
Abstract
The traditional Chinese desert shrub Lycium ruthenicum is widely distributed in arid environments such as north-west China, exhibiting ideal salt tolerance to cope with soil desertification, salinity, and alkalinity. However, the salt-tolerance mechanism of L. ruthenicum, especially of its young seedlings at early vegetative stages, remains largely unknown. In the present study, we collected whole-seedling samples from Lycium ruthenicum at a-pair-leaf stage with and without a mild salt (75 mM sodium chloride) treatment, and then performed transcriptome profiling to compare their gene expression patterns. The de novo assembly achieved 94,651 unigenes with 55,156 annotated. Among them, 199 differentially expressed genes (DEGs) were identified between salt-treated seedlings and control, with 41 up-regulated and 158 down-regulated. These DEGs were highly enriched into gene ontology (GO) classifications ‘metabolic process’ and ‘catalytic activity’, into Clusters of Orthologous Groups (COG) function classifications ‘translation, ribosomal structure and biogenesis’ and ‘energy production and conversion’, and into Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways ‘ribosome’ and ‘oxidative phosphorylation’. Specifically, genes involved in energy metabolism (oxidative phosphorylation) and related energy-consuming metabolisms, including ribosome-associated biogenesis and biosynthesis of organic nitrogen-derived compatible solutes (i.e., arginine and proline), were generally down-regulated. Specific genes involved in abscisic acid (ABA) biosynthesis and signaling pathway were simultaneously up-regulated. Changes in the transcript levels of salt-responsive DEGs selected from the transcriptomic profiling were further validated by real-time quantitative polymerase chain reaction (RT-qPCR) analysis. Based on these results, salinity-adaptive strategies for the L. ruthenicum early seedlings are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The plant Lycium ruthenicum, belonging to the genus Lycium of the Solanaceae family, is mainly distributed in arid environments including northwestern China (e.g., Xinjiang, Qinghai, and Ningxia provinces) and Central Asia. Due to its outstanding traits of salt-resistance, such halophytic shrub has been exploited as biological resource to prevent soil desertification and alleviate soil salinity, thus improving agriculture ecosystem in desert areas (Chen et al. 2008; Dong et al. 2008). As classical salt-tolerant desert species, in every stage of their life cycle, L. ruthenicum would have developed suitable adaptations to osmotic and ionic challenges of saline environments. The soil salinity concentration concerning L. ruthenicum cultivation at northwestern China has been documented approximately 75 mM in average (Fan et al. 2001; Wang et al. 2013). There have existed numerous reports on mild salt-tolerance mechanisms for many plants including Arabidopsis (Jiang et al. 2016; Othman et al. 2017), tomato (Gharbi et al. 2016; Kissoudis et al. 2016), rice (Nam et al. 2015), and forest pine at white spruce seedling stage (Duan and Chang 2017). However, up to date, we still lack knowledge on how L. ruthenicum seedlings deal with such mild salt-stress conditions, especially at their very early vegetative stage.
Adaptation of plants to salinity stress has been evolved to integrate complex molecular processes at multiple levels (Gerszberg and Hnatuszko-Konka 2017). Salt-tolerant plants have developed a series of physiological responses that enable them to overcome salinity damages, including osmotic regulation, oxidative stress protection, ion toxin exclusion, ion homeostasis maintenance, and metabolic rearrangements via phytohormone signaling(s). Activation of proline biosynthesis is known as a common strategy to achieve plant osmotic adjustments under salt stress, and for this purpose, glutamine synthetase (GS)/glutamate dehydrogenase (GDH) shunts are required to supply excess precursor glutamate (Wang et al. 2007; Singh et al. 2016; Annunziata et al. 2017). Reactive oxygen species (ROS) scavengers, including antioxidant enzymes (e.g., catalase, ascorbate peroxidase and superoxide dismutase) and antioxidant metabolites (e.g., ascorbate and glutathione), constitute a major defense system to protect salt-treated plants against oxidative damages (Duan et al. 2012; Baranova et al. 2014). Transcriptomic modulation of genes encoding ion (e.g., Na+ and K+) transporters also contributes to plant salt-tolerance, as reported in Arabidopsis thaliana, tomato, and crops (Gerszberg and Hnatuszko-Konka 2017). Phytohormone abscisic acid (ABA) has been reported to alleviate salt stress for many plant species. For ABA biosynthesis from carotenoids, 9-cis-epoxycarotenoid dioxygenase (NCED) serves as a rate-limiting enzyme, and its transcript (NCED1) abundance usually correlates well with ABA content in salt-treated plants (Horváth et al. 2015; Tian et al. 2015). On the other hand, ABA-responsive genes involved in the phytohormone signaling often interact with various transcription factors, including A20/AN1 zinc-finger proteins (Kang et al. 2017; Dixit et al. 2018), ring-H2 zinc-finger proteins (Chandrasekaran et al. 2014; Brugière et al. 2017), NAC transcription factors (de Oliveira et al. 2011; Xue et al. 2011), homeobox–leucine zipper proteins (Olsson et al. 2004; Lechner et al. 2011; Bhattacharjee et al. 2017), and BTB/POZ/TAZ finger proteins (Lechner et al. 2011), forming ABA-centered regulatory network which in turn affect target protein activities, genetics processes, and primary and energy metabolisms. ABA signal transduction also comprises components of protein kinases and protein phosphatases including mitogen-activated protein kinases (MAPKs) and protein phosphatase 2C (PP2C) (Zhu 2016; Lou et al. 2017).
The high accuracy and sensitivity of RNA-seq makes it one of the most suitable techniques to profile salt stress-responsive gene expression, providing digital gene expression patterns to uncover novel stress-associated strategies. In recent years, RNA-seq has been applied to explore the transcriptomes of L. ruthenicum plants under UV-B stress (Chen et al. 2015), fruit-ripening condition (Peng et al. 2017), and saline-alkaline mixed stress (Chen et al. 2017). So far, the only available transcriptomic analysis of L. ruthenicum plants under salinity stress was conducted using salt-alkali mixed soil (pH 9.0) as media (Chen et al. 2017). There was no available RNA-seq data set on physiological responses of young L. ruthenicum seedlings to mild-salt stress. Transcriptome analysis of young seedlings of sugarcane (Bottino et al. 2013), bread wheat (Takahashi et al. 2015), and tomato (Gerszberg and Hnatuszko-Konka 2017) under mild-salt stress indicates flexible strategies with diverse gene expression profiles. In this study, we performed transcriptomic analysis to explore salt-responsive gene expression profiles for mild-salt-treated L. ruthenicum seedlings at an early vegetative stage.
