
Abstract
Kobresia plant (Kobresia littledalei) is the dominant vegetation type in the Qinghai-Tibet Plateau region where the temperatures can be extremely low and harsh during winter. However, the potential molecular mechanisms that respond to cold remain to be fully elucidated. In this study, we applied the use of high-throughput sequencing technology in investigating the genes involved in Kobresia plant acclimation and response to cold stress. Kobresia plants were grown in pots for 7 days in a 25 °C greenhouse and thereafter subdivided into 6 batches (Kli-0 to Kli-5) that were exposed to cold-treatment in a – 5 °C cryogenic treatment room at varying timelines (0–48 h); With Kli-0 batch being the control (untreated). We sequenced the treated samples and obtained 90,331,944 clean reads. Clustering analysis assigned a total of 214,531 assembled trinity genes. For functional annotation, all the assembled unigenes were aligned against public databases that include NCBI’s Pfam (Pfam protein families), Uniprot (Swiss-Prot), KEGG (Kyoto Encyclopedia of Genes and Genomes database) and KOG (eukaryotic orthologous groups) classification system was used to assign the possible functions of the obtained unigenes. From these, we linked a great number of candidate genes to the cold stress response. Several significant DEGs and metabolic responses were identified and discussed. Further, we identified significant DEG’s from the transcriptome data. AP2/ERF-ERF gene family could be playing a significant role that enhances the survival of K. littledalei to cold stress conditions. In conclusion, our findings herein further the general understanding of Kobresia plants' adaptation and responses to cold stress through the molecular mechanisms involved in signal regulation and cold resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The geographical distribution of plants greatly relies on the changes in climatic conditions. Indeed, the plant’s evolution and geographical distribution patterns are dictated by its adaptation strategies to the existing climatic variables (Byun et al. 2018). The climatic changes have continued to impact on plants’ physiological functions, and this has not only influenced interrelationship among organisms but also between the plants and the abiotic factors within the ecosystems (Lau and Lennon 2012). The shift in the climatic pattern may also result in the alteration of selective pressures on plants, this may equally cause potential influence on the evolutionary processes. Such responses may result to the alteration of ecological processes as well as the species interactions through eco-evolutionary feedbacks mechanism (Franks and Beerling 2009; Shefferson and Salguero-Gomez 2015). Indeed, in some occurrences, evolutionary responses can be so rapid that it is realized within a few generations. On this basis, having a deeper understanding of the mechanism of plant adaptation to climate characteristics and applying modern agricultural practices is important for improved agricultural productivity and a stable ecosystem (Andrae et al. 2019). However, the continuous application of modern agricultural practice has resisted the impact of adverse environmental conditions on plant productivity. It not only promotes the growth and health of plants but also continues to affect the availability of soil nutrients, physical and chemical properties, inhibition of plant diseases, the occurrence of diseases, and soil microbial community (Francioli et al. 2016).
Tibet region occurs on the Qinghai-Tibet Plateau of the Peoples Republic of China. It is within a high altitude and dominated by cold temperatures, strong ultraviolet rays, drought and other extreme and harsh climatic conditions (Piao et al. 2010). Studies on vegetation adapted to climatic conditions similar to the Qinghai-Tibet Plateau environment have identified plant communities with along and well-developed natural selection (Korner 2016; Provart et al. 2003). The germplasm resources of most of the cold environments have a high degree of cold-resistant genetic genes which are valuable resources to the promotion of stable agro-ecological construction within the cold areas such as the Tibet region (Byun et al. 2018). In harsh cold regions, plants develop specialized mechanisms to enhance their survival to the low temperature. Such phenomenon calls for a well-developed gene expression program which enhances the adjustment of the physiological processes such as the metabolic-structural alterations, nutrient contents, cell osmotic potential, and turgor mediated responses like elongation, stomatal, and leaf movement to protect the plant in a complex adverse environment (Baurle 2016; Heidarvand and Amiri 2010). Plants respond to low temperatures by regulating gene expression and protein productions. Indeed, the full genome profiling/ sequencing, mutational and transgenic plant analyses have given some hopes in providing the complex molecular mechanism of plants' response to cold stress (Yoon et al. 2016). Therefore, having accurate information on the distribution of plants and their adaptation strategies to the adverse climatic conditions within the Alpine and high-altitude areas is paramount. This will ensure a clearer understanding of the scientific mechanism of distribution for the alpine vegetation of the high-altitude regions for continued ecological integrity.
The Kobresia littledalei commonly known as the Zang Song Grass is one of the main vegetation types in the Qinghai-Tibet Plateau. It is an herb with an average maturity height of 20–30 cm and has a short rhizome. K. littledalei has numerous economic and ecological functions within the high-altitude Qinghai-Tibet Plateau. Moreover, studies have reported that the physiological adaptation of the K. littledalei to the harsh environment of higher altitudes contributes to the significant changes in its chemical components which consequently confers a higher nutritive value to local herbivores. This makes it necessary to have a deeper understanding of the physiological activities responsible for such nutritional variability within the K. littledalei grass of the Qinghai-Tibet Plateau (Wang et al. 2008). Based on inadequate information on the relationship between the physiological characteristics and the environmental sensitivity and adaptation strategies of the K. littledalei grass within the high altitude, this study applied modern molecular technologies to explore the relationship between the enzymatic activities’ patterns and osmotic regulation of the grass to the cold stress condition within the Qinghai-Tibet Plateau.
