
Abstract
In the present work we synthesized a new series of phenoxyacetohydrazide functional compounds 4a-k and characterized by spectral data. Synthesized compounds were screened in vitro for their antibacterial activity. Compounds 4a, 4j and 4k exhibited inhibitory activity against S. aureus NCIM 5022 with MIC value of 64 µg/ml These compounds also exhibited activity against methicillin resistant S. aureus ATCC 43300 with MIC of 128 µg/ml. Among all the tested compounds 4c and 4j showed highest activity, respectively against B. subtilis NCIM 2545 and K. pneumoniae NCIM 2706. Only one compound i.e. 4d showed activity against another Gram-negative bacteria P. aeruginosa NCIM 2036 with MIC value of 64 µg/ml. Among three tested compounds, 4k exhibited highest inhibitory activity against S. aureus MurD enzyme with IC50 value of 35.80 µM. Further binding interactions of 4a-k with the modelled S. aureus MurD catalytic pocket residues is investigated with the extra-precision molecular docking and binding free energy calculation by MM-GBSA approach. The van der Waals energy term was observed to be the driving force for binding. Further, 50 ns molecular dynamics simulations were performed to validate the stabilities of 4j- and 4k-modelled S. aureus MurD.
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Infections caused by methicillin-resistant Staphylococcus aureus (MRSA) are major public health threat worldwide. MRSA is responsible for morbidity and mortality (Kong et al. 2016; Hamdy et al. 2017) with an alarming rise of resistance to most of the available antibiotics (Macmorran et al. 2017). This instigated new research effort to develop new class of antibacterial agents acting via novel mechanisms of action. Peptidoglycan (PG) an integral part of S. aureus cell envelope and is critical for the maintenance of cell wall structural integrity. Its biosynthetic pathway in bacteria is well understood and considered as an attractive target for the development of novel antibacterial drugs (Bugg et al. 2011; Gautam et al. 2011). Adenosine triphosphate (ATP)-dependent bacterial Mur ligases (MurC-F) catalyze the successive addition of L-alanine, D-glutamic acid (D-Glu) and L-lysine in Gram-positive or meso-2,6-diaminopimelic acid (meso-DAP) in Gram-negative bacteria, and D-alanine-D-alanine to the growing UDP-N-acetyl muramic acid (UDP-MurNAc) (Barreteau et al. 2008; El Zoeiby et al. 2003). Because of their functional essentiality for bacterial survival and absence in eukaryotic cells, Mur ligases are considered as excellent targets for the development of antibacterial agents (Smith et al. 2006).
The MurC to MurF ligases share a common three-domain structure and act by the same reaction mechanism (Smith et al. 2006). The carboxyl group of the nucleotide precursor is activated by ATP leading to the formation of an acyl phosphate intermediate and adenosine diphosphate (ADP). The acyl phosphate in turn is attacked by the amino group of the incoming amino acid resulting in the formation of a high-energy tetrahedral intermediate. This intermediate then breaks down into the product and inorganic phosphate (Bertrand et al. 1997; Perdih et al. 2007). Mur ligases catalytic pocket binding residues among different bacterial strains are well conserved (Gordon et al. 2001; Ikeda et al. 1990; Bouhss et al. 1999). N-Acetylmuramoyl-L-alanine:D-glutamate ligase (MurD), second in the series catalyses the addition of D‐Glu to UDP‐MurNAc‐L‐Ala resulting in the formation of UDP‐Me urNAc‐L‐Ala‐D‐Glu. The cloned MurD from S. aureus showed similarity to the MurD proteins from Streptococcus pyogenes (66%), Haemophilus influenzae (55%), Escherichia coli (54%) and Bacillus subtilis (65%) (El-Sherbeini et al. 1998). Due to the high specificity for D-amino acid, MurD is considered an attractive target for the development of new antibacterial agents (Walsh et al. 1999; Gegnas et al. 1998). This enzyme consists of three distinct globular domains; the N-terminal domain binds UDP moiety of the UDP-N-acetylmuramoyl-L-alanine (UMA) substrate. The central domain is responsible for the binding of ATP, while the C-terminal domain play an important role in the fixation of the condensing amino acid or dipeptide residue.
During last few decades several phosphinates, α-aminophosphonates and N-acetylmuramic acids have been investigated as tetrahedral-like transition state analogs against E. coli MurD (Gobec et al. 2001; Strancar et al. 2006; Auger et al. 1995; Sova et al. 2009). Phosphorylated and non-phosphorylated hydroxyethylamines have also been developed as inhibitors of the whole cascade of intracellular Mur ligases (MurC to MurF) (Sova et al. 2009) with IC50 values in the low micromolar range. In further development, D-glutamic acid analogs and substituted naphthalene-N-sulfonyl-D-Glutamic acids were tested as transition-state analogs of MurD from E. coli (Pratviel-Sosa et al. 1994; Kotnik et al. 2007; Humljan et al. 2008; Perdih et al. 2009). The binding mode analysis of these inhibitors paved the way for leads optimization. Virtual screening approach has also been used to identify novel MurD ligase inhibitors (Zidar et al. 2010). Virtual hit was further optimized by incorporating the D-Glu moiety into the thiazolidin-4-one scaffold or its surrogate resulting in high potency against MurD. Some of these designed inhibitors exhibited weak in vitro antibacterial activity. Discovery of 5-benzylidenethiazolidine-2,4-dione and 5-benzylidenerhodanine-based E. coli MurD inhibitors (Zidar et al. 2011) unveiled inhibitors binding mode which was used successfully for the design of new generation MurD inhibitors. Second generations sulfonamide inhibitors with rigidified D-Glu mimetics have been synthesized and tested (Farlan et al. 2008; Sosic et al. 2011; Perdih et al. 2014). These rigidified analogs of D-Glu displayed improved inhibitory activity against E. coli MurD. Moreover, some of these analogs exhibited promising inhibitory activity against the whole cascade of Mur ligases (MurD-MurF).
The high resolution MurD crystal structures in complex with D-Glu derivatives provided further insights into inhibitors binding modes (Kotnik et al. 2007; Humljan et al. 2008). Binding free energy calculation results clearly indicated non-polar van der Waals interactions as driving force for inhibitors binding with MurD (Perdih et al. 2007; Perdih et al. 2009; Zidar et al. 2011; Perdih et al. 2013. Result from these studies was used as structural basis for the optimisation and improvement in activity against MurD enzyme (Tomasic et al. 2012). Novel MurD inhibitors have also been discovered by the structure-based virtual screenings approaches (Perdih et al. 2009; Turk et al. 2009; Tomasc et al. 2012; Simcic et al. 2014). However, the most of these efforts not yielded MurD inhibitors possessing potent activity against both Gram-positive and Gram-negative bacteria.
In our earlier in silico virtual screening campaign, compound A (Enamine T6806127, CACPD2011a-0001827917) (Fig. 1) exhibited inhibitory activity against S. aureus MurD (SaMurD) with an IC50 of 7 µM (Azam et al. 2019a). This compound also exhibited activity against S. aureus NCIM 5021, S. aureus NCIM 5022 and B. subtilis NCIM 2545 with minimum inhibitory concentration (MIC) of 128 µg/ml for all three tested strains. In this study compound A was selected as the starting point to design new and potent SaMurD inhibitors by introducing different structural modifications. Based on the molecular dynamics result we sought to substitute appropriate groups for linker 2-methylphenyl as it did not show any interactions with the catalytic pocket residues. Further, one of the 1-[1-(3-hydroxyphenyl)ethyl]urea moiety exhibited less interactions during 30 ns of the MD trajectory. As a first step we replaced one of the 1-[1-(3-hydroxyphenyl)ethyl]urea moiety and the linker 2-methylphenyl with different substituted aryl/heteroaryl-2-carbohydrazide groups to improve the binding affinity with SaMurD protein. We also tried different substituted aryl/heteroaryl rings in place of another 1-[1-(3-hydroxyphenyl)ethyl]urea moiety and different linkers between hydrazinyl and aryl/heteroaryl groups to improve the binding affinity. After several structure optimization cycles, phenoxyacetohydrazide derivatives (4a-k) with favorable binding affinity for the modelled SaMurD active site (Azam et al. 2019b) were designed for further study. The target compounds 4a-k was synthesized as depicted in Scheme 1 and characterized by spectral data. All the synthesized compounds were tested for their in vitro antibacterial activity against various pathogenic bacterial strains. Few selected compounds were assayed for their in vitro SaMurD enzyme inhibitory activity to determine their IC50 values. The interaction of designed compounds with modelled SaMurD protein was studied by the extra-precision molecular docking, binding free calculation and molecular dynamics (MD) studies.