Materials and methods
Plant materials and salt treatment
Seeds of L. ruthenicum were collected in Xinjiang province, P. R. China. Intact plump seeds were immersed in 10% (w/v) sodium hypochlorite for 15 min, and then rinsed three times with sterilized double-distilled water. Seeds were germinated in a 150 mL conical flask containing 40 mL Murashige and Skoog (MS) medium in dark for 72 h. Germinated seeds were cultivated on the MS medium for 15 days in a growth chamber with a 16 h/8 h light/dark cycle at 25ºC, 200 µmol m−2 s−1 light intensity, and 50% relative humidity. Seedlings were transferred into plastic tanks with 1/2 strength Hoagland solution, further cultivated for 20 days until the first pair of leaves emerged, and finally transferred to the full-strength Hoagland solution to prepare 2-leaf seedlings (the seedlings with a pair of fully expanded leaves). During seedling cultivation, Hoagland solutions (1/2 or full strength) were refreshed every 4 days. Seedlings at 2-leaf stage were divided into two groups: the salt-treated group with three biological repeats (T1–3) was placed into tanks with 3 L full-strength Hoagland solution containing 75 mM NaCl, and the control group with three biological repeats (CK1–3) was placed into tanks with 3 L NaCl-free full-strength Hoagland solution. We applied 75 mM NaCl in the salt treatment to mimic the average salt concentration determined in wild L. ruthenicum habitat environment (i.e., Xinjiang desert area). Whole seedlings were harvested after 48 h NaCl treatment, quickly frozen in liquid nitrogen, and stored at – 80 ℃till RNA extraction.
RNA extraction, cDNA library construction, and Illumina sequencing
Total RNA of each sample was extracted using Trizol reagent (Invitrogen, USA) according to the manufacturer's protocol. The quality and quantity of RNA samples were examined using Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA) and the integrity was assessed by 1% agarose gel electrophoresis. cDNA libraries for RNA sequencing were constructed according to the protocol of NEBNext Ultra™ RNA Library Prep Kit for Illumina (NEB, USA), using 3 μg total RNA for each sample. Briefly, this involved mRNA purification, mRNA fragmentation, first- and second-strand cDNA synthesis, cDNA 3′-end repairing and adenylation, NEBNext adaptor (sequencing adaptor) ligation, cDNA fragment purification, USER Enzyme (NEB, USA) treatment, and Universal and Index (X) primer-guided high-fidelity PCR. After that, X-attached cDNA fragments were clustered on a cBot Cluster Generation System for library sequencing on Illumina Hiseq platform, resulting in 125–150 bp paired-end reads (raw reads or raw data).
De novo transcriptome assembly and annotation
Raw reads (raw data) with FASTQ format were processed through in-house perl scripts to collect clean reads (clean data) by removing undesirable reads, i.e., sequence adapter-containing reads, ploy-N-containing reads, and other low-quality reads. Q20, Q30, and GC contents were calculated to evaluate data quality. Clean reads with high data quality were subjected to assemble de novo transcriptome without reference genome, using Trinity method (Grabherr et al. 2011). The clean reads were processed to form contigs, and then, these contigs were clustered to obtain transcript sequences, and finally, the longest transcript sequence in each set of transcript assembly was selected as the unigene for further functional annotation. All these unigenes were functionally annotated by homolog analysis (BLAST E-value ≤ 1e−5 and HMMER E-value ≤ 1e−10) against public databases including Gene Ontology (GO) database (Ye et al. 2006, 2018), Clusters of Orthologous Groups (COG) database (Tatusov et al. 2000), euKaryotic Orthologous Groups (KOG) database, eggNOG database, Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa et al. 2008), Swiss-Prot database, Protein family (Pfm) database, and non-redundant (NR) database.
Analysis of differentially expressed genes (DEGs)
Gene expression levels were estimated by mapping clean reads to the Trinity unigenes assembled by RSEM (Li and Dewey 2011). The abundance of all genes was normalized and calculated using uniquely mapped reads by the FPKM (fragments per kb per million fragments) method (Marioni et al. 2008; Mortazavi et al. 2008). Differential expression analysis was performed using the DESeq R package (1.18.0). DESeq provide statistical routes to determine differential expression for digital gene expression, based on a negative binomial distribution model (Anders and Huber 2010; Wang et al. 2010). The resulting P values were adjusted using the Benjamini and Hochberg’s method to control false discovery rates (Benjamini and Hochberg 1995). The cut-off value to define differentially expressed gene (DEG) was the adjusted P value ≤ 0.05 and at least twofold change (i.e., the absolute value of log2 Fold ≥ 1.0) in transcript abundance between two comparison samples. The identified DEGs were hierarchically clustered by Cluster 3.0 (de Hoon et al. 2004), and then subjected to heat-map analysis by Plotly (Montreal, Quebec) software. The distribution of up- and down-regulated DEGs versus unchanged unigenes was visualized by MA and Volcano plots (Love et al. 2014). GO enrichment of DEGs was implemented by the GOseq R package, and KEGG enrichment of DEGs with statistical significance test was processed in the KOBAS software context to identify DEG(s)-involved metabolic and/or signaling pathways (Mao et al. 2005).
Validation of DEGs with real-time quantitative PCR (RT-qPCR)
The expression profile of salt stress-responsive genes was validated by RT-qPCR analysis, using the same RNA samples that were subjected to the present RNA sequencing. The first-strand cDNA was synthesized using the Prime Script First Strand cDNA synthesis kit (Takara, Dalian, China) according to the manufacturer’s protocol. The primers used in the present study were designed using the Primer Premier 5.0 and are listed in Table S1. RT-qPCR was conducted using SYBR Premix Ex Taq™ II kit (Takara, Dalian, China) and run on ABI7500 Real-Time PCR System (Thermo Fisher Scientific, USA). All PCR reactions were performed with three technical replicates and the thermal conditions were as follows: 60 s at 95 °C, followed by 40 cycles of 15 s at 95 °C, 30 s at 60 °C. Dissociation curve analysis was performed to determine the target specificity. The relative transcript abundance of the selected genes versus reference gene β-actin was calculated according to 2–ΔΔCt method (Livak and Schmittgen 2001), and all data were expressed as relative mRNA expression with mean ± SD (standard deviation of the mean). One-way ANOVA followed by the least significant difference (LSD) test was applied to assess the significance of differences between the means (*P < 0.05 and **P < 0.01).
Determination of enzyme activities and metabolite contents
To determine enzyme activities, 1 g (fresh weight) of intact L. ruthenicum seedlings was quickly frozen by liquid nitrogen and grounded to powders with a mortar before adding 5 mL extraction buffer (100 mM Tris–HCl (pH 7.6), 1.0 mM ethylenediaminetetraacetic acid (EDTA), 1.0 mM MgCl2·6H2O, and 10 mM 2-mercaptoethanol). The extracts were centrifuged at 20,000g for 30 min at 4 ℃, and the resulting supernatants were prepared for measuring enzyme activities. Glutamine synthetase (GS; EC 6.3.1.2) activity was determined according to the method of Rhodes et al. (1975), and one GS activity unit was defined as the amount of enzyme producing 1 µmol γ-glutamylhydroxamate per min at 37 ℃. The NADH-dependent amination activity of glutamate dehydrogenase (GDH; EC 1.4.1.2) was determined according to the method of Loulakakis and Roubelakis-Angeiakis (1996), and one NADH-GDH activity unit was defined as the oxidation of 1 µmol of NADH per min at 30ºC. Free proline was extracted from 1 g fresh seedlings in 10 mL 3% (v/v) sulphosalicylic acid at boiling ddH2O for 10 min, then the extract was properly diluted and subjected to content determination using ninhydrin reagent as described before (Wang et al. 2007). ABA was extracted by 80% (v/v) methanol solution from 1 g fresh seedlings, and the ABA content was determined using an enzyme-linked immunosorbent assay (ELISA) (Sun et al. 2012). All the experiments were performed with five biological replicates.