Materials and methods
Plant material and cold treatment
Kobresia plants (Kobresia littledalei) were randomly picked from natural field grounds in Tibet and afterwards transplanted into a nursery bed for recovery. The grass patches were grown in pots for 7 days in a 25 °C greenhouse. Thereafter, the grass pots were sub-divided into 6 batches and moved to a − 5 °C cryogenic treatment room; where each grass pot (Kli-0 to Kli-5) was exposed to different cold treatment timelines.Kli-0, Kli-1, Kli-2, Kli-3, Kli-4, Kli-5 were treated for 0, 0.5, 4, 12, 24, 48 h respectively. Obtained samples were stored at − 80 °C after freezing in liquid nitrogen.
Physiological responses assay
Levels of carbohydrates, proline, and malondialdehyde (MDA) were measured as per previously described protocols (Fan et al. 2015). Plant detoxification enzymes like superoxide dismutase (SOD), peroxidase (POD) were analyzed as described by Fan (Fan et al. 2015). A unit of SOD activity was described as the enzyme quantity that can inhibit NBT reduction by 50% whereas a unit of POD activity was defined as the amount of enzyme causing a change of 0.01 in absorbance per minute. The analysis was conducted in triplicates, and results were expressed as average values (mean ± standard error).
Total RNA extraction, RNA-seq library construction and sequencing
Total RNA was extracted using the RNAprep Pure Kit DP432 (TIANGEN Biotech Co., Ltd., Beijing, China), as per the manufacturer’s instructions. Upon elution, all the RNA samples were assessed for their integrity using an Agilent 2100 Bio Analyzer. RNA samples from the same treatments were mixed and used for the determinations. Library construction was made using VAHTSTM Stranded RNA-seq Library Prep Kit for Illumina (Vazyme Biotech Co.,Ltd, Nanjing, China), as per the manufacturer’s guidelines. Thereafter, paired-end, Hi-seq 2000 Ilumina sequencing was performed.
Bioinformatics analysis of sequenced data
Raw RNA-seq reads were processed to remove low-quality reads (reads of more than 50% bases with Q-value ≤ 20) together with reads comprising of more than 5% ambiguous nucleotides. In addition, adapter sequences were clipped off. Using Trinity, obtained clean reads were de novo assembled to form a collection of non-redundant genes. Overlapping sequences were further merged to produce longer contiguous sequences, after which the reads were mapped back to the contigs using bowtie (bowtie2 software version 2.2.9). The contigs were further assembled to identify sequences without end extension, hence defined as genes. These genes were subjected to alignment (E-value < 10 − 5) against public protein databases; like Pfam (Pfam Protein families), Uniprot (Swiss-Prot), KEGG (Kyoto Encyclopedia of Genes and Genomes database) and KOG (eukaryotic orthologous groups). Blast2GO was used to annotate assembled unigenes to generate GO terms as per their biological process, cellular component, and molecular function. ESTScan was used to predict coding regions and sequence direction of genes without annotation. Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used in determining metabolic pathways of the obtained genes.
Gene expression quantification, differential expression analysis and co-expression network analysis
Trinity analysis platform was used for gene expression quantification and differential analysis. It comprised RSEM (v1.2.6) for transcript abundance estimation and normalization of expression values as FPKM (Fragments per kilobase of transcript per million fragments mapped). Differentially expressed genes were identified with DESeq2 with a filter threshold of adjusted p-value < 0.05and |log2FoldChange|> 1. Calculation of GO terms and pathways enriched in the differentially expressed genes was by hypergeometric test. Weighted Gene Co-expression Network Analysis (WGCNA) was applied in identifying modules of highly correlated genes (Langfelder et al. 2013). Eigengene module was used to summarize the present related gene clusters. The blockwise modules function of the WGCNA package in R was used to generate the modules with a power of six.
Quantitative real-time PCR analysis
Analysis of the transcripts was done using quantitative real-time PCR (qRT-PCR). cDNA was synthesized from 1 μg DNase treated total RNA using M-MLV Reverse Transcriptase (Promega) and poly(dT) oligonucleotides. One-Step SYBR Prime Script RT-PCR Kit (TAKARA, Dalian, China) was used in qRT-PCR, as per the manufacturer’s guidelines. Melting curve analysis was used to verify the qPCR products. Quantification was attained by normalizing the number of target transcript copies to the reference control using the comparative ΔΔCt method. All reactions were performed in triplicates.
