
Abstract
In this work, 3-amino-4-cyano-2-thiophenecarboxamides 1a-k were used as versatile synthons for the preparation of thieno[3,2-d]pyrimidine-7-carbonitriles 2a-k and 4a-d as well as the unexpectedly prepared thieno[3,4-b]pyridine-7-carboxamides 5a-e. Thus, heating thiophene-2-carboxamides 1a-k in formic acid afforded thieno[3,2-d]pyrimidin-4-ones 2a-k. Alternatively, the reaction of compounds 1a-i with 2,2,6-trimethyl-4H-1,3-dioxin-4-one (TMD) in xylene produced the β-keto amides 3a-i. The latter were cyclized to thienopyrimidine-2,4-diones 4a-d by heating with pyrrolidine in toluene using calcium chloride as a desiccant. While refluxing a mixture of β-keto amide derivatives 3a, 3d, 3e, 3f or 3 h and potassium carbonate in ethanol or in ethylene glycol afforded the thieno[3,4-b]pyridine derivatives 5a-e.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Thieno[3,2-d]pyrimidines represent an important class of chemical compounds with diverse biological activities (Abdel-Megid et al. 2016; Ibrahim et al. 1996; Litvinov, 2004, 2006). The most common synthetic methods of thieno[3,2-d]pyrimidin-4-ones, involved cyclization of 3-amino-thiophene-2-carboxylate derivatives using a one-carbon source reagent such as formic acid, triethyl orthoformate (El-Telbany and Hutchins 1985; Hafez et al. 2017; Kim et al. 2014, 2015; Sasikumar et al. 2009), or dimethylformamide dimethylacetal (DMF-DMA); in addition to a primary amine (Ahmad et al. 2016; Brumfield et al. 2012; Carpenter et al. 2006; Sasikumar et al. 2009; Tavares et al. 2006; Warshakoon et al. 2006; Washburn et al. 2014; Zhang et al. 2007; Zheng et al. 2005). Alternatively, the synthesis of thienopyrimidine-4-one derivatives can be achieved by heating 3-amino-thiophene-2-carboxamides either with triethyl orthoformate (El-Hamed et al. 2001; Sasikumar et al. 2010) or formic acid (Ahmed2008; Hertzog et al. 2006; Ryndina et al. 2002).
On the other hand, literature reported the synthesis of thieno[3,2-d]pyrimidine-2,4-dione derivatives via pyrimidine ring closure and were almost exclusively conducted using 2-amino-thiophene-3-carboxylate derivatives as the reaction substrate. (Bakhite 2003; Ibrahim et al. 1996; Litvinov 2004, 2006; Varvounis and Giannopoulos 1996) One of the synthetic strategies that offered a direct route to the key ureido intermediates was based on the reaction of thiophene derivatives with isocyanates. Subsequent base-promoted cyclization of the intermediates yielded the target thienopyrimidines (Fukumi et al. 1989; Meyer et al. 1997, 2001; Press et al. 1991; Romeo et al. 2006; Russell et al. 1988; Shestakov et al. 2014; Sugiyama et al. 1989).
Literature review revealed that limited reports described the synthesis of thieno[3,4-b]pyridine derivatives, which may be attributed to the difficulty of synthesis of their starting precursors (Bakhite 2003; Barker 1977; Björk et al. 1994; Klemm et al. 1970; Litvinov et al. 2005). The first synthetic route to thieno[3,4-b]pyridine was reported by Klemm et al. in 1970. This method was based upon the cyclization of bis-chloromethylpyridine to the dihydrothienopyridine using sodium sulfide, followed by oxidation of the latter with iodobenzene dichloride to the sulfoxide derivative. Dehydration and subsequent aromatization of the latter with alumina afforded thieno[3,4-b]pyridine ring (Klemm et al. 1970).
Recently, new tetrasubstituted thiophene derivatives have been successively prepared by our team (El-Meligie et al. 2020). The method developed by us was a modification of a previously reported procedure by Thomae et al.(2009). The modification consisted in separation of the enethiolate intermediate, which was further reacted with only one molar equivalent of the α-haloketone. The reaction was conducted in green solvents (ethanol, water, or a mixture of them) using mild bases (K2CO3 or TEA). Following this procedure, three series of tetrasubstituted thiophenes were reported in high yields and in pure forms. Among them, 3-amino-4-cyano-2-thiophenecarboxamides 1a-k were documented for the first time.
In continuation to this work, herein we reported the preparation of thieno[3,2-d]pyrimidine-7-carbonitriles 2a–k and 4a–d as well as thieno[3,4-b]pyridine-7-carboxamides 5a-e from 3-amino-4-cyano-2-thiophenecarboxamides 1a–k. Moreover, two new procedures were developed for the synthesis of two series of thieno[3,2-d]pyrimidine-2,4-diones 4a-d and thieno[3,4-b]pyridines 5a-e starting with the β-keto amide derivatives 3.
Experimental
The melting points were determined using Stuart SMP20 apparatus and are uncorrected. The IR spectra were performed using Shimadzu IR-435 spectrophotometer and the values are represented in cm−1. Bruker AVANCETM III 400 MHz spectrophotometer was used for the acquisition of 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra of the new compounds using DMSO-d6 as a solvent. Nuclear magnetic resonance spectra were processed on MestreNova 11.0.4 program using residual solvent peak as the internal standard. Chemical shifts and coupling constants are presented in ppm and in Hz, respectively. Both IR and NMR spectra were carried out at Faculty of Pharmacy, Cairo University, Cairo, Egypt. Elemental analyses were carried out at The Regional Center for Mycology and Biotechnology, Al-Azhar University, Cairo, Egypt. The preparation of thiophene-2-carboxamides 1a–k was performed as previously reported (El-Meligie et al. 2020).
4-Oxo-3-(substituted) phenyl-6-(4-phenylpiperazin-1-yl)-3,4-dihydrothieno[3,2-d] pyrimidine-7-carbonitriles 2a-k
A suspension of thiophene-2-carboxamides 1a–k (2 mmol) in formic acid (15 mL) was heated under reflux with continuous stirring for 5 h. The reaction mixture was then cooled to room temperature, poured onto ice-water (100 mL), then stirred for 30 min. The precipitated solid was filtered, washed with water (2 × 20 mL), then with ethanol (2x20 mL), oven-dried until a constant weight, and crystallized from the suitable solvent.
4-Oxo-3-phenyl-6-(4-phenylpiperazin-1-yl)-3,4-dihydrothieno[3,2-d]pyrimidine-7-carbonitrile (2a)
Yield: 98%, crystallized from toluene; mp 285–286 °C; IR (cm−1): 2210 (C ≡ N); 1678 (C = O). 1H NMR (ppm): 3.41 (t, 4H, J = 5.0 Hz, CH2); 3.91 (t, 4H, J = 5.0 Hz, CH2); 6.82–7.58 (m, 10H, ArH); 8.45 (s, 1H, = CH). 13C NMR (ppm): 47.2, 49.9 (piperazine Cs); 80.7, 107.1, 115.1, 115.7, 119.4, 127.4, 128.9, 129.0, 129.1, 136.8, 150.0, 150.6, 154.4, 158.1, 167.9 (Ar. Cs, C ≡ N, and C = O). Anal Calcd for C23H19N5OS (413.49): C, 66.81; H, 4.63; N, 16.94. Found: C, 67.09; H, 4.78; N, 17.19.
3-(4-Methylphenyl)-4-oxo-6-(4-phenylpiperazin-1-yl)- 3,4-dihydrothieno[3,2-d]pyrimidine-7-carbonitrile (2b)
Yield: 95%, crystallized from ethanol–toluene (1:1) mixture; mp 263-264 °C;IR (cm−1): 2214 (C ≡ N); 1678 (C = O). 1H NMR (ppm): 2.38 (s, 3H, CH3); 3.39-3.41 (m, 4H, CH2); 3.88–3.90 (m, 4H, CH2); 6.82–7.28 (m, 5H, ArH); 7.34 (d, 2H, J = 8.0 Hz, ArH); 7.39 (d, 2H, J = 8.0 Hz, ArH); 8.39 (s, 1H, = CH). 13C NMR (ppm): 20.6 (CH3); 47.2, 49.9 (piperazine Cs); 80.7, 107.1, 115.1, 115.7, 119.4, 127.1, 128.1, 129.0, 129.5, 134.2, 138.6, 150.0, 154.4, 158.0, 167.8 (Ar. Cs, C ≡ N, and C = O). Anal Calcd for C24H21N5OS (427.52): C, 67.43; H, 4.95; N, 16.38. Found: C, 67.68; H, 5.03; N, 16.54.
