeds of the African oil bean (Pentaclethra macrophylla Benth). Food Chem 88:405–410
Paques FW, Macedo GA (2006) Lipases de Látex Vegetais: Propriedades eAplicações Industriais: a review. Quim Nova 29:93–102
PolizelliPP Tiera MJ, Bonilla-Rodriguez GO (2008) Effect of surfactant sand polyethylene glycol on the activity and stability of a lipase from oilseeds of Pachira aquatica. J Am Oil Chem Soc 85:749–753
Panda T, Gowrishankar BS (2005) Production and applications of esterases. Appl Microbiol Biotechnol 67:160–169
Huang AHC, Moreau RA (1978) Lipases in the storage tissues of peanut and other oil seeds during germination. Planta 141:111–116
Arabaci N, Sagiroglu A (2005) Sunflower seed lipase: extraction, purification, and characterization. Prep Biochem Biotech 35:37–51
Sadeghipour HR, Bhatla SC (2003) Light-enhanced oil body mobilization in sunflower seedlings accompanies faster protease action on oleosins. Plant Physiol Biochem 41:309–316
Sammour RH (2005) Purification and partial characterization of an acid lipase in germinating lipid body linseedlings. Turk J Bot 29:177–184
Rakhimov MM, Dzhanbaeva NR, Yuldashev PK (1970) Specificity of the lipase from cotton seed. Chem Nat Compd 6:616–619
Kermasha S, van de Voort FR, Metche M (1986) Characterization of French bean (Phaseolus vulgaris) seed lipase. Can I Food Sci Tech J 19:23–27
Laemmli UK (1970) Cleavage of structural protein during the assembly of head of bacteriophage T4. Nature 227:680–685
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Kiran KR, Monohar B, Divakar S (2001) A central composite rotatable design analysis of lipase catalyzed synthesis of lauroyl lactic acid at bench-scale level. Enzyme Microb Tech 29:122–128
Staubmam R, Ncube I, Gübitz GM, Steiner W, Read JS (1999) Esterase and lipase activity in Jatropha curcas L. seeds. J Biotechnol 75:117–126
IsbilirSS Ozcan MH, Yagar H (2008) Some biochemical properties of lipase from bay laurel (Laurus nobilis L.) seeds. J Am Oil Chem Soc 85:227–233
YesilogluY Baskurt L (2008) Partial purification and characterization of almond seed lipase. Prep Biochem Biotechnol 38:397–410
Kubicka E, Grabska J, Jedrychowski I, Czyz B (2000) Changes of specific activity of lipase and lipoxygenase during germination of wheat and barley. Int J Food Sci Nutr 51:301–304
Liaquat M, Apenten RKO (2000) Synthesis of low molecular weight flavor esters using plant seedling lipases in organic media. J Food Sci 65:295–299
Acknowledgments
This project was supported by the CNPq (Conselho Nacional de Pesquisa e Desenvolvimento).
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
de Barros, M., Macedo, G.A. Biochemical Characterization of Purified Esterase from Soybean (Glycine max) Seed. J Am Oil Chem Soc 92, 37–45 (2015). https://doi.org/10.1007/s11746-014-2573-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11746-014-2573-4