Results
Physiological changes induced by low temperature
To examine physiological responses in Kobresia plants, we measured levels of Sucrose, Fructose, Glucose, soluble starch, soluble protein, soluble total sugar (Fig. 1, Table S1), and proline and malondialdehyde (MDA), Antioxidant enzymes activity of SOD and POD, were also determined (Fig. 2, Table S1). After exposure to − 5 °C cryogenic temperature, soluble total sugar, soluble starch, sucrose, and fructose levels significantly decreased with time from 0.5 h of exposure up to 48 h of exposure. In contrast, Glucose levels rose steadily over from 10.85 mg/g at 0 h to 18.66 mg/g at 48 h while soluble protein remained at almost a constant level (Fig. 1).

Osmoregulation changes under cold treatment

Changes in the content of POD, SOD, Proline, MDA under cold treatment
POD enzymatic activity decreased from 3.510 μ/g at 0 h of exposure to 2.298 μ/g at 48 h of treatment. Correspondingly, SOD activity drastically decreased overtime from 1645 μ/g at 0 h to 835 μ/g at 48 h. Proline and MDA increased significantly over the treatment period from 167.71 ng/g to 280.32 ng/g and 96.53 nmol/g to 143.71 nmol/g, respectively.
Illumina sequencing and reads assembly
In triplicates, the cold-treated Kobresia plant samples; Kli-0, Kli-1, Kli-2, Kli-3, Kli-4 and Kli-5 were sequenced for transcriptome analysis. Paired-end libraries were built from synthesized cDNA, thereafter RNA-seq was performed using Illumina HiSeq2000 platform. From the sequenced data, we obtained more than 4.7 Gb from each treated sample. Low-quality reads and adapters were trimmed and discarded, and a total of 90,331,944 assembled clean reads.
A total of 419,514 transcripts with a length of 306 were produced using Trinity software. For transcription studies without reference genomes, we assembled the sequenced reads to generate contigs of 421 bp average length. Five contigs were obtained; N10, N20, N30, N40 and N50 of sizes 1357, 947, 721, 570, and 460, respectively. Clustering analysis assigned a total of 214,531 assembled trinity genes (Table 1).
Functional annotation and classification
All assembled genes were subjected to an alignment on public databases that include Pfam (Pfam protein families), Uniprot (Swiss-Prot), KEGG (Kyoto Encyclopedia of Genes and Genomes database) and KOG (eukaryotic orthologous groups), using BLASTx alignment algorithm with an of E-value < 10 − 5. A total of 70,960 unigenes had a significant BLASTx hits in at least one of the four above-mentioned databases. 36,454, 62,907, 36,222 and 65,095 were blasted to Pfam, Uniprot, KEGG and KOG, respectively. A total of 16,471 unigenes had hit in all four databases. Among the genes annotated on Pfam, 31,721 genes corresponded to the other three protein databases. Similarly, 62,085 genes that were annotated in the Uniprot databases had hits in the other three tested protein databases (Fig. 3).

Venn diagram of the total number of genes that can be annotated by four databases (GO, Pfam, Uniprot and KEGG)
Gene Ontology (GO) classification system was used to assign the possible functions of genes. A total of 70,960 genes were successfully categorized into 61 functional groups (Fig. 4); which were further separated into three GO terms: ‘biological processes, ‘cellular component’ and ‘molecular function’ using BLAST2GO. Under biological processes, ‘transcription’, ‘regulation of oxidation’ and ‘protein phosphorylation’ were the top three. In cellular component, ‘integral component of membrane’ was leading, followed by ‘nucleus’ and ‘cytoplasm’ whereas the molecular functions had ‘ATP binding’, ‘Iron ion binding’, DNA binding’ and ‘RNA binding’ as the top functions.

Gene Ontology (GO) classification of all putative genes
All the obtained genes were then aligned to the KOG database. They were divided into 47 functional groups (Fig. 5). ‘Translation’, ‘Folding, Sorting and degradation’, Transport and Catabolism’ and ‘Carbohydrate metabolism’ were the top four leading categories. Notable KOG groups involved in plant response activity were ‘Environmental adaptation’, ‘Signal molecules and interaction’ and signal transduction’.

KOG classification of putative proteins
Identification of differentially expressed genes (DEGs)
To analyze any significant difference and alteration in gene expression after cold stress treatment, we used DESEQ2 method (Differential gene expression analysis based on the negative binomial distribution) so as to calculate the gene expression levels. Compared to Kli-0, genes with absolute values of log2 (ratio) ≥ 2 and p-value ≤ 0.001 were counted as DEGs (differentially expressed genes). We had statistics on differential expressed genes. The numbers of DEGs ranges from 118 to 2454 (Fig. 6). GO analysis showed DEGs enrichment in different items (Table S2) such as ‘DNA-binding transcription factor activity’ and ‘response to salicylic acid’ which participate in cold stress response. KEGG analysis showed DEGs enrichment in different items (Table S3) such as ‘Plant hormone signal transduction’ and ‘Starch and sucrose metabolism’ which explain that why the physiological is changing.