3-(4-Ethylphenyl)-4-oxo-6-(4-phenylpiperazin-1-yl)-3,4-dihydrothieno[3,2-d]pyrimidine-7-carbonitrile (2c)
Yield: 86%, crystallized from ethanol–toluene (1:1) mixture; mp 253–254 °C;IR (cm−1): 2210 (C ≡ N); 1678 (C = O). 1H NMR (ppm): 1.22 (t, 3H, J = 7.6 Hz, CH3); 2.68 (q, 2H, J = 7.6 Hz, CH2); 3.39–3.41 (m, 4H, CH2); 3.87–3.89 (m, 4H, CH2); 6.82–7.27 (m, 5H, ArH); 7.37 (d, 2H, J = 8.0 Hz, ArH); 7.42 (d, 2H, J = 8.0 Hz, ArH), 8.40 (s, 1H, = CH). 13C NMR (ppm): 15.5 (CH3); 27.8 (CH2); 47.2, 49.9 (piperazine Cs); 80.7, 107.2, 115.1, 115.7, 119.4, 127.2, 128.4, 129.0, 134.4, 144.8, 150.0, 150.7, 154.5, 158.1, 167.9 (Ar. Cs, C ≡ N, and C = O). Anal Calcd for C25H23N5OS (441.55): C, 68.00; H, 5.25; N, 15.86. Found: C, 67.92; H, 5.41; N, 16.04.
3-(2-Chlorophenyl)-4-oxo-6-(4-phenylpiperazin-1-yl)-3,4-dihydrothieno[3,2-d]pyrimidine-7-carbonitrile (2d)
Yield: 83%, crystallized from toluene; mp 266-267 °C; IR (cm−1): 2210 (C ≡ N); 1670 (C = O). 1H NMR (ppm): 3.41 (t, 4H, J = 5.0 Hz, CH2); 3.91 (t, 4H, J = 5.0 Hz, CH2); 6.82–7.74 (m, 9H, ArH); 8.44 (s, 1H, = CH). 13C NMR (ppm): 47.2, 50.0 (piperazine Cs); 80.9, 106.8, 115.0, 115.7, 119.4, 128.3, 129.0, 130.0, 130.5, 131.4, 131.4, 134.3, 150.0, 150.7, 153.8, 158.3, 168.0 (Ar. Cs, C ≡ N, and C = O). Anal Calcd for C23H18ClN5OS (447.94): C, 61.67; H, 4.05; N, 15.63. Found: C, 61.88; H, 4.23; N, 15.88.
3-(4-Chlorophenyl)-4-oxo-6-(4-phenylpiperazin-1-yl)-3,4-dihydrothieno[3,2-d]pyrimidine-7-carbonitrile (2e)
Yield: 88%, crystallized from toluene; mp 284-285 °C;IR (cm−1): 2210 (C ≡ N); 1670 (C = O). 1H NMR (ppm): 3.40-3.42 (m, 4H, CH2); 3.89–3.91 (m, 4H, CH2); 6.82–7.28 (m, 5H, ArH); 7.57 (d, 2H, J = 8.5 Hz, ArH); 7.63 (d, 2H, J = 8.5 Hz, ArH); 8.44 (s, 1H, = CH). 13C NMR (ppm): 47.2, 49.9 (piperazine Cs); 80.7, 107.0, 115.7, 119.4, 129.0, 129.1, 129.3, 133.6, 135.6, 150.0, 150.4, 154.2, 158.1, 160.1, 167.8 (Ar. Cs, C ≡ N, and C = O). Anal Calcd for C23H18ClN5OS (447.94): C, 61.67; H, 4.05; N, 15.63. Found: C, 61.92; H, 4.18; N, 15.79.
3-(3-Nitrophenyl)-4-oxo-6-(4-phenylpiperazin-1-yl)-3,4-dihydrothieno[3,2-d]pyrimidine-7-carbonitrile (2f)
Yield: 85%, crystallized from acetic acid; mp 277-278 °C;IR (cm−1): 2206 (C ≡ N); 1678 (C = O); 1527, 1350 (NO2). 1H NMR (ppm): 3.40–3.42 (m, 4H, CH2); 3.90–3.92 (m, 4H, CH2); 6.82–8.53 (m, 10H, 9 ArH and = CH). 13C NMR (ppm): 47.2, 50.0 (piperazine Cs); 80.7, 106.9, 115.1, 115.7, 119.4, 122.9, 123.8, 129.0, 130.5, 134.3, 137.6, 147.8, 150.0, 150.3, 154.2, 158.2, 167.9 (Ar. Cs, C ≡ N, and C = O). Anal. Calcd for C23H18N6O3S (458.49): C, 60.25; H, 3.96; N, 18.33. Found: C, 60.44; H, 4.01; N, 18.49.
3-(2,4-Dichlorophenyl)-4-oxo-6-(4-phenylpiperazin-1-yl)-3,4-dihydrothieno[3,2-d]pyrimidine-7-carbonitrile (2 g)
Yield: 85%, crystallized from toluene; mp 268–269 °C; IR (cm−1): 2210 (C ≡ N); 1678 (C = O). 1H NMR (ppm): 3.41 (t, 4H, J = 5.0 Hz, CH2); 3.91 (t, 4H, J = 5.0 Hz, CH2); 6.82–7.95 (m, 8H, ArH); 8.45 (s, 1H, = CH). 13C NMR (ppm): 47.2, 50.0 (piperazine Cs); 80.9, 106.7, 115.0, 115.7, 119.4, 128.5, 129.0, 129.5, 131.8, 132.8, 133.4, 135.2, 150.0, 150.5, 153.6, 158.4, 168.0 (Ar. Cs, C ≡ N, and C = O). Anal Calcd for C23H17Cl2N5OS (482.38): C, 57.27; H, 3.55; N, 14.52. Found: C, 57.43; H, 3.72; N, 14.68.
3-(2-Bromophenyl)-4-oxo-6-(4-phenylpiperazin-1-yl)-3,4-dihydrothieno[3,2-d]pyrimidine-7-carbonitrile (2 h)
Yield: 85%, crystallized from toluene; mp 280–281 °C;IR (cm−1): 2210 (C ≡ N); 1670 (C = O). 1H NMR (ppm): 3.41 (t, 4H, J = 5.0 Hz, CH2); 3.92 (t, 4H, J = 5.0 Hz, CH2); 6.82–7.89 (m, 9H, ArH); 8.43 (s, 1H, = CH). 13C NMR (ppm): 47.2, 50.0 (piperazine Cs); 80.8, 106.9, 115.0, 115.7, 119.4, 122.0, 128.9, 129.0, 130.6, 131.5, 133.1, 135.9, 150.0, 150.7, 153.7, 158.3, 168.0 (Ar. Cs, C ≡ N, and C = O). Anal Calcd for C23H18BrN5OS (492.39): C, 56.10; H, 3.68; N, 14.22. Found: C, 56.42; H, 3.75; N, 14.34.
N-(3-(7-Cyano-4-oxo-6-(4-phenylpiperazin-1-yl)thieno[3,2-d]pyrimidin-3(4H)-yl)phenyl)benzamide (2i)
Yield: 88%, crystallized from ethyl acetate–methanol (1:1); mp 265–266 °C; IR (cm−1): 3302 (NH); 2210 (C ≡ N); 1662 (C = O). 1H NMR (ppm): 3.39–3.41 (m, 4H, CH2); 3.89–3.91 (m, 4H, CH2); 6.81–8.03 (m, 14H, ArH); 8.47 (s, 1H, = CH); 10.52 (s, 1H, NH, D2O exchangeable). 13C NMR (ppm): 47.3, 49.9 (piperazine Cs); 80.7, 107.1, 115.1, 115.7, 119.0, 119.4, 120.6, 122.5, 127.6, 128.4, 129.0, 129.4, 131.7, 134.5, 136.9, 139.9, 150.0, 150.4, 154.3, 158.1, 165.7, 167.8 (Ar. Cs, C ≡ N, and C = O). Anal. Calcd for C30H24N6O2S (532.62): C, 67.65; H, 4.54; N, 15.78. Found: C, 67.23; H, 4.19; N, 15.42.