Determination of photosynthesis-related parameters
The determination of chlorophyll a (chl a) and chlorophyll b (chl b) was performed as described by Zhao and Harris (1992). These chlorophylls were extracted from 1.0 g fresh leaves using 80% acetone (v/v), and after filtered through filter paper, the extracts were applied to measure absorbance at 663 and 645 nm wavelengths using a spectrophotometer (TU-1900, Beijing, China). The chl a and chl b concentrations were calculated according to the method of Inskeep and Bloom (1985), and the total chlorophyll content was defined as the sum of chl a and chl b contents. Net photosynthesis rate (Pn) and net photorespiration rate (Pr) were measured on the pair of fully expanded leaves from six seedlings per treatment (n = 6) at a.m. 9:00–11:00 (i.e., 48 h after the defined NaCl treatment) using an open-flow infrared gas-exchange analyzer (Li-Cor 6400, Li-Cor, Lincoln, NE, USA) under 50% relative humidity and 200 µmol m−2 s−1 photosynthetic photo-flux density (PPFD), and the other conditions (e.g., air temperature, air O2 concentration, and ambient CO2 concentration) during the measurements were all according to the method of Ahammed et al (2018).
Results
Illumina sequencing and de novo transcriptome assembly
We constructed six Illumina libraries, three libraries from 75 mM NaCl-treated samples (T1–3), and the other three libraries from control samples (CK1–3) to reveal transcriptomic profile in the mild NaCl-treated L. ruthenicum early seedlings. The six libraries of CK1, CK2, CK3, T1, T2, and T3 produced 6.84 Gb, 8.56 Gb, 9.05 Gb, 12.60 Gb, 9.97 Gb, and 15.11 Gb of raw data, respectively, and the sequencing quality was validated by Q30 and GC percentages (Table 1). Clean reads from the six libraries were assembled into 7,891,800 contigs in total with N50 length (the shortest sequence length at 50% of the genome) of 116 bp and mean length of 72.65 bp; these contigs were further assembled into transcripts and unigenes, finally providing non-redundant 94,651 unigenes with mean length of 971.90 bp and N50 length of 1467 bp (Table S2). All the unigenes were longer than 200 bp, with 68.80% between 200 and 1000 bp, 21.68% between 1000 and 2000 bp, and 9.52% over 1000 bp (Fig. S1).
Functional annotation and differential expression analysis
Unigenes were annotated by alignment with the currently available public databases. Totally, 55,156 unigenes (58.27%) were functionally annotated in at least one database (Table 2), including 28,248 (29.84%) in GO database, 20,068 (21.20%) in COG database, 32,117 (33.93%) in KOG database, 49,756 (52.57%) in eggNOG database, 21,859 (23.09%) in KEGG database, 30,769 (32.51%) in Swiss-Prot database, 39,223 (41.44%) in Pfam database, and 51,887 (54.82%) in NR database. Specifically, GO annotation classified 28,248 unigenes into three functional categories (Table S3): 22,127 into biological process (BP), 15,283 into cellular component (CC), and 23,536 into molecular function (MF). To identify DEGs in response to salt stress, clean reads obtained from NaCl-treated (T1, T2 and T3) and control (CK1, CK2, and CK3) RNA-seq libraries were mapped to transcriptomic background of all the non-redundant unigenes (Table S4). The reads mapped to non-redundant unigenes were used to calculate FPKM values to assess unigene transcription levels. We totally identified 199 DEGs between salt-treated and control samples (Fig. S2), according to the criteria (adjusted P value ≤ 0.05 and the absolute value of log2 fold change ≥ 1.0).
Functional classification of DEGs and identification of salt-responsive genes
The DEGs identified were subjected to functional classification based on hierarchical cluster (i.e., heat-map) analysis (Fig. 1) and then verified by GO enrichments (Fig. S3). The heat-map analysis suggested six function clusters comprised of almost all DEGs, including down-regulation of genes involved in oxidative phosphorylation, translation, ribosomal structure and biogenesis, and primary carbon and nitrogen metabolisms (Fig. 1a–c) and up-regulation of genes involved in chloroplast biogenesis (i.e., translation and special ribosomal structure in chloroplast) and ABA biosynthesis and the related signaling pathways (Fig. 1d–f).

Expression changes and hierarchical cluster analysis of differentially expressed genes (DEGs) between control (CK1–3) and salt-treated (T1–3) seedlings. a–c Down-regulated DEGs mainly involved in oxidative phosphorylation, translation, ribosomal structure and biogenesis, and primary carbon and nitrogen metabolisms, and d–f up-regulated DEGs involved in chloroplast biogenesis (i.e., translation and special ribosomal structure in chloroplast) and ABA biosynthesis and the related signaling pathways. Cluster analysis of genes was performed using hierarchical clustering with average linkage measurement. Rows represent DEGs, and columns represent control (CK1–3) and salt-treated (T1–3) seedling-samples. NA indicated the gene with no annotated function. Color saturation maker at the right top of each panel reflects the changing fold of the FPKM abundance for each gene
The top ten GO terms in BP category are summarized in Table 3. In this category, the up-regulated DEGs responsive to mild-salt treatment, including NAC domain-containing protein (c148166), protein phosphatase 2C (PP2C) (c143401), and homeobox-leucine zipper protein ATHB-12-like (c138687), were enriched into GO terms functionally associated with plant salinity adaptation, including ‘response to water deprivation’ (GO: 0009414), ‘response to abscisic acid’ (GO: 0009737), ‘abscisic acid-activated signaling pathway’ (GO: 0009738), and ‘hyperosmotic salinity response’ (GO: 0042538); on the other hand, the down-regulated DEGs, i.e., a number of 60S (large) and 40S (small) ribosomal protein subunit-encoding genes, were enriched into GO terms ‘response to oxidative stress’ (GO: 0006979), ‘translation’ (GO: 0006412), and ‘response to salt stress’ (GO: 0009651). The top ten GO terms in CC category were comprised of, without exception, down-regulated DEGs (Table 4), including (1) elongation factor 1-alpha (c130295), enriched in ‘plasma membrane’ (GO: 0005886), ‘chloroplast’ (GO: 0009507), and ‘vacuole’ (GO: 0005773), (2) DSBA-like thioredoxin (c135089), enriched in ‘cytoplasmic part’ (GO: 0044444) and ‘intracellular membrane-bounded organelle’ (GO: 0043231), and (3) 60S and 40S ribosomal protein subunit-encoding genes, predominantly enriched in ‘ribosome’ (GO: 0005840), ‘small ribosomal subunit’ (GO: 0015935), ‘cytosolic ribosome’ (GO: 0022626), and ‘plasma membrane’ (GO: 0005886). For MF category (Table 5), a considerable number of DEGs enriched, as well as their salt-responsive patterns, were the same as those enriched in BP and/or CC categories, including (1) protein phosphatase 2C (PP2C; c143401), enriched in ‘protein serine/threonine phosphatase activity’ (GO: 0004722), (2) elongation factor 1-alpha (c130295), enriched in ‘GTP binding’ (GO: 0005525) and ‘translation elongation factor activity’ (GO: 0003746), and (3) ribosomal protein subunit-encoding genes, mainly enriched in ‘structural constituent of ribosome’ (GO: 0003735). Noticeably, a certain number of DEGs involved in primary metabolisms (e.g., carbon, nitrogen, and energy metabolisms), mostly down-regulated after salt treatment, were enriched within MF category (Table 5), including (1) GAPDH (c145199), enriched in ‘NAD(P) binding’ (GO: 0050661) and ‘NAD-dependent GAPDH activity’ (GO: 0004365), (2) NAD(H)-specific GDH (c144184), enriched in ‘NAD(P)-dependent 2-alkenal reductase activity’ (GO: 0032440) and ‘ATP binding’ (GO: 0005524), (3) mitochondrial ATP synthase subunit alpha (c138547) and beta (c143462), both enriched in ‘ATP binding’ (GO: 0005524), and (4) alkaline and neutral invertase (c151101) (only up-regulated!), enriched in ‘glycopeptide alpha-N-acetylgalactosaminidase activity’ (GO: 0033926). In addition, salt-responsive DEGs were classified into different categories as defined in Clusters of Orthologous Groups of proteins (COG) database and Eukaryotic Orthologous Groups of proteins (KOG) database (Fig. S4, Table S5, Table S6).