Numbers of DEGs under differeent cold treatment times
Plant transcription factor analysis
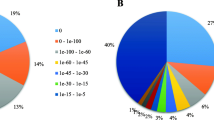
The iTAK transcription factor analysis was performed so as to identify the different genes present during cold stress. From our results, the top five DEGs transcription factors were AP2/ERF-ERF, NAC, WRKY, bHLH, GARP-G2-like transcription factor families that accounted for 22.22, 8.89, 8.89, 5.56 and 4.44%, respectively (Fig. 7a). For all the studied groups, when compared to the control group, there were significant differences in the differentially expressed transcription factor families. For instance, the top five DEGs in Kli-1 group were AP2/ERF-ERF, WRKY, C2H2, Tify, NAC (Fig. 7b), while for Kli-5 study group, AP2/ERF-ERF, NAC, WRKY, bHLH, GARP-G were the top five highly expressed transcription factors (Fig. 7c).

Analysis of transcription factors among differentially expressed genes induced by cold stress in Kobresia littledalei. A Transcription factors analysis of all DEGs after cold treatment. B Transcription factors analysis of DEGs at 0.5 h after cold treatment. C Transcription factors analysis of DEGs at 48 h after cold treatment
Co-expression network analysis and identification
Co-expression networks were made using the WGCNA package (Langfelder and Horvath 2008) (Fig. 8). All co-expressed genes are inter-connected at varying correlation values. From our data, we identified distinct 15 modules, with each module having genes ranging from 110 to 7736. In this group, 11 modules comprised of more than 500 genes (Table 2). Notably, antique-white associated module genes were highly expressed in Kli-1 study group but not in the other groups while genes associated with the dark grey module were highly expressed in Kli-5 study group but with lower expression in the other groups (Fig. 9a, b). The significantly enriched GO terms of antique-white module were ‘protein serine/threonine kinase activity’ and ‘positive regulation of defense response’. As for dark grey module, the significant enriched GO trems were ‘transporter activity’ and ‘calmodulin binding’. We suspect that different genes may function at different stages of adversity processing.

a Validation of the express ion of TRINITY_DN206601_c2_g3 by qPT PCR; b Validation of the expression of TRINITY_DN200900_c1_g2 by qPT PCR

WGCNA analysis of expressed genes under cold treatment a Gene expression pattern in antique-white module b Gene expression pattern in dark grey
Validation of differentially expressed genes by qPT-PCR
To further verify genes that play an important role in responding to cold stress, we selected two of these genes (TRINITY_DN206601_c2_g3 and TRINITY_DN200900_c1_g2) for subsequent qRT-PCR validation. One of two genes, TRINITY_DN206601_c2_g3 is homologous to BAM3_ARATH, and the other is homologous to BSK5_ARATH. BAM3_ARATH and BSK5_ARATH are associated with cold stress. The results showed that both genes were up-regulated under cold stress treatment (Fig. 8). In addition, we randomly selected five genes for qRT-PCR, and the results were consistent with RNA-seq (Table S4).
Discussion
Low temperatures pose a great influence on plant growth, typically because it had seriously affected the metabolism and physiology of plants (Guy et al. 2008). This is because the cellular membranes are liquefied structures and low temperatures can impact on their fluidity, resulting into increased rigidity. Plant cells are able to detect cold stress via low temperature-induced shifts in protein conformation, nucleic acid conformation, membrane and/or specific metabolite concentration. Such stimuli are able to activate cold acclimation responses. Cold acclimation refers to the low-temperature responses and adaptation (Guy et al. 2008; Renaut et al. 2008).
Carbohydrates metabolism under cold stress
Carbohydrates are recognized as key players in plant response and adaptation to cold stress. Plant sugars function as energy reserves, scavengers of free radical, signal molecules and compounds that enhance plant response to biotic and abiotic stress (Guy et al. 2008; Mittler 2002). Specifically, during cold acclimation, there is a buildup of soluble sugars in the plant cells. These accumulated sugars act as osmolytes, compatible solutes, energy reserves and carbon skeletons. Equally, during plant adaptation to low temperatures, the levels of Sucrose, fructose and soluble sugars declined with continuous exposure to cold treatment. The continuous decline of carbohydrates at 0 h of cold treatment to 48 h post-cold treatment may be due to the increase of activity of plant proteolytic enzymes that take effect more when the plants have prolonged exposure to cold treatment.
Changes of antioxidant enzymes under cold stress
Plants' exposure to abiotic stress factors usually gives rise to the accumulation of reactive oxygen species (ROS). These ROS include hydrogen peroxide, superoxide, and hydroxyradicals (Miller et al. 2010; Mittler 2002). High concentrations of ROS can induce injury of cellular structures and related macromolecules, resulting in cell death (Apel and Hirt 2004; Krasensky and Jonak 2012) subsequently, plants at risk of abiotic stress have mounted a defense mechanism against oxidative stress through ROS-scavenging enzymes. These scavenging enzymes include superoxide dismutase (SOD) and peroxidase (POD), among others (Miller et al. 2010). These antioxidant enzyme activities of SOD and POD promote plant tolerance to cold tolerance and promote cell membrane system protection (Alscher et al. 2002).