N-(3-(7-Cyano-4-oxo-6-(4-phenylpiperazin-1-yl)thieno[3,2-d]pyrimidin-3(4H)-yl)-4-methylphenyl)benzamide (2j)
Yield: 96%, crystallized from ethyl acetate–methanol (1:1); mp 179-180 °C; IR (cm−1): 3479 (NH); 2210 (C ≡ N); 1678, 1651 (2 C = O). 1H NMR (ppm): 2.06 (s, 3H, CH3); 3.40–3.42 (m, 4H, CH2); 3.91–3.93 (m, 4H, CH2); 6.82–7.97 (m, 13H, ArH); 8.44 (s, 1H, = CH); 10.44 (s, 1H, NH, D2O exchangeable). 13C NMR (ppm): 16.6 (CH3); 47.3, 50.0 (piperazine Cs); 80.8, 107.2, 115.1, 115.7, 119.5, 119.8, 121.3, 127.6, 128.4, 129.0, 130.4, 130.8, 131.7, 134.6, 135.8, 138.0, 150.0, 150.7, 154.0, 158.4, 165.5, 168.0 (Ar. Cs, C ≡ N, and C = O). Anal. Calcd for C31H26N6O2S (546.65): C, 68.11; H, 4.79; N, 15.37. Found: C, 67.85; H, 4.53; N, 15.52.
N-(3-(7-cyano-4-oxo-6-(4-phenylpiperazin-1-yl)thieno[3,2-d]pyrimidin-3(4H)-yl)-4-methoxyphenyl)benzamide (2 k)
Yield: 80%, crystallized from ethyl acetate–methanol (1:1); mp 250–251 °C; IR (cm−1): 3317 (NH); 2210 (C ≡ N); 1662 (C = O). 1H NMR (ppm): 3.39–3.41 (m, 4H, CH2); 3.78 (s, 3H, OCH3); 3.90–3.92 (m, 4H, CH2); 6.81–7.98 (m, 13H, ArH); 8.38 (s, 1H, = CH); 10.37 (s, 1H, NH, D2O exchangeable). 13C NMR (ppm): 47.3, 50.0 (piperazine Cs); 56.1 (OCH3); 80.9, 107.2, 112.6, 115.1, 115.7, 119.4, 121.5, 122.8, 124.6, 127.5, 128.4, 129.0, 131.5, 132.3, 134.6, 150.0, 150.6, 151.4, 154.1, 158.2, 165.3, 167.9 (Ar. Cs, C ≡ N, and C = O). Anal. Calcd for C31H26N6O3S (562.65): C, 66.18; H, 4.66; N, 14.94. Found: C, 65.89; H, 4.35; N, 14.56.
4-Cyano-3-(3-oxobutanamido)-N-(substituted) phenyl-5-(4-phenylpiperazin-1-yl)thiophene-2-carboxamides 3a-i
A mixture of the appropriate thiophenecarboxamides 1a-i (7 mmol), and TMD (1.11 g, 1 mL, 7.78 mmol) in xylene (25 mL) was heated under reflux for 15 min in a 50-mL round bottom flask fitted with an air condenser. The reaction mixture was then cooled to room temperature, poured onto ethanol (100 mL), and stirred for 30 min. The solid product was filtered, washed with ethanol (2 × 20 mL), oven-dried until a constant weight, and crystallized from ethanol–acetone (1:1) mixture.
4-Cyano-3-(3-oxobutanamido)-N-phenyl-5-(4-phenylpiperazin-1-yl)thiophene-2-carboxamide (3a)
Yield: 86%, mp 212–214 °C; IR (cm−1): 3348, 3170 (2 NH); 2210 (C ≡ N); 1716, 1701, 1693 (3 C = O). 1H NMR (ppm): 2.24 (s, 3H, CH3); 3.36–3.38 (m, 4H, CH2); 3.69 (s, 2H, COCH2); 3.72 (t, 4H, J = 5.0 Hz, CH2); 6.82–7.66 (m, 10H, ArH); 9.34 (s, 1H, NH, D2O exchangeable); 10.36 (s, 1H, NH, D2O exchangeable). 13C NMR (ppm): 30.1 (CH3); 47.4, 49.6 (piperazine Cs); 51.4 (COCH2); 87.0, 114.1, 115.0, 115.8, 119.5, 120.7, 124.0, 128.6, 129.0, 135.5, 138.0, 150.2, 158.3, 164.4, 166.0 (Ar. Cs, C ≡ N, and 2 amide C = O); 203.3 (C = O). Anal Calcd for C26H25N5O3S (487.57): C, 64.05; H, 5.17; N, 14.36. Found: C, 64.32; H, 5.03; N, 14.62.
4-Cyano-N-(4-methylphenyl)-3-(3-oxobutanamido)-5-(4-phenylpiperazin-1-yl) thiophene-2-carboxamide (3b)
Yield: 70%, mp 204–206 °C;IR (cm−1): 3402, 3240 (2 NH); 2202 (C ≡ N); 1724, 1658, 1651 (3 C = O). 1H NMR (ppm): 2.24 (s, 3H, CH3); 2.27 (s, 3H, CH3); 3.33–3.35 (m, 4H, CH2); 3.69 (s, 2H, COCH2); 3.72 (t, 4H, J = 5.0 Hz, CH2); 6.82–7.54 (m, 9H, ArH); 9.26 (s, 1H, NH, D2O exchangeable); 10.34 (s, 1H, NH, D2O exchangeable). 13C NMR (ppm): 20.4 (CH3); 30.1 (CH3CO); 47.4, 49.5 (piperazine Cs); 51.4 (COCH2); 87.0, 114.4, 115.0, 115.8, 119.5, 120.1, 120.8, 129.0, 133.1, 135.3, 135.5, 150.2, 158.1, 164.4, 166.1 (Ar. Cs, C ≡ N, and 2 amide C = O); 203.3 (C = O). Anal Calcd for C27H27N5O3S (501.60): C, 64.65; H, 5.43; N, 13.96. Found: C, 64.91; H, 5.48; N, 14.17.
4-Cyano-N-(4-ethylphenyl)-3-(3-oxobutanamido)-5-(4-phenylpiperazin-1-yl)thiophene-2-carboxamide (3c)
Yield: 87%, mp 196–198 °C;IR (cm−1): 3348, 3147 (2 NH); 2210 (C ≡ N); 1720, 1693, 1627 (3 C = O). 1H NMR (ppm): 1.17 (t, 3H, J = 7.6 Hz, CH3CH2); 2.24 (s, 3H, CH3CO); 2.57 (q, 2H, J = 7.6 Hz, CH3CH2); 3.35–3.37 (m, 4H, CH2); 3.69 (s, 2H, COCH2); 3.72 (t, 4H, J = 5.0 Hz, CH2); 6.82–7.55 (m, 9H, ArH); 9.27 (s, 1H, NH, D2O exchangeable); 10.34 (s, 1H, NH, D2O exchangeable). 13C NMR (ppm): 15.6 (CH3CH2); 27.6 (CH3CH2); 30.1 (CH3CO); 47.4, 49.5 (piperazine Cs); 51.4 (COCH2); 87.0, 114.4, 115.0, 115.8, 119.5, 120.9, 127.8, 129.0, 135.3, 135.7, 139.5, 150.2, 158.1, 164.4, 166.1 (Ar. Cs, C ≡ N, and 2 amide C = O); 203.3 (C = O). Anal Calcd for C28H29N5O3S (515.63): C, 65.22; H, 5.67; N, 13.58. Found: C, 65.38; H, 5.81; N, 13.69.
N-(2-Chlorophenyl)-4-cyano-3-(3-oxobutanamido)-5-(4-phenylpiperazin-1-yl)thiophene-2-carboxamide (3d)
Yield: 79%, mp 176–178 °C; IR (cm−1): 3394, 3217 (2 NH); 2206 (C ≡ N); 1735, 1666, 1651 (3 C = O). 1H NMR (ppm): 2.22 (s, 3H, CH3); 3.35–3.37 (m, 4H, CH2); 3.67 (s, 2H, COCH2); 3.75 (t, 4H, J = 5.0 Hz, CH2); 6.82–7.91 (m, 9H, ArH); 9.17 (s, 1H, NH, D2O exchangeable); 10.45 (s, 1H, NH, D2O exchangeable). 13C NMR (ppm): 30.0 (CH3); 47.3, 49.5 (piperazine Cs); 51.3 (COCH2); 86.8, 114.4, 114.9, 115.8, 119.5, 125.0, 125.9, 126.4, 127.7, 129.0, 129.4, 134.2, 136.1, 150.2, 158.2, 164.6, 166.6 (Ar. Cs, C ≡ N, and 2 amide C = O); 202.3 (C = O). Anal Calcd for C26H24ClN5O3S (522.02): C, 59.82; H, 4.63; N, 13.42. Found: C, 60.07; H, 4.72; N, 13.60.