Chemical structure of compound A as SaMurD inhibitor

Synthesis and structures of titled compounds 4a-k
Experimental
Chemistry
The required chemicals for synthesis were of reagent grade and purified when required. All reactions were monitored by thin layer chromatography (TLC) using Silica Gel F254 plates (Merck, Ltd., Germany). Melting points of synthesized compounds were measured in open capillaries and are uncorrected. The infrared spectra were performed by means of a Perkin Elmer-Spectrum Two (United Kingdom) Fourier transform spectrometer and band positions are presented in units of reciprocal centimetres (cm–1). Proton and 13C-NMR spectra were obtained using a Bruker AV-III 400 spectrometer (Germany) operating at 500 and 125 MHz, respectively. Dimethyl sulfoxide (DMSO-d6) was used as solvent and the chemical shifts are expressed in parts per million (ppm) using DMSO-d6 as internal standard. Mass spectra were recorded either on electrospray ionization source Shimadzu LC-MS2020 (Japan) spectrometer (Japan) or on electron impact (EI) JEOL GCMATE II GC–MS spectrometer (USA) operating at 70 eV. Synthesis of ethyl (2/4-substituted phenoxy)acetates 2a: b.p. 112–114 °C, Lit. b.p. 113 °C (Holla et al. 1992); 2b: b.p. 150–154 °C, Lit. 149–155 °C (Horner et al. 1974); 2c: m.p. 219–221 °C, Lit. m.p. 220–222 °C (Amer et al. 2018); 2d: b.p. 270 °C, Lit. b.p. 272 °C (Liu et al. 2012) and 2-(2/4-disubstituted phenoxy)acetohydrazides 3a: m.p. 108–110 °C; Lit. m.p. 110–111 °C (Fun et al. 2010); 3b: m.p. 155–158 °C, Lit. m.p. 155–157 °C (Chao et al. 1949); 3c: m.p. 190–193 °C (Lit. m.p. 190–192 °C (Amer et al. 2018); 3d: m.p. 178–180 °C, Lit. m.p.180–181 °C (Liu et al. 2012) were carried out by literature methods. Synthetic routes for the titled compounds 4a-k is represented in Scheme 1.
General procedure for the synthesis of N-(2/4-substituted phenoxyacetyl)benzohydrazides(4a-k)
To a solution of appropriate 2-(2/4-substituted phenoxy) acetohydrazides (3a-d) (0.008 mol) in acetonitrile (40 ml), substituted benzoyl chlorides/substituted benzene sulfonyl chlorides/tetrahydrofuran-2-carbonyl chloride (0.008 mol) was added drop-wise with continuous stirring at 0 °C over a period of 15 min. Stirring was further continued overnight at room temperature. The progress of reaction was routinely monitored by TLC. After completion of reaction, mixture was poured into crushed ice and the solid thus obtained was filtered, washed thoroughly with water, dried and recrystallized with suitable solvent to yield titled compounds 4a-k. Synthetic route for the synthesized compounds 4a-k is represented in Scheme 1.
Nʹ-[(2-Chlorophenoxy)acetyl]-2-furohydrazide (4a)
Yield 61% Solvent crystallization: ethylacetate. m.p.: 202–204 °C; Rf = 0.47 (ethylacetate/n-hexane, V/V = 1:4); FT-IR (cm−1): 3323, 3214 (NHNH), 3065 (Ar C–H), 2917 (CH2), 1697, 1681 (> C=O), 1493 (Ar –C=C–), 1251, 1040 (C–O–C), 720 (o-substituted benzene); 1H-NMR (DMSO-d6): δppm 10.19 (s, 2H, 2NH), 7.91 (dd, J = 8.32, 1.81 Hz, 1H), 7.44 (dt, J = 7.79, 1.36 Hz, 1H), 7.32 (dt, J = 8.12, 1.76 Hz, 1H), 7.22 (dd, J = 7.97, 1.63 Hz, 1H), 7.06 (dd, J = 7.97, 1.45 Hz, 1H), 6.85 (dd, J = 7.90, 2.35 Hz, 1H), 6.68 (t, J = 8.3, 2.61 Hz, 1H), 4.76 (s, 2H, OCH2); 13C-NMR (DMSO-d6): δppm 167.44 (> C=O), 157.68 (> C=O), 153.95, 146.56, 146.45, 130.67, 128.83, 122.93, 122.12, 115.39, 114.92, 112.53, 67.16 (OCH2). MS (ESI) m/z: calculated for C13H11ClN2O4 (294.69). Found: 295.15 (M+ + 1).
Nʹ-[2-(4-Methoxyphenoxy) acetyl]furan-2-carbohydrazide (4b)
Yield 63%. Solvent crystallization: methanol. m.p.:148–150 °C; Rf = 0.50 (ethylacetate:n-hexane, V/V = 1:4.5); FT-IR (cm−1): 3394, 3214 (NHNH), 3034 (Ar C–H), 2838 (CH2), 1712, 1657 (> C=O), 1658 (NH amide), 1603, 1470 (Ar –C=C–), 1235, 1040 (C–O–C), 802 (p-substituted benzene); 1H-NMR (DMSO-d6): δppm 10.31 (s, 1H, NH), 10.16 (s, 1H, NH), 7.88 (dd, J = 8.04, 1.82 Hz, 1H, ArH), 7.23 (dd, J = 7.91, 1.84 Hz, 1H), 6.98–6.66 (m, 4H), 6.662 (t, J = 7.96 Hz, 1H), 4.56 (s, 2H, OCH2), 3.70 (s, 3H, OCH3); 13C-NMR spectrum (DMSO-d6): δppm 168.09 (> C=O), 157.72 (> C=O), 154.52, 152.36, 146.68, 146.40, 116.42, 115.31, 115.19, 112.52, 67.47 (OCH2), 56.01 (OCH3). LC–MS (ESI): m/z calculated for C14H14N2O5 (290.27). Found: m/z 291.05 (M+ + 1).
2-Chloro-Nʹ-[2-(2,4-dichlorophenoxy)acetyl]benzohydrazide(4c)
Yield 79%. Solvent crystallization: ethanol. m.p.: 184–188 °C; Rf = 0.52 (ethylacetate:n-hexane, V/V = 1:4); FT-IR (cm−1): 3290, 3208, (NHNH), 3068, 3037, 3025 (Ar C-H), 2825 (CH2), 1716, 1661 (> C=O), 1645, 1615 (NH amide), 1593, 1486 (Ar –C=C–), 1269, 1047 (C–O–C); 1H-NMR (DMSO-d6): δppm 10.45 (s, 2H, 2NH), 7.61 (d, J = 8.34 Hz, 1H), 7.59 (d, J = 7.95 Hz, 1H), 7.57–7.45 (m, 3H), 7.44 (dd, J = 8.40, 2.31 Hz, 1H), 7.15 (dd, J = 7.85, 2.22 Hz, 1H), 4.95 (s, 2H, OCH2); 13C-NMR (DMSO-d6): δppm 166.62 (> C=O), 165.73 (> C=O), 153.06, 134.88, 132.03, 130.90, 130.35, 129.87, 129.84, 128.46, 127.64, 125.66, 123.07, 115.91, 67.10 (OCH2). MS (ESI): m/z calculated for C15H11Cl3N2O3 (373.62). Found: m/z 371.05 (M+.-2).
Nʹ-[2-(2,4-Dichlorophenoxy)acetyl]-3,5-dinitrobenzohydrazide (4d)
Yield 76%. Solvent crystallization: ethanol. m.p.: 155–158 °C; Rf = 0.54 (ethylacetate:n-hexane, V/V = 1:5); FT-IR (cm−1): 3318, 3217 (NHNH), 3076, 3039 (Ar C–H), 2920, 2849 (CH2), 1672,1641 (> C=O), 1632 (NH amide), 1588 (Ar –C=C–), 1480, 1366 (NO2), 1247, 1047 (C–O–C), 866 (C–N); 1H-NMR (DMSO-d6): δppm 9.26 (s, 2H, 2NH), 7.61–7.54 (m, 3H), 7.37 (d, J = 8.13 Hz, 1H), 7.35–6.92 (m, 2H), 4.60 (s, 2H, OCH2); 13C-NMR (DMSO-d6): δppm 170.74 (> C=O), 166.47 (> C=O), 153.09, 129.85, 129.66, 128.49, 128.19, 125.50, 123.00, 115.73, 115.31, 67.53 (OCH2); GC–MS (EI-TOF) m/z calculated for C15H10Cl2N4O5 (429.17). Found: m/z 429.17 (M+).