From our results, SOD activity significantly decreased from 1645 μ/g at 0 h of cold treatment to 835 μ/g at 48 h post-cold treatment; a half-drop in its activity when compared to the control (Fig. 2b). Similarly, the enzymatic activity of POD decreased from 3.510 μ/g at 0 h of low-temperature exposure to 2.298 μ/g at 48 h of cold treatment. The observed decline in enzymatic activities of SOD and POD may be due to the continuous damage of the cell membrane due to the constant exposure to low temperature of – 5 °C. In addition, the observed scenario maybe due to the effect of the low temperature below the optimum range needed for SOD and POD enzymes to function effectively. Furthermore, the low temperature may have influenced RNA transcription and translation, thus reducing POD synthesis. Also, the observed increase in proline under low temperatures to breakdown peroxidase might have influenced the decline in POD activity (Liu et al. 2013).
Changes of free proline content under cold stress
Proline is an organic osmolyte that is widely distributed in plants as a protection material (Verbruggen and Hermans 2008). It plays a very crucial in osmoregulatory in sustaining osmotic balance and maintain the integrity of plant cellular structures. This in turn protects the plant from water loss and dehydration ensuing from cold stress through reduction of water potential of plant cells (Szekely et al. 2008). Moreover, proline has the capability to act as a chaperone in stabilizing the protein structures in addition to playing a role in antioxidant system regulation (Armengaud et al. 2004; Xin and Browse 1998). Under abiotic stress like low temperature, plants accumulate free proline as a response measure against it. This proline accumulation in turn protects the plant against the encountered abiotic stress (Liu et al. 2009), hence the reason we studied proline in this experiment. In our study, the levels of Proline in Kobresia littledalei increased significantly in response to cold stress over the treatment period from 167.71 ng/g under 0 h of treatment to 280.32 ng/g under 48 h treatment (Fig. 2c). Compared to Kli-0 as the control, free proline content gradually rose with time prolongation exposure of low temperature. This observed proline accumulation over prolonged exposure in low temperature indicates its significance in plant responses against cold stress time in maintaining the osmotic pressure at balance. Thus, preventing plant damage due to stress. Our findings are in good agreement with the previously conducted studies that report on proline build-up in plants' response to abiotic stress (Liu et al. 2009).
Changes of malondialdehyde (MDA) content under cold stress
Elongated plant exposure to low temperatures results to damage of cell membranes. Malondialdehyde (MDA) is a well-recognized parameter that indicates the injury extent of cell membrane system and decline in cellular metabolism (Fan et al. 2012; Hodgson and Raison 1991). Generally, MDA is deemed as a final product of lipid peroxidation processes that are present on the cell membrane of the plant (Landi 2017). Further, in ROS scavenging, MDA is a vital intermediate and its presence in plants in high levels is lethal to cells. Thus, MDA production can be used in the evaluation of membrane damage in plants exposed to low temperatures. In this study, MDA concentration significantly increased all through the cold treatment period. Its levels rose from 96.53 nmol/g at 0hours of exposure to cold treatment to 143.71 nmol/g at 48 h post-cold treatment (Fig. 2d). These results imply that the cell membrane was gradually being injured with prolonged exposure time in low temperatures.
Cold-related genes, transcription factors and DEG’s associated with cold stress
Several kinds of transcription families have been confirmed to be widely involved in cold stress responses in plants. These cold-related gene factors are induced in plants when their stimulations come into play when under cold stress. Previously conducted studies have identified GARP, AP2/ERF, MYB76-like, ZAT10-like, DELLA protein GAIP as transcription factors and candidate genes that could regulate or promote cold resistant in plants(Singh et al. 2002). In the present study, AP2/ERF-ERF, NAC, WRKY, bHLH, GARP-G2-like were identified to be involved in the cold stress response in plants. Wholesomely, there were significant differences in the identified differentially expressed transcription factors in short-term cold treatment and long-term cold treatment. C2H2 and Tify transcription factors were found to be significantly expressed in the short-term cold treatment stage only while bHLH, GARP-G2 transcription factors were highly expressed under long-term cold treatment. In this regard, we attribute that there may be different types of transcription factor families associated with cold stress response mechanisms in either short-term cold stress exposure or prolonged exposure. In this case, C2H2 and Tify transcription factors are associated with instant and rapid cold stress response mechanisms while bHLH and GARP-G2-like transcription factors come into play in long-term plant response and adaptation to cold stress exposure. Further, from our transcriptome data, we identified 53 ‘CBF’ associated genes, 318 ‘heat shock’ associated proteins, and 84 ABA-associated proteins. Heat-shock proteins (Hsps) or stress-induced proteins have been reported to be induced in plants as a result of abiotic stress. These plant proteins can be divided into five categories (Hsp100, Hsp90, Hsp70, Hsp60 and small Hsps) according to their molecular weight. It is believed that this diversification of these proteins reflects a response to abiotic stress (Baniwal et al. 2004). In addition, brassinosteroid (BR) is another class of plant steroid hormone that regulates plant development and physiology. In plants, the BR signal is transduced by a receptor kinase-mediated signal transduction pathway called brassinosteroid-signaling kinase (BSK). Studies have identified different homologous BR-signaling kinases such as BSK1, BSK2 BSK3, BSK5. Genetic and transgenic studies have demonstrated that the BSKs represent a small family of kinases that activate BR signaling downstream of BR, which demonstrate that BSKs are the substrates of BR kinase that activate downstream BR signal transduction under stress conditions (Tang et al. 2008).