N-(4-Chlorophenyl)-4-cyano-3-(3-oxobutanamido)-5-(4-phenylpiperazin-1-yl)thiophene-2-carboxamide (3e)
Yield: 85%, mp 222–224 °C; IR (cm−1): 3298, 3190 (2 NH); 2214 (C ≡ N); 1712, 1678, 1639 (3 C = O). 1H NMR (ppm): 2.24 (s, 3H, CH3); 3.35–3.37 (m, 4H, CH2); 3.68 (s, 2H, COCH2); 3.71-3.73 (m, 4H, CH2); 6.82–7.27 (m, 5H, ArH); 7.41 (d, 2H, J = 8.8 Hz, ArH); 7.68 (d, 2H, J = 8.8, ArH); 9.47 (s, 1H, NH, D2O exchangeable); 10.35 (s, 1H, NH, D2O exchangeable). 13C NMR (ppm): 30.0 (CH3); 47.4, 49.6 (piperazine Cs); 51.4 (COCH2); 87.0, 113.3, 114.9, 115.8, 119.5, 122.2, 127.6, 128.5, 129.0, 135.9, 137.1, 150.2, 158.4, 164.5, 165.9 (Ar. Cs, C ≡ N, and 2 amide C = O); 203.3 (C = O). Anal Calcd for C26H24ClN5O3S (522.02): C, 59.82; H, 4.63; N, 13.42. Found: C, 59.96; H, 4.78; N, 13.67.
4-Cyano-N-(3-nitrophenyl)-3-(3-oxobutanamido)-5-(4-phenylpiperazin-1-yl)thiophene-2-carboxamide (3f)
Yield: 90%, mp 216–218 °C; IR (cm−1): 3367, 3259 (2 NH); 2206 (C ≡ N); 1724, 1670, 1643 (3 C = O); 1527, 1354 (NO2). 1H NMR (ppm): 2.24 (s, 3H, CH3); 3.36–3.38 (m, 4H, CH2); 3.69 (s, 2H, COCH2); 3.74 (t, 4H, J = 5.0 Hz, CH2); 6.82–8.65 (m, 9H, ArH); 9.82 (s, 1H, NH, D2O exchangeable); 10.38 (s, 1H, NH, D2O exchangeable). 13C NMR (ppm): 30.1 (CH3); 47.4, 49.6 (piperazine Cs); 51.4 (COCH2); 87.0, 112.1, 114.8, 114.9, 115.8, 118.4, 119.5, 126.6, 129.0, 130.0, 136.8, 139.4, 147.8, 150.2, 158.9, 164.7, 165.8 (Ar. Cs, C ≡ N, and 2 amide C = O); 203.4 (C = O). Anal Calcd for C26H24N6O5S (532.57): C, 58.64; H, 4.54; N, 15.78. Found: C, 58.78; H, 4.67; N, 16.04.
4-Cyano-N-(2,4-dichlorophenyl)-3-(3-oxobutanamido)-5-(4-phenylpiperazin-1-yl)thiophene-2-carboxamide (3 g)
Yield: 69%, mp 213-215 °C; IR (cm−1): 3317, 3221 (2 NH); 2210 (C ≡ N); 1728, 1666, 1635 (3 C = O). 1H NMR (ppm): 2.22 (s, 3H, CH3); 3.35–3.37 (m, 4H, CH2); 3.67 (s, 2H, COCH2); 3.75 (t, 4H, J = 5.0 Hz, CH2); 6.81–7.91 (m, 8H, ArH); 9.20 (s, 1H, NH, D2O exchangeable); 10.45 (s, 1H, NH, D2O exchangeable). 13C NMR (ppm): 30.0 (CH3); 47.3, 49.5 (piperazine Cs); 51.3 (COCH2); 86.8, 114.9, 115.8, 119.5, 126.2, 127.1, 127.8, 128.7, 128.9, 129.0, 129.6, 133.5, 136.4, 150.1, 158.2, 164.6, 166.6 (Ar. Cs, C ≡ N, and 2 amide C = O); 202.5 (C = O). Anal Calcd for C26H23Cl2N5O3S (556.46): C, 56.12; H, 4.17; N, 12.59. Found: C, 56.40; H, 4.23; N, 12.65.
N-(2-Bromophenyl)-4-cyano-3-(3-oxobutanamido)-5-(4-phenylpiperazin-1-yl)thiophene-2-carboxamide (3 h)
Yield: 73%, mp 182–183 °C; IR (cm−1): 3344, 3232 (2 NH); 2210 (C ≡ N); 1724, 1689, 1658 (3 C = O). 1H NMR (ppm): 2.23 (s, 3H, CH3), 3.34–3.36 (m, 4H, CH2); 3.69 (s, 2H, COCH2); 3.75 (t, 4H, J = 5.0 Hz, CH2); 6.82–7.87 (m, 9H, ArH); 9.13 (s, 1H, NH, D2O exchangeable); 10.44 (s, 1H, NH, D2O exchangeable). 13C NMR (ppm): 30.1 (CH3); 47.3, 49.5 (piperazine Cs); 51.4 (COCH2); 86.9, 114.3, 114.9, 115.8, 116.9, 119.5, 125.6, 126.9, 128.2, 129.0, 132.6, 135.6, 136.2, 150.1, 158.2, 164.6, 166.6 (Ar. Cs, C ≡ N, and 2 amide C = O); 202.4 (C = O). Anal Calcd for C26H24BrN5O3S (566.47): C, 55.13; H, 4.27; N, 12.36. Found: C, 55.01; H, 4.39; N, 12.19.
N-(3-Benzamidophenyl)-4-cyano-3-(3-oxobutanamido)-5-(4-phenylpiperazin-1-yl)thiophene-2-carboxamide (3i)
Yield: 86%, mp 210–212 °C; IR (cm−1): 3383, 3275, 3248 (3 NH); 2206 (C ≡ N); 1712, 1674, 1643, 1608 (4 C = O). 1H NMR (ppm): 2.25 (s, 3H, CH3); 3.34–3.36 (m, 4H, CH2); 3.69 (s, 2H, COCH2); 3.72–3.74 (m, 4H, CH2); 6.82–8.23 (m, 14H, ArH); 9.42 (s, 1H, NH, D2O exchangeable); 10.32 (s, 1H, NH, D2O exchangeable); 10.39 (s, 1H, NH, D2O exchangeable). 13C NMR (ppm): 30.0 (CH3); 47.4, 49.6 (piperazine Cs); 51.4 (COCH2); 87.0, 113.2, 114.1, 115.0, 115.8, 116.3, 119.5, 127.6, 128.3, 128.6, 129.0, 131.5, 134.9, 135.5, 135.6, 138.2, 139.4, 150.2, 158.3, 164.4, 165.5, 166.0, (Ar. Cs, C ≡ N, and 3 amide C = O); 203.1 (C = O). Anal Calcd for C33H30N6O4S (606.70): C, 65.33; H, 4.98; N, 13.85. Found: C, 64.98; H, 5.02; N, 13.59.
2,4-Dioxo-3-(substituted) phenyl-6-(4-phenylpiperazin-1-yl)-1,2,3,4-tetrahydrothieno[3,2-d]pyrimidine-7-carbonitriles 4a-d
Pyrrolidine (0.14 g, 0.16 mL, 2 mmol) was added to a stirred suspension of freshly dried CaCl2 (5 g), and β-keto amides 3a, c, e, and f (2 mmol) in anhydrous toluene (50 mL). The reaction mixture was then placed in a water bath and the temperature was gradually raised to 45–50 °C over a period of 2 h. Heating was continued for 70 h at 45–50 °C, followed by further heating for 4 h under reflux. The mixture was then filtered, while hot and the obtained residue was oven-dried, re-suspended in water (50 mL), and stirred at room temperature for 30 min. The suspension was filtered, washed with water (2 × 20 mL), oven-dried until a constant weight, and crystallized from acetic acid.