3-Bromo-Nʹ-[2-(2, 4-dichlorophenoxy)acetyl]benzohydrazide (4e)
Yield 84%. Solvent crystallization: ethanol. m.p.: 198–202 °C; Rf = 0.47 (ethylacetate:n-hexane, V/V = 1:4.5); FT-IR (cm−1): 3268, 3160 (NHNH), 3073, 3003 (Ar C–H), 2924 (CH2), 1697, 1658 (> C=O), 1645 (NH amide), 1548, 1486 (Ar –C = C–), 1251, 1071 (C–O–C); 1H-NMR (DMSO-d6): δppm 10.64 (s, 1H, NH), 10.33 (s, 1H, NH), 8.06 (d, J = 7.6 Hz, 1H), 7.88 (dd, J = 8.2, 1.5 Hz, 1H), 7.69 (dd, J = 7.6, 1.4 Hz, 1H), 7.56 (t, J = 7.7 Hz, 1H), 7.44 (dd, J = 8.05, 1.8 Hz, 1H), 7.06 (dd, J = 8.15, 2.3 Hz, 1H), 6.96 (d, J = 8.32 Hz, 1H), 4.83 (s, 2H, OCH2); 13C-NMR (DMSO-d6): δppm 166.89 (> C=O), 164.51 (> C=O), 153.0, 135.19, 134.82, 131.32, 130.62, 130.57, 129.88, 129.85, 128.44, 127.05, 125.69, 123.07, 67.23 (OCH2); LC–MS (ESI): m/z calculated for C15H11Cl2BrN2O3 (418.07). Found: m/z 416.95 (M+.-1).
4-Chloro-Nʹ-[(2,4-dichlorophenoxy)acetyl]benzohydrazide (4f)
Yield 66%. Solvent crystallization: methanol. m.p.: 204–206 °C; Rf = 0.52 (ethylacetate:n-hexane, V/V = 1:5); FT-IR (cm−1): 3272, 3243 (NHNH), 3066, 3024 (Ar C–H), 2845 (CH2), 1693, 1673 (> C=O), 1651, 1621 (NH amide), 1598, 1488 (Ar –C=C–), 1260, 1078 (C–O–C), 801 (p-substituted benzene); 1H-NMR (DMSO-d6): δppm 10.60 (s, 1H, NH), 10.31 (s, 1H, NH), 7.93–7.89 (m, 2H), 7.61–7.58 (m, 2H), 7.41 (d, J = 8.3 Hz, 1H), 7.39 (dd, J = 8.4, 1.9 Hz, 1H), (d, J = 8.04 Hz, 1H), 4.82 (s, 2H, OCH2); 13C-NMR (DMSO-d6): δppm 166.94 (> C=O), 164.98 (> C=O), 153.04, 137.28, 131.48, 130.05, 129.89, 129.15, 128.47, 125.71, 123.10, 115.96, 67.28 (OCH2); GC–MS (EI-TOF) m/z calculated for C15H11Cl3N2O3 (373.62). Found: m/z 373.62 (M+).
2,4-Dimethoxy-Nʹ-[2-(4-nitrophenoxy)acetyl]benzohydrazide (4g)
Yield 81%. Solvent crystallization: ethanol. m.p.: 185–187 °C; Rf = 0.54 (ethylacetate:n-hexane, V/V = 1:4.5); FT-IR (cm−1): 3324, 3212 (NHNH), 3085, 3073, 3039 (Ar C–H), 2992, 2848 (CH2), 1698, 1658 (> C=O), 1641 (NH amide), 1568, 1462 (Ar –C=C–), 1593, 1342 (NO2), 846 (C–N), 1244, 1050 (C–O–C). 1H-NMR (DMSO-d6): δppm 10.54 (s, 2H, 2NH), 8.25–8.19 (m, 2H), 7.81 (d, J = 8.32, Hz, 1H), 7.22 (d, J = 7.99 Hz, 1H), 7.09 (d, J = 8.4 Hz, 1H), 6.67–6.65 (m, 2H), 4.87 (s, 2H, OCH2), 3.91 (s, 6H, OCH3); 13C-NMR (DMSO-d6): δppm 164.03 (> C=O), 163.86 (> C=O), 163.45, 159.26, 141.78, 133.01, 132.96, 126.28, 126.23, 115.86, 115.50, 113.44, 98.99 (CH2), 66.82 (OCH3), 55.98 (OCH3); LC–MS (ESI): m/z calculated for C17H17N3O7 (375.33). Found: m/z 376.00 (M+ + 1.).
2,4-Dichloro-Nʹ-[2-(4-nitrophenoxy)acetyl]benzohydrazide (4h)
Yield, 71%. Solvent crystallization: methanol. m.p.:218–220 °C; Rf = 0.48 (ethylacetate:n-hexane, V/V = 1:5); FT-IR (cm−1): 3268, 3222 (NHNH), 3089, 3057 (Ar C–H), 2924, 2846 (CH2), 1712, 1665 (> C=O), 1564 (Ar –C=C–), 1595, 1353 (NO2), 1259, 1040 (C–O–C). 1H-NMR (DMSO-d6): δppm 10.52 (s, 2H, 2NH), 8.25–7.89 (m, 2H), 7.74 (d, J = 8.16 Hz 1H), 7.68–7.53 (m, 2H), 7.49 (dd, J = 7.94, 1.34 Hz, 1H), 7.21 (d, J = 8.16 Hz, 1H), 4.98 (s, 2H, OCH2); 13C-NMR spectrum (DMSO-d6): δppm 166.47 (> C=O), 164.96 (> C=O), 135.83, 133.72, 132.18, 131.17, 129.96, 129.70, 129.25, 127.92, 127.32, 126.24, 66.79 (OCH2). LC–MS (ESI): m/z calculated for C15H11Cl2N3O5 (384.17). Found: m/z 382.00 (M+.-2).
Nʹ-[2-(4-Methoxyphenoxy)acetyl]-2-methylbenzohydrazide (4i)
Yield 62%. Solvent crystallization: methanol. m.p.: 88–92 °C; Rf = 0.54 (ethylacetate:n-hexane, V/V = 1:5); FT-IR (cm−1): 3253, 3185 (> NH), 3065 (Ar C–H), 2995, 2831 (CH2), 1689, 1645 (> C=O), 1658 (NH amide), 1509 (Ar –C=C–), 1227, 1040 (C–O–C); 1H-NMR (DMSO-d6): δppm 10.23 (s, 1H, NH), 10.13 (s, 1H, NH), 7.41–7.29 (m, 2H, ArH), 7.28–7.25 (m, 2H, ArH), 6.90 (dd, J = 8.21, 2.74 Hz, 2H), 6.55 (dt, J = 7.81, 1.84 Hz, 2H), 4.68 (s, 2H, OCH2), 3.71 (s, 3H, OCH3), 2.50 (s, 3H, CH3); 13C-NMR spectrum (DMSO-d6): δppm 168.78 (> C=O), 167.84 (> C=O), 154.46, 152.36, 136.47, 135.30, 131.12, 130.49, 127.94, 126.10, 116.34, 115.14, 67.43 (OCH2), 55.95 (OCH3), 19.86 (CH3). LC–MS (ESI): m/z calculated for C17H18N2O4 (314.34). Found: m/z 315.25 (M+ + 1).
Nʹ-(Benzenesulfonyl)-2-(2-chlorophenoxy)acetohydrazide (4j)
Yield 72%. Solvent crystallization: methanol. m.p.:114–116 °C; Rf = 0.51(ethylacetate:n-hexane, V/V = 1:5.5); FT-IR (cm−1): 3339, 3214 (NHNH), 3034, 3003 (Ar C–H), 2940 (CH2), 1704 (> C=O), 1658, 1603 (NH amide), 1486 (Ar –C=C–), 1243, 1048 (C–O–C), 752 (o-substituted benzene); 1H-NMR (DMSO-d6): δppm 10.30 (s, 1H, NH), 10.06 (s, 1H, NH), 7.81 (dt, J = 8.31, 2.45 Hz, 2H), 7.79 (dd, J = 7.84, 1.59 Hz, 1H), 7.77 (dd, J = 8.08, 2.67 Hz, 1H), 7.63–7.52 (m, 3H), 7.49–7.24 (m, 2H), 4.57 (s, 2H, OCH2); 13C-NMR (DMSO-d6): δppm 166.69 (> C=O), 133.63, 130.61, 130.58, 130.01, 129.48, 128.75, 128.68, 128.59, 128.43, 128.14, 66.94 (OCH2). MS (ESI) m/z: calculated for C14H13ClN2O4S (340.78). Found: 339.10 (M+.-1).