Identification of the plant transcription factors and classifying them into different gene families through the iTAK transcription factor analysis ensure the identification of genes dominant during a particular condition such as cold stress (Zheng et al. 2016). The DEGs transcription factors AP2/ERF-ERF, WRKY, C2H2, Tify, NAC, AP2/ERF-ERF, NAC, WRKY, bHLH and GARP-G revealed through iTAK transcription factor analysis indicated the range of genes expressed by K. littledalei under cold stress conditions. The higher percentage of AP2/ERF-ERF (22.22%) indicated the overexpression of the gene cluster under cold conditions and therefore could be playing a significant functional role in the survival of K. littledalei to cold stress. While WGCNA is applicable in most high-dimensional data sets, it has been most widely used in genomic applications. By analyzing gene expression patterns, WGCNA makes modular analysis of genes and expounds the relationship between different modules (Langfelder et al. 2013; Li et al. 2018). It has highly ben recommended to identify miRNAs associated with various responses (Shiah et al. 2014). Using this technique, our study revealed 15 distinct modules, each module having genes ranging from 110 to 7736, with 11 group comprising of more than 500 genes. Furthermore, the dominant antique-white and dark grey associated genes provide a clue on the potential association of these genes to survival strategies in cold stress conditions and calls for further studies to ascertain that.
Through previous studies, the AP2/ERF-ERF gene family that was also found in large quantity under the present study, was found to be a large plant-specific transcription factor family sharing well-conserved DNA-binding domain and includes DRE-binding proteins which are responsible for activating abiotic stress-responsive genes expression through specific binding to the dehydration-responsive element/C-repeat cis-acting element in their promoters (Mizoi et al. 2012). A study by also confirmed that AP2/ERF factors take part in the development of plants, regulation of hormones as well as in plant response to cold conditions (Xiong et al. 2013). Based on its crucial role in the development and survival of the plants, its proposed that a detailed understanding of its molecular basis for development and stresses tolerance in the different plant species is necessary (Cui et al. 2016). Arabidopsis AP2/ERF gene family members such as RAP2.6 enhance plant survival by improving plants’ response to biotic stress, abscisic acid condition, high salt condition, osmotic stress condition, and cold stress conditions. It achieves this by acting as a trans-activator, hence able to bind to the GCC and CE1 cis-elements, being nuclear-localized, its overexpression conferrers hypersensitivity to exogenous abscisic acid condition and abiotic stresses. These findings have suggested that RAP2.6 participates in abiotic stress, possibly through the ABA-dependent pathway.
Conclusion
In summary, this study presents an integrated transcriptome sequencing to identify how Kobresia littledalei mount their responses and initiate acclimation against cold stress. Several significant DEGs and metabolic responses were categorized. By execution of homology analysis of all genes against selected protein databases like Nr, Swiss-Prot, KEGG and COG, we got functional annotations and classifications which provide sufficient resources for future molecular studies. Moreover, information on the KEGG metabolic pathways and transcription factors will facilitate the discovery of other cold-resistant genes. AP2/ERF-ERF gene family could be playing a significant role that enhances the survival of K. littledalei to cold-stress conditions. The dominant antique-white and dark grey associated genes may be produced as a response strategy to the survival of the plant in cold stress conditions, hence there is a need for further studies to ascertain the finding from the present study. Importantly, our findings herein further the general understanding of Kobresia plants adaptation and responses to cold stress through the molecular mechanisms involved in signal regulation and cold resistance.
Author contribution statement
GQ, GB, YL, LW, and MC conceived and supervised the work. WW and YL performed the RNA-seq. SC and QT planted samples and did the treatment. GB, YL, and LW performed the data analysis. GQ, GB, YL, LW, and MC wrote the article with contributions from all other authors.