2,4-Dioxo-3-phenyl-6-(4-phenylpiperazin-1-yl)-1,2,3,4-tetrahydrothieno[3,2-d]pyrimidine-7-carbonitrile (4a)
Yield: 80%, mp > 300 °C; IR (cm−1): 3132 (NH); 2214 (C ≡ N); 1708, 1658 (2 C = O). 1H NMR (ppm): 3.38-4.00 (m, 4H, CH2); 3.83–3.85 (m, 4H, CH2); 6.82-7.47 (m, 10H, ArH); 12.15 (s, 1H, NH). 13C NMR (ppm): 47.2, 50.0 (piperazine Cs); 76.2, 94.1, 114.1, 115.7, 119.4, 127.8, 128.6, 129.0, 129.1, 135.9, 150.0, 152.2, 156.8, 157.1, 168.6 (Ar. Cs and CN). Anal Calcd for C23H19N5O2S (429.50): C, 64.32; H, 4.46; N, 16.31. Found: C, 64.17; H, 4.39; N, 16.56.
3-(4-Ethylphenyl)-2,4-dioxo-6-(4-phenylpiperazin-1-yl)-1,2,3,4-tetrahydrothieno[3,2-d]pyrimidine-7-carbonitrile (4b)
Yield: 71%, mp > 300 °C; IR (cm−1): 3182 (NH); 2210 (C ≡ N); 1732, 1654 (2 C = O). 1H NMR (ppm): 1.22 (t, 3H, J = 7.6 Hz, CH3); 2.65 (q, 2H, J = 7.6 Hz, CH2); 3.38 (t, 4H, J = 5.0 Hz, CH2); 3.85 (t, 4H, J = 5.0 Hz, CH2); 6.82-7.29 (m, 9H, ArH); 12.18 (s, 1H, NH). 13C NMR (ppm): 15.5 (CH3); 27.8 (CH2); 47.2, 50.1 (piperazine Cs); 75.4, 94.3, 115.6, 119.4, 119.4, 128.1, 128.8, 129.0, 129.1, 132.8, 132.9, 143.6, 150.0, 156.9, 168.8 (Ar. Cs and CN). Anal Calcd for C25H23N5O2S (457.55): C, 65.63; H, 5.07; N, 15.31. Found: C, 65.49; H, 5.21; N, 15.43.
3-(4-Chlorophenyl)-2,4-dioxo-6-(4-phenylpiperazin-1-yl)-1,2,3,4-tetrahydrothieno[3,2-d]pyrimidine-7-carbonitrile (4c)
Yield: 60%, mp > 300 °C; IR (cm−1): 3132 (NH); 2214 (C ≡ N); 1708, 1658 (2 C = O). 1H NMR (ppm): 3.35–3.37 (m, 4H, CH2); 3.83 (t, 4H, J = 5.0 Hz, CH2); 6.81–7.50 (m, 9H, ArH); 12.15 (s, 1H, NH). 13C NMR (ppm): 15.5 (CH3); 27.8 (CH2); 47.2, 49.9 (piperazine Cs); 80.7, 107.0, 115.7, 119.4, 129.0, 129.1, 129.3, 133.6, 135.6, 150.0, 150.4, 154.2, 158.1, 160.1, 167.8 (Ar. Cs and CN). Anal Calcd for C23H18ClN5O2S (463.94): C, 59.54; H, 3.91; N, 15.10. Found: C, 59.76; H, 4.15; N, 15.37.
3-(3-Nitrophenyl)-2,4-dioxo-6-(4-phenylpiperazin-1-yl)-1,2,3,4-tetrahydrothieno[3,2-d]pyrimidine-7-carbonitrile (4d)
Yield: 69%, mp > 300 °C; IR (cm−1): 3178 (NH); 2210 (C ≡ N); 1732, 1654 (2 C = O); 1535, 1357 (NO2). 1H NMR (ppm): 3.39 (t, 4H, J = 5.0 Hz, CH2); 3.88 (t, 4H, J = 5.0 Hz, CH2); 6.82-8.30 (m, 9H, ArH); 12.34 (s, 1H, NH). 13C NMR (ppm): 47.2, 50.1 (piperazine Cs); 75.4, 94.1, 113.7, 115.6, 119.4, 123.2, 124.6, 129.0, 130.1, 136.4, 136.6, 146.4, 148.0, 150.0, 151.1, 156.6, 168.9 (Ar. Cs and CN). Anal Calcd for C23H18N6O4S (474.50): C, 58.22; H, 3.82; N, 17.71. Found: C, 58.01; H, 3.96; N, 17.88.
3-Acetyl-2-hydroxy-4-imino-N-(substituted)-phenyl-5-(4-phenylpiperazin-1-yl)-1,4-dihydrothieno[3,4-b]pyridine-7-carboxamides 5a-e
Method A (for the synthesis of compounds 5a, b, d, e)
A mixture of the appropriate 3-oxobutanamido thiophenecarboxamides 3a, d, f, and h (3 mmol), and K2CO3 (0.83 g, 6 mmol) in ethanol (25 mL) was heated under reflux with continuous stirring for 6 h. The reaction mixture was cooled to room temperature and filtered. The obtained solid product was washed with ethanol (2 × 20 mL), oven-dried until a constant weight, and crystallized from acetic acid.
Method B (for the synthesis of compound 5c)
A solution of K2CO3 (0.83 g, 6 mmol) in water (2 mL) was added to a stirred solution of 3-oxobutanamido thiophenecarboxamide 3e in ethylene glycol (25 mL) maintained at 120 °C. The reaction mixture was then heated for 10 min, cooled to room temperature, and poured onto water (75 mL). The formed solid was filtered, washed with water (2 × 50 mL), oven-dried until a constant weight, and crystallized from acetic acid.
3-Acetyl-2-hydroxy-4-imino-N-phenyl-5-(4-phenylpiperazin-1-yl)-1,4-dihydrothieno[3,4-b]pyridine-7-carboxamide (5a)
Yield: 63%, mp 273–275 °C; IR (cm−1): 3356, 3332, 3317 (3 NH); 1658, 1651 (2 C = O). 1H NMR (ppm): 2.53 (s, 3H, CH3); 2.90–3.16 (m, 4H, CH2); 3.45–3.75 (m, 4H, CH2); 6.82–7.69 (m, 10H, ArH); 8.21 (s, 1H, NH or OH, D2O exchangeable); 9.92 (s, 1H, NH, D2O exchangeable); 10.08 (s, 1H, NH, D2O exchangeable); 10.76 (s, 1H, NH or OH, D2O exchangeable). 13C NMR (ppm): 32.7 (CH3); 47.6, 54.6 (piperazine Cs); 97.2, 98.1, 109.7, 115.8, 119.5, 121.1, 121.2, 123.9, 128.6, 129.1, 138.4, 141.8, 150.5, 155.0, 161.6, 162.4 (Ar. Cs and C = O); 199.9 (C = O). Anal Calcd for C26H25N5O3S (487.57): C, 64.05; H, 5.17; N, 14.36. Found: C, 64.31; H, 5.28; N, 14.19.
3-Acetyl-N-(2-chlorophenyl)-2-hydroxy-4-imino-5-(4-phenylpiperazin-1-yl)-1,4-dihydrothieno[3,4-b]pyridine-7-carboxamide (5b)
Yield: 65%, mp 284–286 °C; IR (cm−1): 3394, 3348, 3313 (3 NH); 1658, 1651 (2 C = O). 1H NMR (ppm): 2.95–3.05 (m, 4H, CH2); 3.04 (s, 3H, CH3); 3.35–3.50 (s, 2H, CH2); 3.60–3.77 (m, 4H, CH2); 7.03–8.22 (m, 9H, ArH); 8.26 (s, 1H, NH or OH, D2O exchangeable); 10.04 (s, 1H, NH, D2O exchangeable); 10.68 (s, 1H, NH, D2O exchangeable); 11.32 (s, 1H, NH or OH, D2O exchangeable). 13C NMR (ppm): 33.7 (CH3); 49.2, 55.7 (piperazine Cs); 98.5, 100.0, 111.7, 117.2, 121.0, 127.4, 127.5, 128.2, 128.8, 130.0, 130.3, 135.7, 143.4, 151.6, 156.1, 162.4, 162.7, 163.4 (Ar. Cs and C = O); 201.6 (C = O). Anal Calcd for C26H24ClN5O3S (522.02): C, 59.82; H, 4.63; N, 13.42. Found: C, 59.67; H, 4.71; N, 13.68.