Intensively, the present study applied KEGG analysis to enrich salt-responsive DEGs into specific metabolic pathways (Fig. S5; Table S7). According to FDR values, the top five KEGG pathways are listed in Table 6, including ‘oxidative phosphorylation’ (ko02262), ‘purine and pyrimidine metabolism’ (ko00940), ‘glycolysis and carbon metabolism’ (ko01623), ‘arginine and proline metabolism’ (ko01581), and ‘RNA transport and degradation’ (ko12614). All the five KEGG pathways were comprised of down-regulated DEGs, including energy metabolism-related genes such as ATPase subunit-encoding gene (c138547), purine/pyrimidine metabolism-related gene such as nucleoside-diphosphate kinase (NDPK)-encoding gene (c136077), and carbon/nitrogen metabolisms-related genes such as enolase-encoding gene (c147375), glutamine synthetase (GS)-encoding gene (c143766) and ornithine decarboxylase (ODC)-encoding gene (c145406). On the other hand, Table 6 also indicates that the only three KEGG pathways were associated with up-regulated DEGs including NCED-encoding gene (c116077) in ‘carotenoid biosynthesis’ pathway (ko09840) and PP2C-encoding gene (c143401) in ‘plant hormone signal transduction’ pathway (ko14497).
Validation of salt-responsive genes by RT-qPCR and physiological assays
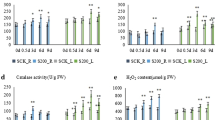
Based on GO, COG/KOG, and KEGG enrichments, we have identified salt-responsive genes as potential contributors to the salt-adaptation of L. ruthenicum seedlings. Most of them were down-regulated after salt treatment, including energy metabolism-related genes (e.g., ATPase subunit-encoding gene), ribosomal structure and biogenesis (translation)-related genes (e.g., 60S and 40S ribosomal protein subunit-encoding genes and RNA helicase-encoding gene), carbon/nitrogen metabolism-related genes (e.g., enolase-encoding gene, GAPDH-encoding gene, and GS-, GDH- and ODC-encoding genes with special functions in proline biosynthesis), and purine/pyrimidine metabolism-related gene (e.g., NDPK-encoding gene). Meanwhile, a certain number of them were up-regulated, including NCED-encoding gene with a critical role in biosynthesis of carotenoid (precursor for ABA biosynthesis), PP2C-encoding gene involved in plant hormone (e.g., ABA) signal transduction, and chloroplast biogenesis-related genes (e.g., RPL-, dnaJ 11- and dnaJ 8-encoding genes in the chloroplast) presumably involved in maintaining photosynthesis efficiency. The expression profiles of all these salt-responsive genes, selected from our RNA-seq data set, have been RT-qPCR validated (Fig. 2). Meanwhile, in the particular physiological assays for the salt-treated seedlings, we observed (1) sharp decrease in GS and NAD(H)-GDH activities with no obvious change in proline content (Fig. 3a, b), confirming the down-regulation of GS- and GDH-encoding genes, (2) a significant increase in ABA content (Fig. 3c), confirming the up-regulation of NCED-encoding gene, and (3) no obvious change in photosynthesis-related parameters including chlorophyll contents, photosynthesis efficiency, and photorespiration rate (Table 7), indicating potential contribution of these chloroplast biogenesis-related genes to the L. ruthenicum seedling photosynthesis.

qRT-PCR validation of 18 salt-responsive DEGs. The DEGs subjected to qRT-PCR validation were identified in the L. ruthenicum seedling response to mild NaCl treatment by KOG and KEGG enrichments. Values are means ± SD (n = 3). Transcript levels were normalized to β-actin, and the signals ‘**’ denote difference between control and NaCl-treated samples at p < 0.01

Changes in enzyme GS and NADH-GDH activities (a) and proline (b) and ABA (c) contents after salt treatment. Enzyme proteins and the related metabolites were extracted from 1 g fresh weight of early seedlings. Enzymatic activity unit was defined as µmol·min−1·mg−1 protein for GS and NADH-GDH. Values are means ± SD (n = 5), and signals ‘*’ and ‘**’ denote difference between control and NaCl-treated samples at p < 0.05 and p < 0.01, respectively
Discussion
Plants have developed efficient mechanisms to overcome abiotic stresses. A number of salinity- or salt-induced responses that confer plants such stress tolerance have been intensively studied in many model plants (e.g., Arabidopsis, tobacco, rice, and tomato), as previously reviewed (Julkowska and Testerink 2015; Zhu 2016; Gerszberg and Hnatuszko-Konka 2017), including energy-status alterations, nutrient-metabolism modulations, inter/intracellular ion-reorganizations, compatible osmoregulations, antioxidant system activations, and, particularly, a series of phytohormone-induced signaling processes. Nevertheless, to date, it is still largely unclear if and how such anti-stress strategies contribute to developing stress tolerance for non-model plants, e.g., L. ruthenicum, which are naturally exposed to multiple abiotic stress conditions, including salinity. Recently, RNA-seq analysis revealed some interesting stress-adaptive mechanisms for the early seedlings of Haloxylon ammodendron (Fan et al. 2018), Desmostachya bipinnata (Asrar et al. 2018), Sporobolus virginicus (Tada et al. 2019) and garlic (Wang et al. 2019). These include inhibition of respiratory photosynthetic electron transport chain to reduce ROS reactions, down-regulation of general energy metabolism, and PP2C-centered up-regulation of salt-responsive genes through ABA-dependent pathways. The present study exploited L. ruthenicum, in which the response to mild-salt stress was activated during seed germination (Han et al. 2014), as plant materials to investigate salt-tolerance mechanisms at its early seedling stage when the halophytic plant underwent autotrophy-to-heterotrophy transitions during post-germination growth.