2-Chloro-5-{[2-(4-methoxyphenoxy)acetohydrazido]sulfonyl}benzoic acid (4k)
Yield 67%. Solvent crystallization: ethanol. m.p.: 196–200 °C; Rf = 0.48 (ethylacetate:n-hexane, V/V = 1:6); FT-IR (cm−1): 3315, 3222 (NHNH), 3065, 3018 (Ar C–H), 2838 (CH2), 1704, 1650 (> C=O), 1650 (NH amide), 1439 (Ar –C=C–), 1235, 1032 (C–O–C). 1H-NMR (DMSO-d6): δppm 10.28 (s, 1H, OH), 10.27 (s, 1H, NH), 10.12 (s, 1H, NH), 8.19 (d, J = 8.24 Hz, 1H), 7.86 (d, J = 8.32 Hz, 1H), 7.40 (dd, J = 7.88, 1.98 Hz, 1H), 6.95–6.75 (m, 4H), 4.54 (s, 2H, OCH2), 3.69 (s, 3H, OCH3); 13C-NMR spectrum (DMSO-d6): δppm 167.64 (> C=O), 167.41 (> C=O), 154.35, 154.33, 152.08, 138.56, 137.14, 132.09, 131.79, 116.23, 116.03, 115.76, 67.26 (OCH2), 55.85 (OCH3); GC–MS (EI-TOF) m/z calculated for C16H15ClN2O7S (414.81). Found: m/z 414.81 (M+).
Determination of minimum inhibitory concentration (MIC)
In vitro antibacterial activity of synthetic compounds 4a-k was assessed according to the guidelines of the Clinical Laboratories Standard Institute (CLSI 2007) against Gram-positive and Gram-negative bacterial strains of S. aureus (NCIM 5021 and NCIM 5022), methicillin resistant S. aureus (MRSA strain 43300), B. subtilis (NCIM 2545), E. coli (NCIM 2567), K. pneumoniae (NCIM 2706) and P. aeruginosa (NCIM 2036). Triplicate analyses were performed for each selected strain by microdilution broth technique using Mueller Hinton medium (Hi-media). Compounds 4a-k was screened for their antibacterial activity dissolved in sterile dimethyl sulfoxide (DMSO). Ciprofloxacin and gentamicin were used as positive control in sterile DMSO, while sterile DMSO was used as a negative control. The 96 well microtitre plates were incubation at 37 °C for 24 h, agitated and read for the absorbance. The final DMSO concentration in assays well was < 1% and the final volume for MIC protocols was 100 µL. The absorbance was measured at a wavelength of 600 nm. The MIC values are summarized Table 1 and expressed as microgram per millilitre (µg/ml).
Enzyme inhibitory activity of S. aureus MurD ligase
Three compounds 4d, 4j and 4k showing promising inhibitory activity against the tested strains of S. aureus are selected for SaMurD ligase inhibition assay. The inhibitory activity of three compounds was assessed against Sa MurD enzyme (ProFoldin, USA) using the malachite green assay with slight modifications (Auger et al. 1998; Lanzetta et al. 1979). All of the experiments were carried out in duplicate. The final volume of 50 µL mixture contained: 40 mM KCl, 50 mM TrisHCl, pH 8.0, 0.005% Triton X-100, 0.1 mM UDP-MurNAc-L-Ala, 0.5 mM D-Glu, 1 mM ATP, 1 mM DTT, 1 mM MgCl2, 500 nM purified SaMurD and 0.6 µL of each test compound different concentrations dissolved in dimethylsulfoxide (DMSO). In all experiments the final concentration of DMSO was 5% (v/v). The prepared mixture was incubated at 37 °C for 60 min and then quenched using the Dye MPA3000. Mixture was further incubated for 5 min and absorbance at 650 nm was determined and percentage inhibition was calculated. Percent inhibitions were also calculated omitting the test compounds and with 5% DMSO. GraphPad PRISM was used to determine IC50 values (Fig. 2).

Graphs used for IC50 value determinations of compounds 4d (red), 4j (green) and 4k (blue) against SaMurD enzyme
Computational analysis
Molecular docking and binding free energy calculation (MM-GBSA)
The 3D-structure of compounds 4a-k were sketched in the builder panel of Maestro 11.5 (Schrodinger software suite 2018-1, LLC, New York, NY) and subsequently optimized with LigPrep (Sastry et al. 2013). The extra-precision molecular docking was performed using Glide without applying any constraints. In order to accurately rank the binding strength binding free energy of compounds 4a-k was calculated by prime MM-GBSA approach (Jacobson et al. 2004) using VSGB 2.0 energy (Li et al. 2011) and OPLS3e force field (Roos et al. 2019). In this study we used the earlier prepared grid box of homolg modelled SaMurD protein (Azam et al. 2019b). The low energy conformations of all ligands were docked in the catalytic pocket and their binding free energies were determined using the protocols described earlier (Azam et al. 2019b).
MD simulation
The extra-precision-docked structure of compound 4k/SaMurD modelled protein complex was used as starting structure for MD simulation using the Desmond software with OPLS3e force field (Roos et al. 2019). The complex was solvated in an orthorhombic box with explicit TIP4P (Jorgensen and Madura 1985; Lawrence and Skinner 2003) water with a 10 Å buffer region between protein atoms and box sides. Eleven counter sodium ions were added to neutralize the solvated system. The modelled system contains approximately 49,403 atoms, 14,095 water molecules with a final box volume of 1,197,217 Å3. The whole system was then minimized with OPLS3e (Roos et al. 2019) force field using 200 steepest descent and 1000 steps of conjugate gradient until 25 kcal/mol/Å gradient threshold was reached. Particle-mesh-Ewald method (Essmann et al. 1995) at a tolerance of 1e-09 was used for the long range electrostatic interactions. A cutoff radius of 9 Å was applied for van der Waals and Coulomb interactions. A 50 ns simulation was performed for this minimized system under an isothermal-isobaric ensemble (NPT) at a constant temperature of 300 K and a constant pressure of 1 bar were used to maintain during simulation temperature and pressure were maintained, respectively with Nose–Hoover thermostat (Martyna et al. 1992) and Martyna–Tobias–Klein barostat (Martyna et al. 1994). For the bonded, near nonbonded, and far nonbonded interactions a multiple time step RESPA integration algorithm was used, with time steps of 2, 2, and 6 fs, respectively. For the overall simulation 2 fs time step was selected and data were collected every 100 ps. The 3D structures and MD trajectory were analyzed visually with the Maestro graphical interface.
Results and discussion
Chemistry
The synthetic routes for title compounds 4a-k is outlined in Scheme 1. The intermediate ethyl (2/4-substituted phenoxy) acetates 2a-d is prepared by treatment of 2/4-substituted phenols (1a-d, Scheme 1) with 2-chloroethylacetate in dry acetone (Holla et al. 1992; Horner et al. 1974; Amer et al. 2018; Liu et al. 2012). These acetates (2a-d) are reacted with hydrazine hydrate (99% w/w) in boiling absolute ethanol to furnish 2-(2/4-disubstituted phenoxy)acetohydrazides 3a-d (Fun et al. 2010; Chao et al. 1949; Amer et al., 2018; Liu et al. 2012). The acetohydrazides 3a-d was then stirred with appropriate substituted acid chlorides in acetonitrile to furnish titled compounds 4a-k in good yields (Hariskishore et al. 2013).
Synthesized compounds 4a-k structures were confirmed by FT-IR, 1H-NMR, 13C-NMR and mass spectroscopy. In the IR spectra of phenoxyacetohydrazides 4a-k the appearance of band in the range 3208–3339 cm−1 indicated the presence of hydrazide –NHNH– group, while the bands appeared in the range 1641–1716 cm−1 is due to carbonyl groups. In these compounds the C–O–C absorption bands appeared in the region 1032–1259 cm−1. In the 1H-NMR spectra of compounds 4a-k two singlet signals appeared in the region δ 9.26–10.64 ppm indicated the presence of two protons of NHNH function. The singlet signals appeared in the region δ 4.54–4.98 ppm is ascribed to two protons of –OCH2 group. Further, the splitting patterns for the aromatic hydrogens agree with the substitution pattern of respective compounds. The chemical shift values and coupling constant (J values) were assigned accordingly with the position of aromatic hydrogens. Aromatic hydrogens on coupling with ortho and meta hydrogens exhibited characteristic splitting patterns like double doublet (dd), triplet of doublet (dt), triplet (t) and doublet (d). In compound 4a the two protons H-3 and H-6 of 2-chlorophenyl ring appeared as double doublets, respectively at δ 7.22 and 7.06 ppm with average coupling constant of δ 7.97 and 1.54 Hz. Two triplet of doublet (dt) signals at δ 7.44 and 7.32 ppm with average J values of 7.95 and 1.56 Hz were attributed, respectively for H-4 and H-5 hydrogens of the same ring. Two hydrogens H-3′ and H-5′ of the furan ring were observed at δ 7.91 and 6.85 ppm, respectively with average J values of 8.11 and 1.67 Hz. H-4′ hydrogen of furan ring exhibited triplet signal at δ 6.68 ppm with J values of 8.3 and 2.61 Hz. Other synthesized compounds also exhibited splitting patterns according to the substitution pattern. In the 13C-NMR spectra of compounds 4a-k the carbonyl carbon signals appeared in the region δ 157.68–170.74 ppm while the signals appeared in the region δ 66.79–67.53 ppm is due to the -OCH2- fragment carbon. Aromatic carbon signals appeared at the expected region. In the mass spectra, compounds 4a-k exhibited molecular ion peaks which are in agreement with their molecular formula (see experimental also Supplementary Figs. S1–S8).