References
Alscher RG, Erturk N, Heath LS (2002) Role of superoxide dismutases (SODs) in controlling oxidative stress in plants. J Exp Bot 53(372):1331–1341
Andrae JW, McInerney FA, Tibby J, Henderson ACG, Hall PA, Marshall JC et al (2019) Variation in leaf wax n-alkane characteristics with climate in the broad-leaved paperbark (Melaleuca quinquenervia). Org Geochem 130:33–42. https://doi.org/10.1016/j.orggeochem.2019.02.004
Apel K, Hirt H (2004) Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol 55:373–399. https://doi.org/10.1146/annurev.arplant.55.031903.141701
Armengaud P, Thiery L, Buhot N, Grenier-De March G, Savoure A (2004) Transcriptional regulation of proline biosynthesis in Medicago truncatula reveals developmental and environmental specific features. Physiol Plant 120(3):442–450. https://doi.org/10.1111/j.0031-9317.2004.00251.x
Baniwal SK, Bharti K, Chan KY, Fauth M, Ganguli A, Kotak S et al (2004) Heat stress response in plants: a complex game with chaperones and more than twenty heat stress transcription factors. J Biosci 29(4):471–487. https://doi.org/10.1007/bf02712120
Baurle I (2016) Plant heat adaptation: priming in response to heat stress. F1000Res. https://doi.org/10.12688/f1000research.7526.1
Byun MY, Cui LH, Lee J, Park H, Lee A, Kim WT, Lee H (2018) Identification of rice genes associated with enhanced cold tolerance by comparative transcriptome analysis with two transgenic rice plants overexpressing DaCBF4 or DaCBF7, isolated from Antarctic flowering plant Deschampsia Antarctica. Front Plant Sci 9:601. https://doi.org/10.3389/fpls.2018.00601
Cui L, Feng K, Wang M, Wang M, Deng P, Song W, Nie X (2016) Genome-wide identification, phylogeny and expression analysis of AP2/ERF transcription factors family in Brachypodium distachyon. BMC Genomics 17(1):636. https://doi.org/10.1186/s12864-016-2968-8
Fan WJ, Zhang M, Zhang HX, Zhang P (2012) Improved tolerance to various abiotic stresses in transgenic sweet potato (Ipomoea batatas) expressing spinach betaine aldehyde dehydrogenase. PLoS ONE 7(5):e37344. https://doi.org/10.1371/journal.pone.0037344
Fan JB, Chen K, Amombo E, Hu ZR, Chen L, Fu JM (2015) Physiological and molecular mechanism of nitric oxide (NO) involved in bermudagrass response to cold stress. PLoS ONE 10(7):e0132991. https://doi.org/10.1371/journal.pone.0132991
Francioli D, Schulz E, Lentendu G, Wubet T, Buscot F, Reitz T (2016) Mineral vs. Organic amendments: microbial community structure, activity and abundance of agriculturally relevant microbes are driven by long-term fertilization strategies. Front Microbiol 7:1446. https://doi.org/10.3389/fmicb.2016.01446
Franks PJ, Beerling DJ (2009) CO2-forced evolution of plant gas exchange capacity and water-use efficiency over the Phanerozoic. Geobiology 7(2):227–236. https://doi.org/10.1111/j.1472-4669.2009.00193.x
Guy C, Kaplan F, Kopka J, Selbig J, Hincha DK (2008) Metabolomics of temperature stress. Physiol Plant 132(2):220–235. https://doi.org/10.1111/j.1399-3054.2007.00999.x
Heidarvand L, Amiri RM (2010) What happens in plant molecular responses to cold stress? Acta Physiol Plant 32(3):419–431. https://doi.org/10.1007/s11738-009-0451-8
Hodgson RA, Raison JK (1991) Lipid peroxidation and superoxide dismutase activity in relation to photoinhibition induced by chilling in moderate light. Planta 185(2):215–219. https://doi.org/10.1007/BF00194063
Korner C (2016) Plant adaptation to cold climates. F1000Res. https://doi.org/10.12688/f1000research.9107.1
Krasensky J, Jonak C (2012) Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J Exp Bot 63(4):1593–1608. https://doi.org/10.1093/jxb/err460
Landi M (2017) Commentary to: “improving the thiobarbituric acid-reactive-substances assay for estimating lipid peroxidation in plant tissues containing anthocyanin and other interfering compounds” by Hodges et al. Planta (1999) 207:604–611. Planta 245(6):1067–1067. https://doi.org/10.1007/s00425-017-2699-3
Langfelder P, Mischel PS, Horvath S (2013) When is hub gene selection better than standard meta-analysis? PLoS ONE 8(4):e61505. https://doi.org/10.1371/journal.pone.0061505
Lau JA, Lennon JT (2012) Rapid responses of soil microorganisms improve plant fitness in novel environments. Proc Natl Acad Sci U S A 109(35):14058–14062. https://doi.org/10.1073/pnas.1202319109
Li J, Zhou D, Qiu W, Shi Y, Yang JJ, Chen S et al (2018) Application of weighted gene co-expression network analysis for data from paired design. Sci Rep 8(1):622. https://doi.org/10.1038/s41598-017-18705-z