According to the previous reports (Möller 2001; Golldack et al. 2014; Szypulska et al. 2017), plants often decreased their energy metabolisms to reduce ROS production under NaCl stress. Our RNA-seq data sets also revealed salt-induced down-regulation of energy-metabolism-related unigenes by hierarchical clustering (Fig. 1a, b), GO enrichment (Table 5), COG enrichment (Table S5), KOG enrichment (Table S6), and KEGG enrichment (Table 6), including mitochondrial ATP synthase subunit-encoding genes, cytochrome b/c oxidase (terminal enzymes in mitochondrial respiratory chain) subunit-encoding genes, NADH dehydrogenase-encoding gene, and NAD/FAD oxidoreductase-encoding genes. Similar evidence has been reported in salinity-induced proteomic changes for barley sprouts (Szypulska et al. 2017) and for early seedling growth of extremophyte Haloxylon ammodendron under drought (Fan et al. 2018). Modulation of energy metabolisms has been regarded as an effective solution to help plants adapt stress environments. Besides down-regulation of energy metabolisms, up-regulation of energy metabolisms was also considered as plant anti-stress responses, exclusively along with activated flux of carbon catabolism including TCA cycle, as recently reported in D. bipinnata under moderate salinity (Asrar et al. 2018), grape plant under physiologically damaging salinity (Das and Majumder 2019), and winter wheat seeds germinated under salt stress (Liu et al. 2019). In contrast to the latest reports, here, we observed a down-regulation of energy metabolism to help L. ruthenicum early seedlings adapt a mild-salt environment, which was further supported by a consensus down-regulation of carbon-catabolism-related genes encoding fructose-bisphosphate aldolase, GAPDH, and enolase (Fig. 1a, b, Tables 5, 6, Tables S5, S6). The lower flux of carbon catabolism would favor to limit ROS production, especially without up-regulation of any ROS-scavenging genes, as the case in our RNA-seq data sets. On the other hand, some energy-consuming biochemical processes, e.g., biosynthesis of compatible solute proline, were observed to be limited in the salt-treated L. ruthenicum seedlings, as indicated by simultaneous down-regulation of genes encoding NAD(H)-specific GDH, GS, and ODC (Fig. 1a, b). Over-accumulation of proline through GS-GDH pathway has been accepted as a common strategy for plants to tolerate salinity stress (Singh et al. 2016; Annunziata et al. 2017; Gerszberg and Hnatuszko-Konka 2017). However, such increase in proline and related enzyme activities did not occur in the present study (Fig. 3a, b). Other compatible solutes, except for sugars, were also not found to be over-accumulated for the L. ruthenicum early seedlings after salt treatment. Interestingly, according to our unpublished data, we observed a significant increase in sugar contents in the salt-treated L. ruthenicum early seedlings (especially in their leaves), which could result from sustained (almost unchanged) photosynthetic capacity (Table 7) and the reduced carbon catabolism (as discussed above). Therefore, the present study suggests a new anti-salinity strategy that L. ruthenicum early seedlings preferentially adopt sugars (somewhat energy-saving form of compatible solutes during post-germination stage), instead of the other energy-consuming form of compatible solutes including proline, to achieve necessary osmoregulation under energy-limited conditions that would be specially required to avoid ROS damages.
Accordingly, such energy-limited status also led to inhibition of biogenesis processes including de novo protein biosynthesis, as indicated in our transcriptome enrichments, e.g., a general down-regulation of ribosomal protein subunit-encoding genes (Fig. 1a–c; Tables 3, 4, 5). Ribosomal proteins have been identified as potential contributors to facilitating plants tolerance against various abiotic stresses (Ludwig and Tenhaken 2001; Mukhopadhyay et al. 2011; Cheng et al. 2015; Moin et al. 2016; Zhang et al. 2016; Bai et al. 2017). Suppression of ribosomal L2 gene was the early evidence regarding a novel mechanism for plants (soybean) to cope with oxidative stress (e.g., H2O2 treatment) (Ludwig and Tenhaken 2001). Studies in recent years confirmed this stress-responsive role for more ribosomal proteins, including 50S ribosomal proteins L1 and L12 in drought-treated wheat cultivars (Cheng et al. 2015), ribosomal protein S12 in drought-treated cotton seedlings (Zhang et al. 2016), and 50S ribosomal protein L10 in oat leaves at long-term salinity stress treatment (Bai et al. 2017). Nevertheless, the previous reports based on high-throughput transcriptome and/or proteome sequencing only associated a single or several ribosomal protein(s) with stress responses. Unlike those occasional responses, the present study provided the first evidence on the salt-induced repression of transcript abundances for a large number of ribosomal proteins. The significantly lower levels of ribosomal proteins in general, as a result of the mild-salt treatment, well verified an energy-saving strategy for L. ruthenicum post-germination seedlings to adapt various salt-induced stresses including oxidative stress. In contrast, such general repression of biogenesis did not occur in chloroplast, as indicated by up-regulation of 30S ribosomal protein S4 (chloroplastic)-encoding gene (Fig. 1f; Table S6). This line of evidence with the other two results, i.e., (1) up-regulation of chloroplastic chaperone proteins dnaJ 8 and dnaJ 11 (Fig. 1d, e; Table S5) and (2) photosynthetic capacity maintenance (Table 7), all indicated the functional importance of chloroplast (photosynthetic organ) to support salt-tolerance of L. ruthenicum early seedlings. Thus, we proposed a chloroplast-priority strategy for the L. ruthenicum seedlings to cope with salinity conditions.
Plant hormones including abscisic acid (ABA) and their initiated signaling processes constitute crucial defense system against abiotic stresses including salinity. ABA has been accepted as a sensitive indicator (also as a signal molecule) to induce plant salinity tolerance, and the level of ABA usually increased quickly upon salt treatments (Julkowska and Testerink 2015; de Zelicourt et al. 2016; Zhu 2016; Gerszberg and Hnatuszko-Konka 2017; Silva et al. 2018; Tian et al. 2018). The present transcriptomic evidence suggested a significant up-regulation of NCED (Fig. 1f; Table 6, Tables S5–7), and NCED encodes a special epithelial membrane enzyme that is rate-limiting to control ABA biosynthesis (Horváth et al. 2015; Tian et al. 2015; Sales et al. 2017). The increase in transcript abundance of NCED (Fig. 2p) was correlated with the elevation of endogenous ABA content (Fig. 3c), thus ensuring the essential role of ABA in the salt tolerance of L. ruthenicum early seedlings. Furthermore, through enrichment analysis, we found a wide array of DEGs simultaneously which up-regulated after the present salt treatment, encoding transcription factors as well as some particular signal-transducer proteins (Fig. 1d–f, Tables 3, 5, 6, Tables S5–7), including NAC (NAM, ATAF1, ATAF2, and CUC2) domain-containing proteins, homeobox-leucine zipper proteins, TAZ zinc finger (BTB/POZ domain), ring-H2 zinc finger, AN1/A20-like zinc finger, protein phosphatase 2C(PP2C), and mitogen-activated protein kinase kinase kinase (MAPKKK). More interestingly, all these salt-responsive genes identified here were exclusively associated with the ABA signaling pathway. For plants to cope with salinity stress, NAC transcription factors have been demonstrated as ABA-mobilized trans-elements to function in ROS regulation (de Oliveira et al. 2011; Xue et al. 2011); homeobox-leucine zippers with multiple isoforms have been verified to participate in ABA signaling-induced sodium exclusion (Shin et al. 2004; Lechner et al. 2011; Wang et al. 2015; Bhattacharjee et al. 2017). Zinc fingers including TAZ-type, ring-H2-type, and AN1/A20-type contributed extensively to ABA signaling-mediated redox balance and ion homeostasis (Kim et al. 2004; Lechner et al. 2011; Chandrasekaran et al. 2014; Brugière et al. 2017; Kang et al. 2017; Dixit et al. 2018). PP2C is a key helper in PYL–ABA–PP2C complex to sense ABA signal (Zhu 2016; Lou et al. 2017). MAP kinases including MAPKKK, previously identified as components in jasmonate (JA) signaling, were recently elucidated as essential modules’ functioning downstream of ABA signaling (de Zelicourt et al. 2016; Zhu 2016; Choi et al. 2017; Abid et al. 2018; Matsuoka et al. 2018). Thus, we proposed a strategy of ABA signaling-centered regulation to allow L. ruthenicum early seedlings endure salinity environment. Muchate et al. (2016) have assumed ABA as the most important regulator to induce plant salt tolerance, as compared with the other phytohormones [e.g., auxin, gibberellic acid (GA), JA, etc.], due to its ability to be rapidly accumulated upon salinity stress especially at early seedling stage. The present study did not find involvement of any other phytohormone. These results might support the assumption raised by Muchate et al. (2016). However, the physiological significance for such ABA signaling-centered regulation remained unclear and would be an interesting topic to further study.