Determination of minimum inhibitory concentration (MIC)
The antibacterial activities of the synthesized compounds were screened by two-fold broth micro-dilution method, as per the procedures of the Clinical and Laboratory Standards Institute (CLSI 2007). The result of antibacterial screening is represented in Table 1. Tested compounds MIC values are compared with the standard drugs ciprofloxacin and gentamicin. In vitro antibacterial screening of tested phenoxyacetohydrazides 4a, 4c, 4d, 4f, 4j, and 4k exhibited promising inhibitory activity against Gram-positive bacteria S. aureus NCIM 5021 with MIC values in the range of 32–64 µg/ml in comparison to the standard drugs ciprofloxacin (MIC, 2 µg/ml) and gentamcin (MIC 8 µg/ml). Tested compounds 4a, 4j and 4k also exhibited inhibitory activity against S. aureus NCIM 5022 with MIC value of 64 µg/ml in each case. Three compounds i.e. 4a, 4j and 4k exhibited activity against methicillin resistant S. aureus ATCC 43300 with MIC of 128 µg/ml, while all other tested compounds were observed to be either weakly active (MIC 256 µg/ml) or inactive (MIC > 256 µg/ml) compared to the standard drugs ciprofloxacin (MIC, 32 µg/ml) and gentamicin (MIC, 20 µg/ml). The low activity of these compounds is probably due to the failure of bacterial cell wall penetration.
Among all the tested compounds, phenoxyacetohydrazides 4e, 4f and 4k displayed MIC of 64 µg/ml against B. subtilis NCIM 2545. Compound 4j exhibited maximum activity against this strain with an MIC of 32 µg/ml, whereas other tested compounds were observed to be either less active (128 to 256 µg/ml) or inactive (MIC > 256 µg/ml). Further, compounds 4a, 4c, 4i and 4j displayed promising inhibitory activity against Gram-negative K. pneumoniae NCIM 2706 (Table 1, MIC, 64, 32, 64, and 64 µg/ml, respectively). Among all tested compound, 4c exhibited highest activity against this bacterial strain with MIC value of 32 µg/ml in comparison to the standard drugs ciprofloxacin (2 µg/ml) and gentamicin (1 µg/ml). Only one compound, i.e. 4d showed activity against another Gram-negative bacteria P. aeruginosa NCIM 2036 (MIC value of 64 µg/ml), while all other tested compounds were observed to be either less active (MIC, 128–256 µg/ml) or inactive (MIC > 256 µg/ml). On the other hand, compounds 4e, 4f, 4j and 4k displayed promising activity against E. coli NCIM 2567 (MIC, 32–64 µg/ml). Against this strain the highest activity was observed for compounds 4f, 4j and 4k with MIC value of 32 µg/ml in all cases. It is evident from Table 1 that compounds 4j and 4k displayed inhibitory activity against all the tested strains of Gram-positive and Gram-negative bacteria. Improvement in activity against S. aureus NCIM 5021was observed in compounds 4a, 4c, 4d, 4f, 4j and 4k (MIC 32–64 µg/ml) in comparison to compound A (MIC 128 µg/ml). Further, compared to compound A (MIC 128 µg/ml) improvement in inhibitory activity against S. aureus NCIM 5022 was also observed for compounds 4a, 4j and 4k (MIC 64 µg/ml). Also in comparison to compound A (MIC 256 µg/ml), these three compounds also exhibited a lower MIC of 128 µg/ml against methicillin resistant S. aureus ATCC 43300.
Enzyme inhibitory activity of S. aureus MurD ligase
In vitro inhibitory potency of three selected compounds 4d, 4j and 4k was carried out using SaMurD enzyme (ProFoldin, USA) (Table 2). The tested compounds showed IC50 values in the range 35.80–98.66 µM, compound 4k being the highest active (IC50 35.80 µM). Remaining two compounds namely 4d and 4j inhibited SaMurD with IC50 values of 37.82 and 98.66 µM, respectively. It is evident from above result that the presence of an electron withdrawing substituent on the phenyl ring attached to either –SO2 or > C=O group is important for the SaMurD inhibitory activity as evident by the lower activity of compound 4j (IC50, 98.66 µM) lacking electron withdrawing substituent on this phenyl ring.
Computational analysis
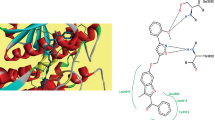
Interaction energies of compounds 4a-k with the catalytic pocket residues of modelled S. aureus MurD enzyme were computed to get insight into their binding modes and affinity. Analysis of extra-precision docking poses showed hydrogen bonding, π–π stacking and π-cation stacking salt bridge interactions with conserved residues Lys19, Lys26, His48, Gly144, Gly147, Val149, Trp425 and Phe 431 (Fig. 3 also Supplementary Fig. S9). The extra-precision docking and binding free energy calculation results are summarized in Tables 3 and 4, respectively.

Plots represent interactions of catalytic pocket residues of modelled SaMurD with compounds (a) 4j and (b) 4k
Sulfonylhydrazide 4j with IC50 of 98.66 µM against SaMurD protein occupied all three domains of the catalytic pocket and exhibited two hydrogen bonding interactions. Precisely, oxygen atom of carbonyl group attached to the 2-chlorophenoxymethylene fragment in compound 4j accepted hydrogen bond from the protonated NH2 of Lys19 (NH⋯O=C < , 1.90 Å), while the –NH– function attached to this carbonyl group formed hydrogen bond with the carbonyl oxygen of Gly147 (> C=O⋯HN–, 1.82 Å) (Fig. 3a). The phenyl ring of 4-chlorophenoxymethyle fragment established π-cation interaction with the protonated NH2 of Lys328. The lower inhibitory activity of compound 4j against SaMurD may be due to fewer interactions with the catalytic pocket residues. The high active sulfonylhydrazide 4k (IC50 35.80 µM) exhibited four hydrogen bonding interactions, two more than 4j (Fig. 3b). The NH of -CONHNH- fragment formed hydrogen bond with carbonyl oxygen of Gly147 (> C=O⋯NH, 2.19 Å), while > C=O oxygen of the same fragment established hydrogen bonding interaction with the NH of Lys19 (NH⋯O=C < , 2.47 Å). The oxygen atom attached to the 4-methoxyphenyl ring also accepted hydrogen bond from the NH of Lys19 (NH⋯O-C6H4-4-OCH3 2.59 Å), while the oxygen atom of –SO2– function attached to the 4-chloro-3-carboxy phenyl moiety accepted a hydrogen bond from the NH of Val149 (NH⋯O=S(=O)–, 2.12 Å). The carboxylate oxygen of 4-chloro-3-carboxy phenyl moiety exhibited two salt bridge interaction one each with Ly26 and Ly152. This compound is further stabilized in the catalytic pocket by two π–π stacking interactions one each with Tyr22 and Phe431 residues. Compound 4d (IC50 37.82 µM) belonging to the hydrazide series exhibited three hydrogen bonding interactions one each with Asn78, Gly80 and Gly144 (Supplementary Fig. S9). 2,4-Dichlorophenyl ring of this compound also established a π–π stacking interaction with the phenyl ring of Phe431. A π-cation interaction was also observed between the nitrogen atom of one of nitro groups and Glu101. It is evident that compounds belonging to the hydrazide series exhibited less interactions with the catalytic pocket residues compared to the sulfonylhydrazide 4k (Supplementary Fig. S9).