Liu WY, Wang MM, Huang J, Tang HJ, Lan HX, Zhang HS (2009) The OsDHODH1 gene is involved in salt and drought tolerance in rice. J Integr Plant Biol 51(9):825–833. https://doi.org/10.1111/j.1744-7909.2009.00853.x
Liu W, Yu K, He T, Li F, Zhang D, Liu J (2013) The low temperature induced physiological responses of Avena nuda L., a cold-tolerant plant species. Sci World J 2013:658793. https://doi.org/10.1155/2013/658793
Miller G, Suzuki N, Ciftci-Yilmaz S, Mittler R (2010) Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell Environ 33(4):453–467. https://doi.org/10.1111/j.1365-3040.2009.02041.x
Mittler R (2002) Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci 7(9):405–410. https://doi.org/10.1016/s1360-1385(02)02312-9
Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K (2012) AP2/ERF family transcription factors in plant abiotic stress responses. Biochim Biophys Acta 1819(2):86–96. https://doi.org/10.1016/j.bbagrm.2011.08.004
Piao S, Ciais P, Huang Y, Shen Z, Peng S, Li J et al (2010) The impacts of climate change on water resources and agriculture in China. Nature 467(7311):43–51. https://doi.org/10.1038/nature09364
Provart NJ, Gil P, Chen W, Han B, Chang HS, Wang X, Zhu T (2003) Gene expression phenotypes of Arabidopsis associated with sensitivity to low temperatures. Plant Physiol 132(2):893–906. https://doi.org/10.1104/pp.103.021261
Renaut J, Hausman JF, Bassett C, Artlip T, Cauchie HM, Witters E, Wisniewski M (2008) Quantitative proteomic analysis of short photoperiod and low-temperature responses in bark tissues of peach (Prunus persica L. Batsch). Tree Genet Genomes 4(4):589–600. https://doi.org/10.1007/s11295-008-0134-4
Shefferson RP, Salguero-Gomez R (2015) Eco-evolutionary dynamics in plants: interactive processes at overlapping time-scales and their implications. J Ecol 103(4):789–797. https://doi.org/10.1111/1365-2745.12432
Shiah SG, Hsiao JR, Chang WM, Chen YW, Jin YT, Wong TY et al (2014) Downregulated miR329 and miR410 promote the proliferation and invasion of oral squamous cell carcinoma by targeting Wnt-7b. Cancer Res 74(24):7560–7572. https://doi.org/10.1158/0008-5472.CAN-14-0978
Singh K, Foley RC, Onate-Sanchez L (2002) Transcription factors in plant defense and stress responses. Curr Opin Plant Biol 5(5):430–436. https://doi.org/10.1016/s1369-5266(02)00289-3
Szekely G, Abraham E, Cseplo A, Rigo G, Zsigmond L, Csiszar J et al (2008) Duplicated P5CS genes of Arabidopsis play distinct roles in stress regulation and developmental control of proline biosynthesis. Plant J 53(1):11–28. https://doi.org/10.1111/j.1365-313X.2007.03318.x
Tang W, Kim TW, Oses-Prieto JA, Sun Y, Deng Z, Zhu S et al (2008) BSKs mediate signal transduction from the receptor kinase BRI1 in Arabidopsis. Science 321(5888):557–560. https://doi.org/10.1126/science.1156973
Verbruggen N, Hermans C (2008) Proline accumulation in plants: a review. Amino Acids 35(4):753–759. https://doi.org/10.1007/s00726-008-0061-6
Wang CT, Cao GM, Wang QL, Jing ZC, Ding LM, Long RJ (2008) Changes in plant biomass and species composition of alpine Kobresia meadows along altitudinal gradient on the Qinghai-Tibetan Plateau. Sci China Series C-Life Sci 51(1):86–94. https://doi.org/10.1007/s11427-008-0011-2
Xin Z, Browse J (1998) Eskimo1 mutants of Arabidopsis are constitutively freezing-tolerant. Proc Natl Acad Sci U S A 95(13):7799–7804. https://doi.org/10.1073/pnas.95.13.7799
Xiong AS, Jiang HH, Zhuang J, Peng RH, Jin XF, Zhu B et al (2013) Expression and function of a modified AP2/ERF transcription factor from Brassica napus enhances cold tolerance in transgenic Arabidopsis. Mol Biotechnol 53(2):198–206. https://doi.org/10.1007/s12033-012-9515-x
Yoon DH, Lee SS, Park HJ, Lyu JI, Chong WS, Liu JR et al (2016) Overexpression of OsCYP19-4 increases tolerance to cold stress and enhances grain yield in rice (Oryza sativa). J Exp Bot 67(1):69–82. https://doi.org/10.1093/jxb/erv421
Zheng Y, Jiao C, Sun H, Rosli HG, Pombo MA, Zhang P et al (2016) iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol Plant 9(12):1667–1670. https://doi.org/10.1016/j.molp.2016.09.014
Acknowledgements
This work was made possible through Investigation on the protection of germplasm resources and the exploition of genetic technology of kobresia myosuroides (Science and Technology Department of Tibet) (grant No. XZ202001ZY0016N).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Huang.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Qu, G., Baima, G., Liu, Y. et al. Adaptation and response of Kobresia littledalei to cold stress conditions. Acta Physiol Plant 43, 92 (2021). https://doi.org/10.1007/s11738-021-03246-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-021-03246-w