In summary, we reported strategies of L. ruthenicum early seedlings to adapt salt stress based on transcriptome profile of salt-responsive genes. The strategies were summarized as (1) decreasing energy metabolism to reduce ROS reactions; (2) decreasing biogenesis processes to save energy to ensure chloroplast priority (i.e., to maintain photosynthetic capacity); and (3) ABA signaling-centered regulation to achieve ROS-scavenging, redox balance, as well as ion homeostasis. The transcriptome evidences presented here would be useful information to better understand physiological and molecular processes involved in salt tolerance of the L. ruthenicum early seedlings.
Author contribution statement
YY and YZ conceived the idea, obtained project funds, design experiments, and supervised all the research activities including final editing of the manuscript. YY, HW, and JG performed the bioinformatics analysis and data organization including figure drawing. YY drafted and edited the main text. HW and JG performed all the biological experiments in this work. ZT, JL, and LD contributed to preparing plant materials and some laboratory work. All authors have read and approved the final manuscript.
Abbreviations
- AASCL:
-
Amino acid solute carrier like
- ABA:
-
Abscisic acid
- BP:
-
Biological process
- CC:
-
Cellular component
- COG:
-
Clusters of Orthologous Groups
- DEG:
-
Differentially expressed gene
- EDTA:
-
Ethylenediaminetetraacetic acid
- EF:
-
Elongation factor
- ELISA:
-
Enzyme-linked immunosorbent assay
- FPKM:
-
Fragments per kb per million fragments
- GA:
-
Gibberellic acid
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GDH:
-
Glutamate dehydrogenase
- GO:
-
Gene ontology
- GS:
-
Glutamine synthetase
- JA:
-
Jasmonate
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- KOG:
-
EuKaryotic Orthologous Groups
- MAPKKK:
-
Mitogen-activated protein kinase kinase kinase
- MAPKs:
-
Mitogen-activated protein kinases
- MF:
-
Molecular function
- NADH-UQ:
-
NADH-ubiquinone oxidoreductase
- NCED:
-
9-Cis-epoxycarotenoid dioxygenase
- NDPK:
-
Nucleoside-diphosphate kinase
- NR:
-
Non-redundant
- ODC:
-
Ornithine decarboxylase
- Pfm:
-
Protein family
- PP2C:
-
Protein phosphatase 2C
- RT-qPCR:
-
Real-time quantitative PCR
- ROS:
-
Reactive oxygen species
- RPL:
-
Ribosome protein large unit (60S)
- RPS:
-
Ribosome protein small unit (40S
References
Abid MA, Liang C, Malik W, Meng Z, Tao Z, Meng Z, Ashraf J, Guo S, Zhang R (2018) Cascades of ionic and molecular networks involved in expression of genes underpin salinity tolerance in cotton. J Plant Growth Regul 37:668–679
Ahammed GJ, Li X, Zhang G, Zhang H, Shi J, Pan C, Yu J, Shi K (2018) Tomato photorespiratory glycolate-oxidase-derived H2O2 production contributes to basal defense against Pseudomonas syringae. Plant Cell Environ 41:1126–1138
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11:106
Annunziata MG, Ciarmiello LF, Woodrow P, Maximova E, Fuggi A, Carillo P (2017) Durum wheat roots adapt to salinity remodeling the cellular content of nitrogen metabolites and sucrose. Front Plant Sci 7:2035
Asrar H, Hussain T, Gul B, Khan MA, Nielsen BL (2018) Differential protein expression reveals salt tolerance mechanisms of Desmostachya bipinnata at moderate and high levels of salinity. Funct Plant Biol 45:793–812
Bai J, Qin Y, Liu J, Wang Y, Sa R, Zhang N, Jia R (2017) Proteomic response of oat leaves to long-term salinity stress. Environ Sci Pollut Res 24:3387–3399
Baranova YN, Akanov EN, Gulevich AA, Kurenina LV, Danilova SA, Khaliluev MR (2014) Dark respiration rate of transgenic tomato plants expressing FeSOD1 gene under chloride and sulfate salinity. Russ Agr Sci 1:14–17
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple hypothesis testing. J R Stat Soc B 57:289–300
Bhattacharjee A, Sharma R, Jain M (2017) Over-expression of OsHOX24 confers enhanced susceptibility to abiotic stresses in transgenic rice via modulating stress-responsive gene expression. Front Plant Sci 8:628
Bottino MC, Rosario S, Grativol C, Thiebaut F, Rojas CA, Farrineli L, Hemerly AS, Gomes Ferreira PC (2013) High-throughput sequencing of small RNA transcriptome reveals salt stress regulated microRNAs in sugarcane. PLoS ONE 8(3):e59423
Brugière N, Zhang WJ, Xu QZ, Scolaro EJ, Lu C, Kahsay RY, Kise R, Trecker L, Williams RW, Hakimi S, Niu XP, Lafitte R, Habben JE (2017) Overexpression of RING domain E3 ligase ZmXerico1 confers drought tolerance through regulation of ABA homeostasis. Plant Physiol 175:1350–1369
Chandrasekaran U, Xu W, Liu AZ (2014) Transcriptome profiling identifies ABA mediated regulatory changes towards storage filling in developing seeds of castor bean (Ricinus communis L.). Cell Biosci 4:33
Chen H, Pu L, Cao J, Ren X (2008) Current research state and exploitation of Lycium ruthenicum Murr. Heilongjiang Agric Sci 5:155–157
Chen HK, Feng Y, Wang LN, Yonezawa TC, Crabbe MJ, Zhang X, Zhong Y, (2015) Transcriptome profiling of the UV-B stress response in the desert shrub Lycium ruthenicum. Mol Biol Rep 42:639–649
Chen JH, Zhang DZ, Zhang C, Xu ML, Yin WL (2017) Physiological characterization, transcriptomic profiling, and microsatellite marker mining of Lycium ruthenicum. J Zhejiang Univ-Sci B 18(11):1002–1021
Cheng Z, Dong K, Ge P, Bian Y, Dong L, Deng X, Li X, Yan Y (2015) Identification of leaf proteins differentially accumulated between wheat cultivars distinct in their levels of drought tolerance. PLoS ONE 10(5):e0125302
Choi S, Lee S, Na Y, Jeung S, Kim SY (2017) Arabidopsis MAP3K16 and other salt-inducible MAP3Ks regulate ABA response redundantly. Mol Cells 40(3):230–242
Das P, Majumder AL (2019) Transcriptome analysis of grapevine under salinity and identification of key genes responsible for salt tolerance. Funct Integr Genom 19:61–73
de Hoon MJL, Imoto S, Nolan J, Miyano S (2004) Open source clustering software. Bioinformatics 20:1453–1454
de Oliveira TM, Cidade LC, Gesteira AS, Coelho Filho MA, Soares Filho WS, Costa CMG (2011) Analysis of the NAC transcription factor gene family in citrus reveals a novel member involved in multiple abiotic stress responses. Tree Genet Genom 7:1123–1134