To accurately rank the binding strength of compounds 4a-k with modelled SaMurD protein the binding free energy (ΔGbind) was computed by the MM-GBSA approach (Table 4). However, in the present investigation ΔGbind did not exhibit any correlation either with the MIC or IC50 values. The ΔGbind values are observed to be in range between − 67.28 to − 112.23 kcal/mol. It is evident from binding free energy components that van der Waals (ΔGvdW, − 28.53 to − 76.36 kcal/mol) and electrostatic interaction (ΔGCoul, − 12.42 to − 92.60 kcal/mol) energy terms are major contributors for the inhibitors binding. Except for 4b and 4k, electrostatic solvation energy (ΔGSolv, 4.21 to 78.92 kcal/mol) term disfavors, while non-polar solvation (ΔGLipo, − 14.98 to − 30.87 kcal/mol) energy term moderately favors the binding to the SaMurD protein. Compared to ΔGCoul, the high negative value of ΔGvdW indicated it as the driving force for binding. This is observed to be in agreement with the earlier reports (Perdih et al. 2007,2009,2014,2013; Zidar et al. 2011) and also with the value of Glide Emodel (Gemodel, − 54.14 to − 94.85 kcal/mol) possessing significant weighting of the force field. s
To validate the stability of docking complex and to get insight about molecular mechanism of binding, we performed a 50 ns MD simulation for 4k/SaMurD complex. It is evident from Fig. 4a that root mean square deviations (RMSDs) of protein all Cα, backbone and heavy atoms increased sharply (3.91, 3.97, 4.18 Å, respectively) during first 5 ns of MD simulation and then observed to stable with average fluctuations of 1.08, 1.05 and 1.03 Å, respectively during rest of the simulation trajectory. This shows less conformational changes in protein structure during simulation. Ligand–protein interactions were observed in the region Ala18-Lys52 and Ser327-Glu432, while 4k did not show contacts in the region Val153-Asp326 (Fig. 4b also Supplementary Figs. S10 and S11). The root mean square fluctuations (RMSF) in Cα, backbone and heavy atoms of ligand contacting residues was observed in the range 0.65–2.17, 0.5–2.11, 1.01–2.30 Å, indicating less fluctuations in protein structure during simulation. During simulation the maximum RMSF in Cα, backbone and heavy atoms was observed in amino acid stretch Ser44-Gln45 (3.61–3.18, 3.25–3.49 and 3.41–4.29 Å, respectively) present on loop (Asp39-His48). The values for Cα, backbone and heavy atoms of binding residues of catalytic pocket was found to be in the range 0.68 to 1.27 and 0.69 to 1.28 Å, respectively (Fig. 4b). This indicated no significant conformational changes in these residues. The maximum RMSFs for Cα (2.38–3.17 Å) and backbone (2.66–3.16 Å) atoms was observed in residues Glu232-Leu234 present on the loop (Tyr223-Thr238) and connecting two β-sheets (Leu239-Phe241).

Plot represent (a) RMSD (Å) of the simulated positions of Cα, backbone and heavy atoms of 4k/SaMurD complex protein protein compared to the initial structure. (b) RMSF profile of active site residues of SaMurD complex during MD simulation of 4k/SaMurD complex (c) different interactions of compound 4k with active site residues of SaMurD during 50 ns simulation trajectory
It is evident from analysis of MD trajectory that compound 4k occupied all three domains of the catalytic pocket and exhibited hydrogen bonding, salt bridge, π–π stacking and π-cation interactions (Fig. 4c and also Supplementary Fig. S10). Accurately, one of the –NH– function linked to the –O–CH2CO–NH–NH– fragment formed a strong hydrogen bonding interaction with Gly147 (90% of the MD trajectory), while another NH function linked to the –SO2– group established a strong hydrogen bonding interaction (82% of the MD trajectory) with Ser429. A strong hydrogen bonding interaction (99% of the MD trajectory) was also observed between carbonyl oxygen of the same fragment and Trp425 located in the C-terminal domain. The phenyl ring of this residue also exhibited a π–π stacking interaction with the phenyl ring of 4-chloro-3-carboxyphenyl moiety. One of the oxygen atoms of SO2 group accepted two direct hydrogen bond one each from the central domain residue Val149 (48% of the MD trajectory) and the N-terminal residue Lys19 (21% of the MD trajectory). Another oxygen atom of this group accepted water mediated hydrogen bond (38% of the MD trajectory) from Val149. The carboxylate oxygen of 4-chloro-3-carboxyphenyl moiety accepted two moderate frequency hydrogen bonds, a direct bond from the side chain NH of Lys19 (24% of the MD trajectory) and a water mediated bond from the side chain NH of Glu23 (39% of the MD trajectory). Glu23 also established moderate frequency water mediated hydrogen bond (30% of the MD trajectory) with the carbonyl oxygen of carboxylate function. Compound 4k also exhibited low frequency hydrogen bonding and salt bridge interactions (10–16% of the MD trajectory) with Lys152, Asn145, Tyr428 and Phe 431. It is evident from above result that extensive hydrogen bonding interactions play a crucial role for the stabilization of compound 4k within the catalytic pocket of SaMurD protein.
The rGyr 2.21–3.84 Å of ligand (Supplementary Fig. S12), which measures the extendedness, indicated the stability of compound 4k within the catalytic pocket during 50 ns MD simulation. The RMSD value of ligand fluctuated within a narrow range of 2.21–3.84 Å during 25–50 ns of MD simulation, further indicating stability and less conformational flexibility of 4k. An increase in the solvent accessible surface area (SASA) of inhibitor 4k was observed up to 25 ns (96–300 Å2) which is then stabilized in the lower window of 85.3–16.4 Å2 showing no significant change in the binding pocket volume during simulation. Further, polar surface area (PSA) of 4k stabilized in the narrow range of 125–145 Å2 during last 25 ns of simulation trajectory, indicating the burial of inhibitor within catalytic pocket. After superposition similar orientations was observed between conformations of 4k best docking pose and pose after MD simulation with RMSD of 1.25 Å (Supplementary Fig. S13). This indicated the validity and rationality of the extra-precision docking model. In order to compare the binding modes, we also performed a 50 ns MD simulation for 4j/SaMurD complex using the protocols as described in experimental section. It is evident from supplementary Fig. S14a that RMSDs of protein Cα and backbone all atoms increased upto 6.25 and 6.24 Å, respectively during first 12 ns of MD simulation and then fluctuated, respectively in the range 5.54–3.22 and 5.57–3.23 Å during 12–23 ns of the simulation trajectory. After 23 ns RMSDs of protein Cα and backbone all atoms stabilized in the range 3.18–4.81 Å, indicating slightly higher fluctuations compared to the fluctuations observed for Cα and backbone atoms in case of 4k/SaMurD complex simulation trajectory. During simulation Ala423 from C-terminal domain showed medium frequency hydrogen bonding interactions with both oxygen atoms of the sulphonyl group (32 and 26% of MD trajectory) (Supplementary Fig. S14b-d). One of the oxygen atoms of the sulphonyl group exhibited a low frequency water mediated hydrogen bonding interaction with Asp354. A strong frequency π-π stacking is also observed between phenyl rings of the chlorophenoxy moiety and Trp425. It is evident that interactions observed in case of compound 4j are comparatively less compared to 4k and probably may be the reason for the lower inhibitory activity of 4j against SaMurD protein.
Conclusion
In the present investigation we synthesized a new series of phenoxyacetohydrazide derivatives 4a-k and characterized them by spectral data. Synthesized compounds were screened in vitro for their antibacterial activity against Gram-positive S. aureus (NCIM 5021, NCIM 5022 and methicillin-resistant isolate 43300), B. subtilis (NCIM 2545) and Gram-negative E. coli (NCIM 2567), K. pneumoniae (NCIM 2706) and P. aeruginosa (NCIM 2036) bacteria. Tested compounds 4a, 4c, 4d, 4f, 4j and 4k displayed promising inhibitory activity against Gram-positive S. aureus NCIM 5021 (MIC 32–64 µg/ml), while compounds 4a, 4j and 4k exhibited inhibitory activity against S. aureus NCIM 5022 with MIC value of 64 µg/ml in each case. These compounds also exhibited activity against methicillin resistant S. aureus ATCC 43300 with MIC of 128 µg/ml. Among all the tested compounds 4j showed maximum inhibitory activity against B. subtilis NCIM 2545 with MIC value 32 µg/ml. Whereas compound 4c exhibited highest activity against Gram-negative K. pneumoniae NCIM 2706 with MIC value of 32 µg/ml. Only compound 4d showed activity against another Gram-negative bacteria P. aeruginosa NCIM 2036 with MIC 64 µg/ml. Further, it is evident from result that sulfonylhydrazides (4j and 4k) is more active against the tested strains of both Gram-positive and Gram-negative bacteria compared to hydrazides 4a-i. The high activity of sulfonylhydrazides may be attributed to the presence of -SO2 group in these compounds. The inhibitory activity of three compounds 4d, 4j and 4k against SaMurD was performed using the malachite green assay. Compound 4k exhibited highest inhibitory activity with IC50 value of 35.80 µM, while other two tested compounds 4d and 4j showed IC50 values of 37.82 and 98.66 μM. These three tested compounds exhibited low inhibitory activity against SaMurD enzyme, however the bacterial cell penetration of these compounds is better compared to compound A as evident by their MIC values against the tested strains of S. aureus. Further molecular mechanism of binding of 4a-k with the catalytic pocket residues of modelled SaMurD is investigated with the extra-precision molecular docking and binding free energy calculation by MM-GBSA approach. The van der Waals (ΔGvdW, − 28.53 to − 76.36 kcal/mol) and electrostatic (ΔGCoul, − 12.42 to − 92.60 kcal/mol) interaction energy terms are observed to be major contributors for binding to the SaMurD enzyme. Further, the high negative value of ΔGvdW indicated it as the driving force for binding. 50 ns molecular dynamics simulations were performed for 4j and 4k/SaMurD complexes to validate the stability of their docking poses. Similar orientations was observed between conformations of 4k best docking pose and pose after MD simulation indicating the validity and rationality of the extra-precision docking model.