3-Acetyl-N-(4-chlorophenyl)-2-hydroxy-4-imino-5-(4-phenylpiperazin-1-yl)-1,4-dihydrothieno[3,4-b]pyridine-7-carboxamide (5c)
Yield: 60%, mp 294–296 °C; IR (cm−1): 3371, 3340, 3309 (3 NH); 1651, 1643 (2 C = O). 1H NMR (ppm): 2.52 (s, 3H, CH3); 2.85–3.25 (m, 4H, CH2); 3.40–3.85 (m, 4H, CH2); 6.82-7.73 (m, 9H, ArH); 8.18 (s, 1H, NH or OH, D2O exchangeable); 10.02 (s, 1H, NH, D2O exchangeable); 10.04 (s, 1H, NH, D2O exchangeable); 10.76 (s, 1H, NH or OH, D2O exchangeable). 13C NMR (ppm): 32.70 (CH3); 47.5, 54.6 (piperazine Cs); 96.8, 98.0, 109.5, 115.7, 119.4, 122.4, 127.4, 128.4, 129.0, 137.4, 142.0, 150.4, 154.9, 161.6, 162.0, 162.6 (Ar. Cs and C = O); 199.8 (C = O). Anal Calcd for C26H24ClN5O3S (522.02): C, 59.82; H, 4.63; N, 13.42. Found: C, 59.70; H, 4.72; N, 13.74.
3-Acetyl-2-hydroxy-4-imino-N-(3-nitrophenyl)-5-(4-phenylpiperazin-1-yl)-1,4-dihydrothieno[3,4-b]pyridine-7-carboxamide (5d)
Yield: 45%, mp 293–295 °C; IR (cm−1): 3402, 3360, 3348 (3 NH); 1658, 1639 (2 C = O); 1531, 1350 (NO2). 1H NMR (ppm): 2.53 (s, 3H, CH3); 2.90–3.25 (m, 4H, CH2); 3.40–3.95 (m, 4H, CH2); 6.83–8.69 (m, 9H, ArH); 8.17 (s, 1H, NH or OH, D2O exchangeable); 10.04 (s, 1H, NH, D2O exchangeable); 10.35 (s, 1H, NH, D2O exchangeable); 10.77 (s, 1H, NH or OH, D2O exchangeable). 13C NMR (ppm): 32.7 (CH3); 47.6, 54.5 (piperazine Cs); 96.5, 98.0, 109.4, 114.4, 115.8, 117.8, 119.5, 126.1, 129.0, 129.6, 139.8, 142.4, 147.6, 150.4, 150.4, 154.8, 161.7, 162.8 (Ar. Cs and C = O); 199.7 (C = O). Anal Calcd for C26H24N6O5S (532.57): C, 58.64; H, 4.54; N, 15.78. Found: C, 58.91; H, 4.63; N, 15.96.
3-Acetyl-N-(2-bromophenyl)-2-hydroxy-4-imino-5-(4-phenylpiperazin-1-yl)-1,4-dihydrothieno[3,4-b]pyridine-7-carboxamide (5e)
Yield: 64%, mp 287–289 °C; IR (cm−1): 3379, 3348, 3298 (3 NH); 1658, 1651 (2 C = O). 1H NMR (ppm): 2.52 (s, 3H, CH3); 2.90–3.30 (m, 4H, CH2); 3.40–3.90 (m, 4H, CH2); 6.82-7.72 (m, 9H, ArH); 8.21 (s, 1H, NH or OH, D2O exchangeable); 9.82 (s, 1H, NH, D2O exchangeable); 9.96 (s, 1H, NH, D2O exchangeable); 10.76 (s, 1H, NH or OH, D2O exchangeable). 13C NMR (ppm): 32.7 (CH3); 47.5, 54.6 (piperazine Cs); 96.6, 98.0, 109.6, 115.8, 119.4, 121.1, 128.1, 128.2, 129.0, 129.2, 132.7, 135.8, 141.8, 150.4, 154.9, 161.7, 162.0, 162.6 (Ar. Cs and C = O); 199.8 (C = O). Anal Calcd for C26H24BrN5O3S (566.47): C, 55.13; H, 4.27; N, 12.36. Found: C, 55.37; H, 4.40; N, 12.47.
Results and discussion
In this work, thieno[3,2-d]pyrimidin-4-ones 2a–k were synthesized in excellent yields (80-98%) by heating the respective thiophene-2-carboxamides 1a–k in formic acid (Scheme 1). The IR spectra of compounds 2a–k revealed the disappearance of NH2/NH bands of the precursor thiophene derivatives. Other characteristic bands of compounds 2a-k were observed at 2214–2206 cm−1, and at 1678–1662 cm−1 corresponding to C ≡ N, and C = O groups, respectively. The 1H NMR and 13C NMR spectra offered further confirmation of the structure of compounds 2a–k. The 1H NMR spectra of compounds 2a–k showed a characteristic singlet signal at δ 8.38–8.54 ppm corresponding to C(2)-H. Furthermore, no exchangeable protons were detected for compounds 2a–h, whereas derivatives 2i–k showed an exchangeable signal at δ 10.37–10.52 ppm corresponding to the CONH proton. Moreover, the 13C NMR spectra of compounds 2a–k showed the expected number of signals for each derivative.

Synthesis of fused thiophene derivatives 2-5
β-Keto amides comprise an important class of organic reagents that has widespread use in the construction of various heterocyclic rings (Alagesan et al. 2018; El-Meligie et al. 2019). In the synthetic pathway to β-keto amides 3a–i, compounds 1a–i were reacted with 2,2,6-trimethyl-4H-1,3-dioxin-4-one (TMD) in xylene. It is noteworthy that the commercially available TMD was of 90–95% concentration; therefore, optimization of the amount of the reagent used was necessary. When the reagent was used in a stoichiometric amount to the amine 1a, TLC analysis of the crude 3a revealed an incomplete reaction. Optimum ratio of TMD reagent relative to 1a was found to be 1.11 to 1 molar equivalents. Moreover, as a precautionary measure, an air condenser was used to bar the evolving acetone from returning to the reaction medium, thereby preventing the consumption of the acetyl ketene intermediate A in the formation of the side product B (Scheme 2).

Potential side product formation in TMD reaction
The structure of compounds 3a–i was confirmed by spectral analysis. IR spectra of compounds 3a–i showed two bands at 3402–3147 cm−1 corresponding to the NH functions. Moreover, characteristic absorption bands at 2214–2202 cm−1 and 1735-1627 cm−1 confirmed the presence of C ≡ N and C = O groups, respectively. 1H NMR spectra showed two new singlet signals at δ 2.22–2.25 ppm and at δ 3.67–3.69 ppm corresponding to the CH3 and CH2 protons of the β-keto acyl moiety, respectively. Moreover, the disappearance of the D2O-exchangeable singlet signal of the NH2 protons of the parent compounds and the concomitant appearance of a new D2O-exchangeable signal at δ 10.32–10.45 ppm confirmed the introduction of the β-keto acyl moiety at the amino group of the parent precursor. 13C NMR spectra revealed the appearance of two new aliphatic carbon signals at δ 30.0-51.4 ppm, in addition to a signal at δ 202.3–203.4 ppm corresponding to the ketonic C = O carbon.
The prepared β-keto amides 3a–i were used as precursors for the synthesis of thieno[3,2-d]pyrimidine-2,4-diones 4a–d and the infrequently reported thieno[3,4-b]pyridines 5a–e. The reported procedures are of practical and theoretical values, since no previous reports was published describing similar procedures.
Reaction of β-keto amides 3a, c, e, and f with pyrrolidine in toluene using CaCl2 as a desiccant afforded the thieno[3,2-d]pyrimidine-2,4-diones 4a–d. The concept of using β-keto amide derivatives 3 as substrates for pyrimidine ring closure stemmed from the work published in 2010 by Wei et al. (Wei et al. 2010), who reported the synthesis of asymmetrical urea derivatives F via thermally induced fragmentation of enamino amide D to the respective isocyanate E (Scheme 3); and the earlier observation by Wiley that substituted amides G add to isocyanates E to form N-acyl urea derivatives H.(Wiley, 1949a, 1949b).

Synthesis of asymmetrical urea derivatives using β-keto amides
Based on the above findings, it stood to reason that subjecting β-keto amide derivatives 3 to conditions similar to those reported by Wei et al. (Wei et al. 2010), may potentially generate an isocyanate intermediate which can be subsequently captured by the ortho carboxamide group to afford the thieno[3,2-d]pyrimidine-2,4-dione derivatives 4 according to Scheme 4.