Dixit A, Tomar P, Vaine E, Abdullah H, Hazen S, Dhankher OP (2018) A stress-associated protein, AtSAP13, from Arabidopsis thaliana provides tolerance to multiple abiotic stresses. Plant Cell Environ 41:1171–1185
Dong JZ, Yang JJ, Wang Y (2008) Resources of Lycium species and related research progress. China Mater Med 33:2020–2027
Duan M, Chang SX (2017) Nitrogen fertilization improves the growth of lodgepole pine and white spruce seedlings under low salt stress through enhancing photosynthesis and plant nutrition. Forest Ecol Manag 404:197–204
Duan M, Feng HL, Wang LY, Li D, Meng QW (2012) Overexpression of thylakoidal ascorbate peroxidase shows enhanced resistance to chilling stress in tomato. J Plant Physiol 169:867–877
Fan L, Wang G, Hu W, Pantha P, Tran KN, Zhang H, An L, Dassanayake M, Qiu QS (2018) Transcriptomic view of survival during early seedling growth of the extremophyte Haloxylon ammodendron. Plant Physiol Biochem 132:475–489
Fan ZL, Ma YJ, Ma YJ (2001) Salinized soils and their improvement and utilization in west China. Arid Zone Res 18(3):1–6 (in Chinese)
Gerszberg A, Hnatuszko-Konka K (2017) Tomato tolerance to abiotic stress: a review of most often engineered target sequences. Plant Growth Regul 83:175–198
Gharbi E, Martínez J, Benahmed H, Fauconnier M, Lutts S, Quinet M (2016) Salicylic acid differently impacts ethylene and polyamine synthesis in the glycophyte Solanum lycopersicum and the wild-related halophyte Solanum chilense exposed to mild salt stress. Physiol Plant 158:152–167
Golldack D, Li C, Mohan H, Probst N (2014) Tolerance to drought and salt stress in plants: unraveling the signaling networks. Front Plant Sci 5:151
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng QD, Chen ZH, Mauceli E, Hacohen N, Gnirke A, Rhind N, Palma FD, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Han DH, Li SJ, Wang EJ, Meng HM, Chen Y, Zhang Y (2014) Effect of exogenous calcium on seed germination and seedling physiological characteristics of Lycium ruthenium. China J Chin Mater Med 39:34–39
Horváth E, Csiszár J, Gallé Á, Poór P, Szepesi Á, Tari I (2015) Hardening with salicylic acid induces concentration-dependent changes in abscisic acid biosynthesis of tomato under salt stress. J Plant Physiol 183:54–63
Inskeep WP, Bloom PR (1985) Extinction coefficients of chlorophyll a and b in N,N-dimethylformamide and 80% acetone. Plant Physiol 77:483–485
Jiang K, Moe-Lange J, Hennet L, Feldman LJ (2016) Salt stress affects the redox status of Arabidopsis root meristems. Front Plant Sci 7:81
Julkowska MM, Testerink C (2015) Tuning plant signaling and growth to survive salt. Trends Plant Sci 20(9):586–594
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36(Database issue):D480–D484
Kang M, Lee S, Abdelmageed H, Reichert A, Lee H-K, Fokar M, Mysore KS, Allen RD (2017) Arabidopsis stress associated protein 9 mediates biotic and abiotic stress responsive ABA signaling via the proteasome pathway. Plant Cell Environ 40:702–716
Kim S, Choi H, Ryu HJ, Park JH, Kim MD, Kim SY (2004) ARIA, an Arabidopsis arm repeat protein interacting with a transcriptional regulator of abscisic acid-responsive gene expression, is a novel abscisic acid signaling component. Plant Physiol 136:3639–3648
Kissoudis C, Sunarti S, van de Wiel C, Visser R, van der Linden CG, Bai YL (2016) Responses to combined abiotic and biotic stress in tomato are governed by stress intensity and resistance mechanism. J Exp Bot 67:5119–5132
Lechner E, Leonhardt N, Eisler H, Parmentier Y, Alioua M, Jacquet H, Leung J, Genschik P (2011) MATH/BTB CRL3 receptors target the homeodomain-leucine zipper ATHB6 to modulate abscisic acid signaling. Dev Cell 21:1116–1128
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform 12:323
Liu D, Han C, Deng X, Liu Y, Liu N, Yan Y (2019) Integrated physiological and proteomic analysis of embryo and endosperm reveals central salt stress response proteins during seed germination of winter wheat cultivar Zhengmai 366. BMC Plant Biol 19:29
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25(4):402–408
Lou D, Wang H, Liang G, Yu D (2017) OsSAPK2 confers abscisic acid sensitivity and tolerance to drought stress in rice. Front Plant Sci 8:993
Loulakakis KA, Roubelakis-Angeiakis KA (1996) The seven NAD(H)-glutamate dehydrogenase isoenzymes exhibit similar anabolic and catabolic activities. Physiol Plant 96:29–35
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550
Ludwig A, Tenhaken R (2001) Suppression of the ribosomal L2 gene reveals a novel mechanism for stress adaptation in soybean. Planta 212:792–798
Mao X, Cai T, Olyarchuk JG, Wei L (2005) Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21:3787–3793
Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y (2008) RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18:1509–1517
Matsuoka D, Soga K, Yasufuku T, Nanmori T (2018) Control of plant growth and development by overexpressing MAP3K17, an ABA-inducible MAP3K, in Arabidopsis. Plant Biotechnol 35:171–176
Moin M, Bakshi A, Saha A, Kumar MU, Reddy AR, Rao KV, Siddiq EA, Kirti PB (2016) Activation tagging in indica rice identifies ribosomal proteins as potential targets for manipulation of water-use efficiency and abiotic stress tolerance in plants. Plant Cell Environ 39:2440–2459
Möller IM (2001) Plant mitochondria and oxidative stress: electron transport, NADPH turnover, and metabolism of reactive oxygen species. Annu Rev Plant Physiol Plant Mol Biol 52:561–591
Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
Muchate NS, Nikalje GC, Rajurkar NS, Suprasanna P, Nikam TD (2016) Plant salt stress: adaptive responses, tolerance mechanism and bioengineering for salt tolerance. Bot Rev 82:371–406
Mukhopadhyay P, Reddy MK, Singla-Pareek SL, Sopory SK (2011) Transcriptional downregulation of rice rpL32 gene under abiotic stress is associated with removal of transcription factors within the promoter region. PLoS ONE 6(11):e28058
Nam MH, Bang E, Kwon TY, Kim Y, Kim EH, Cho K, Park WJ, Kim B, Yoon IS (2015) Metabolite profiling of diverse rice germplasm and identification of conserved metabolic markers of rice roots in response to long-term mild salinity stress. Int J Mol Sci 16:21959–21974