Abbreviations
- DAP:
-
2,6-Diaminopimelic acid
- D-Glu:
-
D-glutamic acid
- MurC:
-
UDP-N-acetylmuramoyl-L-alanine ligase
- MRSA:
-
Methicillin-resistant Staphylococcus aureus
- MIC:
-
Minimum inhibitory concentration
- MBC:
-
Minimum bactericidal concentration
- MurD:
-
UDP-N-acetylmuramoyl-L-alanine:D-glutamate ligase
- MurE ligase:
-
UDP-N-acetylmuramoyl-L-alaninyl-D-glutamate-2,6-diaminopimelate (or L-lysine) ligase
- MurF ligase:
-
UDP-N-acetylmuramoyl-L-alanine-D-glutamyl-lysine-D-alanyl-D-alanine ligase
- MurNAc:
-
N-Acetyl muramic acid; UDP: uridine-5′-diphosphate
- UMA:
-
Uridine-5′-diphosphate-N-acetylmueamoyl-l-alanine
- RMSD:
-
Root mean square deviation
- UMAG:
-
UDP-N-acetylmuramoyl-L-alanine-D-glutamate
References
Amer HA, Ali OM, Salama AA, El-gendy MS, Houssin OK (2018) Synthesis of some new 1,3,4-oxadiazole derivatives bearing sugars and α-aminophosphonate derived from 4-nitrophenol as anticancer agents. Nat J Physiol Pharm Pharmacol 8:1275–1286. https://doi.org/10.5455/njppp.2018.8.0416408052018
Auger G, van Heijenoort J, Blanot D (1995) Synthesis of N-acetylmuramic acid derivatives as potential inhibitors of the D-glutamic acid-adding enzyme. J Prakt Chem 337:351–357. https://doi.org/10.1002/prac.19953370176
Auger G, Martin L, Bertrand J, Ferrari P, Fanchon E, Vaganay S, Petillot Y, van Heijenoort J, Blanot D, Dideberg O (1998) Large-scale preparation, purification, and crystallization of UDP-N-acetylmuramoyl-L-alanine: D-glutamate ligase from Escherichia coli. Prot Express Purif 13:23–29. https://doi.org/10.1006/prep.1997.0850
Azam MA, Jupudi S (2019a) Structure-based virtual screening to identify inhibitors against Staphylococcus aureus MurD enzyme. Struct Chem 30:2123–2133. https://doi.org/10.1007/s11224-019-01330-z
Azam MA, Jupudi S, Saha N, Paul RK (2019) Combining molecular docking and molecular dynamics studies for modelling Staphylococcus aureus MurD inhibitory activity. SAR QSAR Environ Res 30:1–20. https://doi.org/10.1080/1062936X.2018.1539034
Barreteau H, Kova A, Boniface A, Sova M, Gobec S, Blanot D (2008) Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol Rev 32:168–207. https://doi.org/10.1111/j.1574-6976.2008.00104.x
Bertrand JA, Auger G, Fanchon E, Martin L, Blanot D, van Heijenoort J, Dideberg O (1997) Crystal structure of UDP-N-acetylmuramoyl-L-alanine: D-glutamate ligase from Escherichia coli. Embo J 16:3416–3425. https://doi.org/10.1093/emboj/16.12.3416
Bouhss A, Dementin S, Parquet C, Mengin-Lecreulx D, Bertrand JA, Le Beller D, Dideberg O, van Heijenoort J, Blanot D (1999) Role of the ortholog and paralog amino acid invariants in the active site of the UDP-MurNAc-Lalanine:D-glutamate ligase (MurD). Biochemistry 38:12240–12247. https://doi.org/10.1021/bi990517r
Bugg TDH, Braddick D, Dowson CG, Roper DI (2011) Bacterial cell wall assembly: still an attractive antibacterial target. Trends Biotechnol 29:167–173. https://doi.org/10.1016/j.tibtech.2010.12.006
Chao JCC, Sah PPT, Oneto JF (1949) Derivatives of 2,4-dichlorophenoxyacetic hydrazide. Recl Trav Chim Pays-Bas 68:506–508. https://doi.org/10.1002/recl.19490680604
Clinical and Laboratory Standard Institute (CLSI) (2007) Methods for dilution antibacterial susceptibility test for bacteria that grow aerobically, 7th ed. Approved Standard (MA7-A7), Clinical and Laboratory Standard Institute: Wayne, 27, pp. 133.
El Zoeiby A, Sanschagrin F, Levesque RC (2003) Structure and function of the Mur enzymes: development of novel inhibitors. Mol Microbiol 47:1–12. https://doi.org/10.1046/j.1365-2958.2003.03289.x
El-Sherbeini M, Geissler WM, Pittman J, Yuan X, Wong KK, Pompliano DL (1998) Cloning and expression of Staphylococcus aureus and Streptococcus pyogenes murD genes encoding uridinediphosphate N-acetylmuramoyl-L-alanine:D-glutamate ligases. Gene 210:117–125. https://doi.org/10.1016/s0378-1119(98)00059-6
Essmann U, Perera L, Berkowit ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577–8593. https://doi.org/10.1063/1.470117
Farlan R, Kovac A, Blanot D, Gobec S, Pecar S, Obreza A (2008) Design and synthesis of novel N-benzylidenesulfonohydrazide inhibitors of MurC and MurD as potential antibacterial agents. Molecules 13:11–30. https://doi.org/10.3390/molecules13010011
Fun HK, Quah CK, Isloor AM, Sunil D, Shetty P (2010) 2-(2-chlorophenoxy)acetohydrazide. Acta Crystallogr E Struct Rep 66:31–32. https://doi.org/10.1107/S1600536809051356
Gautam A, Vyas R, Tewari R (2011) Peptidoglycan biosynthesis machinery: a rich source of drug targets. Crit Rev Biotechnol 31:295–336. https://doi.org/10.3109/07388551.2010.525498
Gegnas LD, Waddell ST, Chabin RM, Reddy S, Wong KK (1998) Inhibitors of the bacterial cell wall biosynthesis enzyme MurD. Bioorg Med Chem Lett 8:1643–1648. https://doi.org/10.1016/s0960-894x(98)00285-6
Gobec S, Urleb U, Auger G, Blanot D (2001) Synthesis and biochemical evaluation of some novel N-acyl phosphono- and phosphinoalanine derivatives as potential inhibitors of the D-glutamic acid-adding enzyme. Pharmazie 56:295–297 PMID:11338666
Gordon E, Flouret B, Chantalat L, van Heijenoort J, Mengin-Lecreulx D, Dideberg O (2001) Crystal structure of UDP-N-acetylmuramoyl-l-alanyl-d-glutamate:meso-Diaminopimelate Ligase from Escherichia coli. J Biol Chem 276:10999–11006. https://doi.org/10.1074/jbc.M009835200
Hamdy RF, Hsu AJ, Stockmann C, Olson JA, Bryan M, Hersh AL, Tamma PD, Gerber JS (2017) Epidemiology of methicillin-resistant Staphylococcus aureus Bacteremia in children. Pediatrics 139:e20170183. https://doi.org/10.1542/peds.2017-0183
Harikishore A, Leow ML, Niang M, Rajan S, Pasunooti KK, Preiser PR, Liu X, Yoon HS (2013) Adamantyl derivative as a potent inhibitor of Plasmodium FK506 binding protein 35. ACS Med Chem Lett 4: 1097–1101.https://www.ncbi.nlm.nih.gov/pubmed/24900611
Holla BS, Udupa KV (1992) Synthesis, spectral studies and biological activities of some N-bridged heterocycles derived from 3-arylaminomethyl-4-amino-5-mercapto-1,2,4-triazoles. Farmaco 47:305–318. https://doi.org/10.1002/chin.199408235
Horner J, QueHee SS, Sutherland RG (1974) Esterification of (2,4-dichlorophenoxy)acetic acid-A quantitative comparison of esterification techniques. Anal Chem 46:110–112. https://doi.org/10.1021/ac60337a048
Humljan J, Kotnik M, Contreras-Martel C, Blanot D, Urleb U, Dessen A, Solmajer T, Gobec S (2008) Novel naphthalene-N-sulfonyl-D-glutamic acid derivatives as inhibitors of MurD, a key peptidoglycan biosynthesis enzyme. J Med Chem 51:7486–7494. https://doi.org/10.1021/jm800762u
Ikeda M, Wachi M, Jung HK, Ishino F, Matsunashi M (1990) Homology among MurC, MurD, MurE and MurF proteins in Escherichia coli and that between Escherichia coli MurG and a possible MurG protein in Bacillus subtilis. J Gen Appl Microbiol 36:179–187. https://doi.org/10.2323/jgam.36.179
Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, Friesner RA (2004) A hierarchical approach to all-atom protein loop prediction. Proteins 55:351–367. https://doi.org/10.1002/prot.10613