Potential pyrimidine ring annulation via enamino amide fragmentation
In an initial attempt to prepare compound 4a, a mixture of 3a and a slight molar excess of piperidine was heated under reflux for 2 h in accordance with the experimental procedures described by Wei et al.(2010). Unfortunately, the desired product 4a was not obtained, yet the starting compound 3a was quantitatively converted to 3-aminothiophene-2-carboxamide 1a. Promising results, however, were achieved when pyrrolidine was used as the secondary amine, giving rise to the target thienopyrimidine 4a in 30% yield (Table 1). The observed hydrolysis of β-keto amide 3a could be attributed to the moisture susceptibility of the isocyanate intermediate. Therefore, CaCl2 was added to the reaction medium as a desiccant to prevent the hydrolysis of the isocyanate. Under these conditions, the target thieno[3,2-d]pyrimidine-2,4-diones 4a–d were obtained in moderate to good yields ranging from 60 to 80% (Table 1).

This cyclization reaction may have proceeded according to the mechanism proposed in Scheme 5, whereby the formed enamine was fragmented to isocyanate intermediate that was subsequently captured by the carboxamide group (Pathway A). Another plausible explanation for the formation of 4a–d was nucleophilic 1,2-addition of the carboxamide nitrogen on the enamino amide, followed by proton extraction of the spatially adjacent enamine, and subsequent departure of the 1-(prop-1-en-2-yl)pyrrolidine moiety (Pathway B).

Possible mechanism for the formation of compounds 4a-d
The structure of compounds 4a–d was confirmed by spectral and elemental analyses. The absorption bands at 3187–3132 cm−1, 2210–2214 cm−1, and 1732–1654 cm−1 in the IR spectra of 4a–d indicated the presence of NH, C ≡ N, and two C = O groups, respectively. The 1H NMR spectra of these compounds revealed the disappearance of the two CONH signals along with the CH3COCH2 protons of the parent 3a, c, e, and f. Moreover, there is no trace of the NH2 and CONH protons of the parent compounds 1 (that would otherwise have indicated the cleavage of the acetoacetyl group). On the other hand, the spectrum revealed the appearance of a new characteristic singlet signal at δ 12.15-12.34 ppm corresponding to pyrimidine NH proton.
Another new method for the synthesis of thieno[3,4-b]pyridine was developed. Thus, heating a mixture of β-keto amide derivatives 3a, d, f and h and K2CO3 in ethanol under reflux conditions afforded the target derivatives 5a, b, d, and e.
Our initial goal, however, was to assess the applicability of base-promoted pyrimidine ring closure of ortho diamides in compounds 3 to achieve the synthesis of thienopyrimidine derivatives 5* (Scheme 1). Therefore, a mixture of β-keto amide 3a and NaOH was heated either in ethanol or in dioxane. However, this reaction condition resulted in quantitative hydrolysis of 3a to its parent thiophene 1a. Nevertheless, when K2CO3 was used, the thienopyridine derivative 5a was obtained.
Following the same procedure, thienopyridines 5b, d, and e were synthesized in 45-65% yield. However, the β-keto amide 3e failed to cyclize to the respective thienopyridine under these reaction conditions. When ethanol was replaced by higher boiling point solvent such as ethylene glycol, thienopyridine 5c was obtained in 60% yield.
The chemical structure of compounds 5a–e was confirmed using IR, 1H NMR, 13C NMR, and elemental analyses. IR spectra of the compounds revealed the disappearance of the C ≡ N group of the parent β-keto amide derivatives 3a, d, e, f, and h. This ruled out the formation of 5*. Moreover, their 1H NMR spectra showed four exchangeable proton signals between δ 8.17–11.32 ppm.
Three possible tautomers were suggested for compounds 5a-e (Scheme 6). The existence of tautomer 1 was excluded due to absence of any aliphatic signal corresponding to 3-CH proton or carbon of pyridine in the NMR spectra. Tautomers 2 (the keto form) and 3 (the enol form) are possible tautomers; however, the 1H NMR spectra of the compounds did not show a signal corresponding to the 2 protons of amino group that are present in tautomer 2. On the other hand, 1H NMR spectra distinctly revealed the existence of four exchangeable signals, each integrated for one proton. Therefore, we assumed that tautomer 3 (the enol form) is more appropriate. The stability of tautomer 3 might be attributed to the intramolecular H-bond between the enolic OH and the adjacent carbonyl.

Possible tautomeric forms of compounds 5a-e
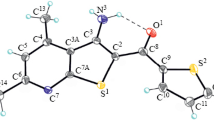
Heteronuclear H–C 2D spectra were acquired for further confirmation of the proposed structure of thienopyridine 5e. HSQC spectrum revealed that proton signals at δ 7.24, 7.42, 7.50, and 7.71 ppm showed H–C correlations with the carbon signals appearing at δ 128.2, 128.1, 129.2, and 132.7 ppm, respectively. These protons also showed HMBC correlations with the named carbon signals in addition to two more signals appearing at δ 121.1 and 135.8 ppm. These observations allowed for assigning the carbon signals at δ 121.1, 135.8, 129.2, 128.2, 128.1, 132.7 ppm to C(31), C(18), C(35), C(34), C(33), and C(32), respectively. Moreover, the singlet proton signal at δ 9.82 ppm which showed no HSQC correlations, exhibited HMBC correlations with both carbon signal at δ 121.1, and 129.2 ppm that were assigned to C(31) and C(35), respectively. In addition, HMBC correlation of this proton with the carbon signal at δ 161.7 ppm, which could be assigned to C(15), allowed for assigning this proton signal to H(17) Table 2).
The newly formed pyridine moiety was also confirmed by the observed HSQC and HMBC correlation patterns: Aliphatic proton signal at δ 2.52 ppm showed an HSQC correlation with carbon signal at δ 32.7 ppm, in addition to an HMBC correlation with carbon signal at δ 199.8 ppm, which confirmed the presence of an acyl group. Moreover, this proton signal showed an additional HMBC correlation with the sp2 carbon appearing at δ 98.0 ppm, which in turn, did not correlate with any proton in the HSQC spectrum, but showed three additional HMBC correlations with the exchangeable protons at δ 8.21, 9.96, and 10.77 ppm.
Conclusion
In summary, two series of thieno[3,2-d]pyrimidines and a series of thieno[3,4-b]pyridines were synthesized starting from 3-amino-4-cyano-2-thiophenecarboxamides. Two novel procedures were developed and optimized for the synthesis of thienopyrimidine-2,4-diones 4 and thieno[3,4-b]pyridine derivatives 5 using β-keto amides 3. The thieno[3,4-b]pyridine ring is infrequently documented in the literature; therefore, the reaction conditions were optimized to provide an appropriate synthetic route and reaction conditions to this ring system.