Olsson A, Engström P, Söderman E (2004) The homeobox genes ATHB12 and ATHB7 encode potential regulators of growth in response to water deficit in Arabidopsis. Plant Mol Biol 55:663–677
Othman AB, Ellouzi H, Planchais S, Vos DD, Faiyue B, Carol P, Abdelly C, Savouré A (2017) Phospholipases dζ1 and dζ2 have distinct roles in growth and antioxidant systems in Arabidopsis thaliana responding to salt stress. Planta 246:721–735
Peng Y, Ma HQ, Chen SW (2017) De novo characterization of the Lycium ruthenicum transcriptome and analysis of its digital gene expression profiles during fruit development and ripening. Arch Biol Sci 69(1):181–190
Rhodes D, Rendon GA, Stewart GR (1975) The control of glutamine synthetase level in Lemna minor L. Planta 125:201–211
Sales L, Ohara H, Ohkawa K, Saito T, Todoroki Y, Srilaong V, Kondo S (2017) Salt tolerance in apple seedlings is affected by an inhibitor of ABA 8’-Hydroxylase CYP707A. J Plant Growth Regul 36:643–650
Shin D, Koo YD, Lee J, Lee H, Baek D, Lee S, Cheon CI, Kwak SS, Lee SY, Yun DJ (2004) Athb-12, a homeobox-leucine zipper domain protein from Arabidopsis thaliana, increases salt tolerance in yeast by regulating sodium exclusion. Biochem Biophys Res Commun 323:534–540
Silva N, de Souza GA, Pimenta TM, Brito F, Picoli E, Zsögön A, Ribeiro DM (2018) Salt stress inhibits germination of Stylosanthes humilis seeds through abscisic acid accumulation and associated changes in ethylene production. Plant Physiol Biochem 130:399–407
Singh M, Singh VP, Prasad SM (2016) Responses of photosynthesis, nitrogen and proline metabolism to salinity stress in Solanum lycopersicum under different levels of nitrogen supplementation. Plant Physiol Biochem 109:72–83
Sun L, Sun YF, Zhang M, Wang L, Ren J, Cui MM, Wang YP, Ji K, Li P, Li Q, Chen P, Dai SJ, Duan CR, Wu Y, Leng P (2012) Suppression of 9-cis-epoxycarotenoid dioxygenase, which encodes a key enzyme in abscisic acid biosynthesis, alters fruit texture in transgenic tomato. Plant Physiol 158:283–298
Szypulska E, Jankowski K, Weidner S (2017) ABA pretreatment can limit salinity-induced proteome changes in growing barley sprouts. Acta Physiol Plant 39:190
Tada Y, Kawano R, Komatsubara S, Nishimura H, Katsuhara M, Ozaki S, Terashima S, Yano K, Endo C, Sato M, Okamoto M, Sawada Y, Hirai MY, Kurusu T (2019) Functional screening of salt tolerance genes from a halophyte Sporobolus virginicus and transcriptomic and metabolomic analysis of salt tolerant plants expressing glycine-rich RNA-binding protein. Plant Sci 278:54–63
Takahashi F, Tilbrook J, Trittermann C, Berger B, Roy SJ, Seki M, Shinozaki K, Tester M (2015) Comparison of leaf sheath transcriptome profiles with physiological traits of bread wheat cultivars under salinity stress. PLoS ONE 10(8):e0133322
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28(1):33–36
Tian X, Wang Z, Zhang Q, Ci H, Wang P, Yu L, Jia G (2018) Genome-wide transcriptome analysis of the salt stress tolerance mechanism in Rosa chinensis. PLoS ONE 13(7):e0200938
Tian XW, Ji J, Wang G, Jin C, Guan CF, Wu DY, Li ZD (2015) Cloning and expression analysis of 9-cis-epoxycarotenoid dioxygenase gene 1 involved in fruit maturation and abiotic stress response in Lycium chinense. J Plant Growth Regul 34:465–474
Wang GL, Ren XQ, Liu JX, Yang F, Wang YP, Xiong AS (2019) Transcript profiling reveals an important role of cell wall remodeling and hormone signaling under salt stress in garlic. Plant Physiol Biochem 135:87–98
Wang H, Lin J, Li XG, Chang Y (2015) Genome-wide identification of pear HD-Zip gene family and expression patterns under stress induced by drought, salinity, and pathogen. Acta Physiol Plant 37:189
Wang L, Zhao ZY, Zhang K, Tian CY (2013) Reclamation and utilization of saline soils in arid northwestern China: a promising halophyte drip-irrigation system. Environ Sci Technol 47:5518–5519
Wang LK, Feng ZX, Wang X, Wang XW, Zhang XG (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26(1):136–138
Wang ZQ, Yuan YZ, Ou JQ, Lin QH, Zhang CF (2007) Glutamine synthetase and glutamate dehydrogenase contribute differentially to proline accumulation in leaves of wheat (Triticum aestivum) seedlings exposed to different salinity. J Plant Physiol 164:695–701
Xue GP, Way HM, Richardson T, Drenth J, Joyce PA, McIntyre CL (2011) Overexpression of TaNAC69 leads to enhanced transcript levels of stress up-regulated genes and dehydration tolerance in bread wheat. Mol Plant 4:697–712
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34(Web Server issue):W293–W297
Ye J, Zhang Y, Cui H, Liu J, Wu Y, Cheng Y, Xu H, Huang X, Li S, Zhou A, Zhang X, Bolund L, Chen Q, Wang J, Yang H, Fang L, Shi C (2018) WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res 46(Web Server issue):W71–W75
de Zelicourt A, Colcombet J, Hirt H (2016) The role of MAPK modules and ABA during abiotic stress signaling. Trends Plant Sci 21(8):677–685
Zhang H, Ni Z, Chen Q, Guo Z, Gao W, Su X, Qu Y (2016) Proteomic responses of drought-tolerant and drought-sensitive cotton varieties to drought stress. Mol Genet Genom 291:1293–1303
Zhao KF, Harris PJ (1992) The effects of iso-osmotic salt and water stresses on the growth of halophytes and non-halophytes. J Plant Physiol 139:761–763
Zhu JK (2016) Abiotic stress signaling and responses in plants. Cell 167(2):313–324
Acknowledgements
This work was supported by Xinjiang Key Laboratory of Special Species Conservation and Regulatory Biology Project (Grant no. XJDX1414-2015-01) and the National Natural Science Foundations of China (Grant nos. 31371669 and 31101595). We sincerely thank Dr. Xuemin Wang (Department of Biology, University of Missouri, United States; Donald Danforth Plant Science Center, United States) for his helpful suggestions in the research and English language editing.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by P. Wojtaszek.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.





Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Wang, H., Guo, J., Tian, Z. et al. Transcriptome profiling of mild-salt responses in Lycium ruthenicum early seedlings to reveal salinity-adaptive strategies. Acta Physiol Plant 42, 63 (2020). https://doi.org/10.1007/s11738-020-03048-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-020-03048-6