Jorgensen WL, Madura JD (1985) Temperature and size dependence for Monte–Carlo simulations of TIP4P water. Mol Phys 56:1381–1392. https://doi.org/10.1080/00268978500103111
Kong EF, Johnson JK, Jabra-Rizk MA (2016) Community-Associated Methicillin-Resistant Staphylococcus aureus: an enemy amidst Us. PLoS Pathog 12:e1005837. https://doi.org/10.1371/journal.ppat.1005837
Kotnik M, Humljan J, Contreras-Martel C, Oblak M, Kristan K, Herve M, Blanot D, Urleb U, Gobec S, Dessen A, Solmajer T (2007) Structural and functional characterization of enantiomeric glutamic acid derivatives as potential transition state analogue inhibitors of MurD ligase. J Mol Biol 370:107–115. https://doi.org/10.1016/j.jmb.2007.04.048
Lanzetta PA, Alvarez LJ, Reinach PS, Candia O (1979) An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem 100:95–97. https://doi.org/10.1016/0003-2697(79)90115-5
Lawrence CP, Skinner JL (2003) Flexible TIP4P model for molecular dynamics simulation of liquid water. Chem Phys Lett 372:842–847. https://doi.org/10.1016/S0009-2614(03)00526-8
Li J, Abel R, Zhu K, Cao Y, Zhao S, Friesner RA (2011) The VSGB 2.0 Model: a next generation energy model for high resolution protein structure modelling. Proteins 79:2794–2812. https://doi.org/10.1002/prot.23106
Liu G, Gao J (2012) 2-(4-methoxyphenoxy)acetohydrazide. Acta Crystallogr E Crystallogr Comm 68:1969. https://doi.org/10.1107/S16005368120243
Macmorran E, Harch S, Athan E, Lane S, Tong S, Crawford L, Krishnaswamy S, Hewagama S (2017) The rise of methicillin resistant Staphylococcus aureus: now the dominant cause of skin and soft tissue infection in Central Australia. Epidemiol Infect 145:2817–2826. https://doi.org/10.1017/S0950268817001716
Martyna GJ, Klein ML, Tuckerman M (1992) Nose-Hoover chains: The canonical ensemble via continuous dynamics. J Chem Phys 97:2635–2643. https://doi.org/10.1063/1.463940
Martyna GJ, Tobias DJ, Klein ML (1994) Constant-pressure molecular dynamics algorithms. J Chem Phys 101:4177–4189. https://doi.org/10.1063/1.467468
Perdih A, Kotnik M, Hodoscek M, Solmajer T (2007) Targeted molecular dynamics simulation studies of binding and conformational changes in E. coli MurD. Proteins 68:243–254. https://doi.org/10.1002/prot.21374
Perdih A, Bren U, Solmajer T (2009) Binding free energy calculations of N-sulphonyl-glutamic acid inhibitors of MurD ligase. Mol Model 15:983–996. https://doi.org/10.1021/jm800762u
Perdih A, Wolber G, Solmajer T (2013) Molecular dynamics simulation and linear interaction energy study of D-Glu-based inhibitors of the MurD ligase. J Comput Aided Mol Des 27:723–738. https://doi.org/10.1007/s10822-013-9673-3
Perdih A, Hrast M, Barreteau H, Gobec S, Wolber G, Solmajer T (2014) Benzene-1,3-dicarboxylic acid 2,5-dimethylpyrrole derivatives as multiple inhibitors of bacterial Mur ligases (MurC-MurF). Bioorg Med Chem 22:4124–4134. https://doi.org/10.1016/j.bmc.2014.05.058
Pratviel-Sosa F, Acher F, Trigalo F, Blanot D, Azerad R, van Heijenoort J (1994) Effect of various analogues of D-glutamic acid on the D-glutamate-adding enzyme from Escherichia coli. FEMS Microbiol Lett 115:223–228. https://doi.org/10.1111/j.1574-6968.1994.tb06642.x
Roos K, Wu C, Damm W, Reboul M, Stevenson JM, Lu C, Dahlgren MK, Mondal S, Chen W, Wang L, Abel R, Friesner RA, Harder ED (2019) OPLS3e: Extending force field coverage for drug-like small molecules. J Chem Theory Comput 15:1863–1874. https://doi.org/10.1021/acs.jctc.8b01026
Sastry MG, Adzhigirey M, Day T, Annabhimoju R, Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27:221–234. https://doi.org/10.1007/s10822-013-9644-8
Simcic M, Pureber K, Kristan K, Urleb U, Kocjan D, Grdadolnik SG (2014) A novel 2-oxoindolinylidene inhibitor of bacterial MurD ligase: enzyme kinetics, protein-inhibitor binding by NMR and a molecular dynamics study. Eur J Med Chem 83:92–101. https://doi.org/10.1016/j.ejmech.2014.06.021
Smith CA (2006) Structure, function and dynamics in the mur family of bacterial cell wall ligases. J Mol Biol 362:640–655. https://doi.org/10.1016/j.jmb.2006.07.066
Sosic I, Barreteau H, Simcic M, Sink R, Cesar J, Zega A, Grdadolnik SG, Contreras-Martel C, Dessen A, Amoroso A, Joris B, Blanot D, Gobec S (2011) Second-generation sulfonamide inhibitors of D-glutamic acid-adding enzyme: activity optimisation with conformationally rigid analogues of D-glutamic acid. Eur J Med Chem 46:2880–2894. https://doi.org/10.1016/j.ejmech.2011.04.011
Sova M, Kovac A, Turk S, Hrast M, Blanot D, Gobec S (2009) Phosphorylated hydroxyethylamines as novel inhibitors of the bacterial cell wall biosynthesis enzymes MurC to MurF. Bioorg Chem 37:217–222. https://doi.org/10.1016/j.bioorg.2009.09.001
Strancar K, Blanot D, Gobec S (2006) Design, synthesis and structure–activity relationships of new phosphinate inhibitors of MurD. Bioorg Med Chem Lett 16:343–348. https://doi.org/10.1016/j.bmcl.2005.09.086
Tomasic T, Sink R, Zidar N, Fic A, Contreras-Martel C, Dessen A, Patin D, Blanot D, Premru-Mllerm M, Gobec S, Zega A, Kikelj D, Masic LP (2012) Dual inhibitor of MurD and MurE ligases from Escherichia coli and Staphylococcus aureus. ACS Med Chem Lett 3:626–630. https://doi.org/10.1021/ml300047h
Turk S, Kovac A, Boniface A, Bostock JM, Chopra I, Blanot D, Gobec S (2009) Discovery of new inhibitors of the bacterial peptidoglycan biosynthesis enzymes MurD and MurF by structure-based virtual screening. Bioorg Med Chem 17:1884–1889. https://doi.org/10.1016/j.bmc.2009.01.052
Walsh AW, Falk PJ, Thanassi J, Discotto L, Pucci MJ, Ho HT (1999) Comparison of the D-glutamate-adding enzymes from selected gram-positive and gram-negative bacteria. J Bacteriol 181:5395–5401. https://doi.org/10.1128/JB.181.17.5395-5401.1999
Zidar N, Tomasic T, Sink R, Rupnik V, Kovac A, Turk S, Patin D, Blanot D, Contreras Martel C, Dessen A, Muller Premru M, Zega A, Gobec S, Masic L, Kikelj D (2010) Discovery of novel 5-benzylidenerhodanine and 5-benzylidenethiazolidine-2,4-dione inhibitors of MurD ligase. J Med Chem 53:6584–6594. https://doi.org/10.1021/jm100285g
Zidar N, Tomasic T, Sink R, Kovac A, Patin D, Blanot D, Contreras-Martel C, Dessen A, Premru MM, Zega A, Gobec S, Masic LP, Kikelj D (2011) New 5-benzylidenethiazolidin-4-one inhibitors of bacterial MurD ligase: Design, synthesis, crystal structures, and biological evaluation. Eur J Med Chem 46:5512–5523. https://doi.org/10.1016/j.ejmech.2011.09.017
Acknowledgements
We would like to thank the Science and Engineering Research Board (SERB), Government of India for the financial support (No. EMR/2016/002981).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary file 2 (MPG 85856 kb)
Supplementary file 3 (MPEG 13856 kb)
Rights and permissions
About this article

Cite this article
Jupudi, S., Azam, M.A. & Wadhwani, A. Design, synthesis and molecular modelling of phenoxyacetohydrazide derivatives as Staphylococcus aureus MurD inhibitors. Chem. Pap. 75, 1221–1235 (2021). https://doi.org/10.1007/s11696-020-01380-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-020-01380-2