References
Abdel-Megid M, Elmahdy KM, Elkazak AM, Seada MH, Mohamed OF (2016) Chemistry of thienopyrimidines and their biological applications. J Pharm Appl Chem 2(3):103–127. https://doi.org/10.18576/jpac/020301
Ahmad S, Washburn WN, Hernandez AS, Bisaha S, Ngu K, Wang W, Murphy BJ (2016) Synthesis and antiobesity properties of 6-(4-chlorophenyl)-3-(4-((3,3-difluoro-1-hydroxycyclobutyl)methoxy)-3-methoxyphenyl)thieno[3,2- d]pyrimidin-4(3 H)-one (BMS-814580): a highly efficacious melanin concentrating hormone receptor 1 (MCHR1) Inhibitor. J Med Chem 59(19):8848–8858. https://doi.org/10.1021/acs.jmedchem.6b00676
Ahmed GA (2008) Heterocyclic synthesis with thiophene-2-carboxamide. Phosphorus Sulfur Silicon Relat Elements 183:74–81
Alagesan A, Sangeetha M, Sekar G (2018) Organic and biomolecular chemistry. Org Biomol Chem 16:7068–7083. https://doi.org/10.1039/C8OB01423J
Bakhite EA-G (2003) Recent trends in the chemistry of thienopyridines. Phosphorus Sulfur Silicon Relat Elements 178(5):929–992. https://doi.org/10.1080/10426500307855
Barker JM (1977) The thienopyridines. Adv Heterocycl Chem. https://doi.org/10.1016/S0065-2725(08)60730-8
Björk P, Hörnfeldt A-B, Gronowitz S (1994) The syntheses and NMR spectra of the twelve isomeric thieno[b]fused naphthyridines. J Heterocycl Chem 31(5):1161–1169. https://doi.org/10.1002/jhet.5570310511
Brumfield S, Korakas P, Silverman LS, Tulshian D, Matasi JJ, Qiang L, Li C (2012) Synthesis and SAR development of novel mGluR1 antagonists for the treatment of chronic pain. Bioorg Med Chem Lett 22(23):7223–7226. https://doi.org/10.1016/j.bmcl.2012.09.048
Carpenter AJ, Al-Barazanji KA, Barvian KK, Bishop MJ, Britt CS, Cooper JP, Swain WR (2006) Novel benzimidazole-based MCH R1 antagonists. Bioorg Med Chem Lett 16(19):4994–5000. https://doi.org/10.1016/j.bmcl.2006.07.054
El-Hamed MKA, Kandeel MM, Roshdy SMA, El-Telbany F (2001) Synthesis of some fused thienopyrimidine derivatives of potential antimicrobial activity. Bull Fac Pharm 39:11–21
El-meligie SEM, Khalil NA, El-nassan HB, Ibraheem AAM (2019) A Review on the Synthetic Routes to β -keto amides. Curr Org Chem 23:2005–2015
El-Meligie SEM, Khalil NA, El-Nassan HB, Ibraheem AAM (2020) Synthesis of new 3-amino-4-cyanothiophene derivatives. Chem Papers https://doi.org/10.1007/s11696-020-01070-z
El-Telbany F, Hutchins RO (1985) Synthesis of a novel series of 3-substituted [1]benzothieno[3,2-d]pyrimidine derivatives. J Heterocycl Chem 22(2):401–403. https://doi.org/10.1002/jhet.5570220236
Fukumi H, Sugiyama M, Sakamoto T (1989) A novel heterocyclic compound. Synthesis and reactivities of an oxazolo[3,2-a]thieno[3,2-d]-pyrimidine derivative. Chem Pharm Bull 37(5):1197–1200
Hafez HN, Alsalamah SA, El-Gazzar A-RBA (2017) Synthesis of thiophene and N-substituted thieno[3,2-d] pyrimidine derivatives as potent antitumor and antibacterial agents. Acta Pharmaceutica 67(3):275–292. https://doi.org/10.1515/acph-2017-0028
Hertzog DL, Al-Barazanji KA, Bigham EC, Bishop MJ, Britt CS, Carlton DL, Zhou H (2006) The discovery and optimization of pyrimidinone-containing MCH R1 antagonists. Bioorg Med Chem Lett 16(18):4723–4727. https://doi.org/10.1016/j.bmcl.2006.07.008
Ibrahim YA, Elwahy AHM, Kadry AM (1996) Thienopyrimidines: synthesis, reactions, and biological activity. Adv Heterocycl Chem 65:235–281. https://doi.org/10.1016/S0065-2725(08)60297-4
Kim Y, Kim J, Kim S, Ki Y, Seo SH, Tae J, Choo H (2014) Novel thienopyrimidinones as mGluR1 antagonists. Eur J Med Chem 85:629–637. https://doi.org/10.1016/j.ejmech.2014.08.027
Kim Y, Kim M, Park M, Tae J, Baek D-J, Park K, Choo H (2015) Synthesis of novel dihydropyridothienopyrimidin-4,9-dione derivatives. Molecules 20(3):5074–5084. https://doi.org/10.3390/molecules20035074
Klemm LH, Johnson WO, White DV (1970) Chemistry of thienopyridines. X Syntheses of thieno[3,4-b] and thieno[3,4-c] pyridines. J Heterocycl Chem 7(2):463–464. https://doi.org/10.1002/jhet.5570070244
Litvinov VP (2004) Thienopyrimidines: synthesis, properties, and biological activity. Russ Chem Bull 53(3):487–516. https://doi.org/10.1023/B:RUCB.0000035630.75564.2b
Litvinov VP (2006) The chemistry of thienopyrimidines. In: Katritzky AR (ed) Advances in heterocyclic chemistry. Academic Press, London. https://doi.org/10.1016/S0065-2725(06)92003-0
Litvinov VP, Dotsenko VV, Krivokolysko SG (2005) Thienopyridines: synthesis, properties, and biological activity. Russ Chem Bull 54(4):864–904. https://doi.org/10.1007/s11172-005-0333-1
Meyer MD, Altenbach RJ, Basha FZ, Carroll WA, Drizin I, Elmore SW, Kerwin JF (1997) Synthesis and pharmacological characterization of 3-[2-((3a R,9b R)- cis -6-Methoxy- 2,3,3a,4,5,9b-hexahydro-1 H - benz[e]isoindol-2-yl)ethyl]pyrido[3‘,4‘:4,5]thieno[3,2-d]pyrimidine- 2,4(1 H,3 H)-dione (A-131701): a uroselective α 1A adrenoceptor. J Med Chem 40(20):3141–3143. https://doi.org/10.1021/jm970364a
Meyer MD, Altenbach RJ, Bai H, Basha FZ, Carroll WA, Kerwin JF, Drizin I (2001) Structure–activity studies for a novel series of bicyclic substituted hexahydrobenz[e]isoindole α 1A adrenoceptor antagonists as potential agents for the symptomatic treatment of benign prostatic hyperplasia. J Med Chem 44(12):1971–1985. https://doi.org/10.1021/jm000541z
Press J, Russell R, McNally J, Rampulla R, Falotico R, Scott C, Tobia J (1991) Thiophene systems 12. Analogues of ketanserin and ritanserin as selective 5-HT2 antagonists. Eur J Med Chem 26(8):807–813. https://doi.org/10.1016/0223-5234(91)90007-A
Romeo G, Materia L, Marucci G, Modica M, Pittalà V, Salerno L, Minneman KP (2006) New pyrimido[5,4-b]indoles and [1]benzothieno[3,2-d]pyrimidines: high affinity ligands for the α1-adrenoceptor subtypes. Bioorg Med Chem Lett 16(24):6200–6203. https://doi.org/10.1016/j.bmcl.2006.09.034
Russell RK, Press JB, Rampulla RA, McNally JJ, Falotico R, Keiser JA, Tobia A (1988) Thiophene systems 9. Thienopyrimidinedione derivatives as potential antihypertensive agents. J Med Chem 31(9):1786–1793. https://doi.org/10.1021/jm00117a019
Ryndina SA, Kadushkin AV, Solov NP, Granik VG (2002) Application of the Thorpe–Ziegler reaction for the synthesis of functionalized thiophenes, thienopyrimidines, and thienotriazines. Russ Chem Bull 51(5):854–859. https://doi.org/10.1023/A:1016049204323
Sasikumar TK, Qiang L, Burnett DA, Greenlee WJ, Li C, Heimark L, Reggiani A (2009) Tricyclic thienopyridine–pyrimidones/thienopyrimidine–pyrimidones as orally efficacious mGluR1 antagonists for neuropathic pain. Bioorg Med Chem Lett 19(12):3199–3203. https://doi.org/10.1016/j.bmcl.2009.04.104
Sasikumar TK, Qiang L, Burnett DA, Greenlee WJ, Li C, Grilli M, Reggiani A (2010) A-ring modifications on the triazafluorenone core structure and their mGluR1 antagonist properties. Bioorg Med Chem Lett 20(8):2474–2477. https://doi.org/10.1016/j.bmcl.2010.03.004
Shestakov AS, Prezent MA, Kartsev VG, Shikhaliev KS (2014) Synthesis of thieno[3,2-d]pyrimidin-4-ones and alkylation thereof. Eur Chem Bull 3(7):713–718
Sugiyama M, Sakamoto T, Tabata K, Endo K, Ito K, Kobayashi M, Fukumi H (1989) Condensed thienopyrimidines. I. Synthesis and gastric antisecretory activity of 2,3-dihydro-5H-oxazolothienopyrimidin-5-one derivatives. Chem Pharm Bull 37(8):2091–2102. https://doi.org/10.1248/cpb.37.2091
Tavares FX, Al-Barazanji KA, Bishop MJ, Britt CS, Carlton DL, Cooper JP, Zhou H-Q (2006) 6-(4-Chlorophenyl)-3-substituted-thieno[3,2-d]pyrimidin-4(3 H)-one-based melanin-concentrating hormone receptor 1 antagonist. J Med Chem 49(24):7108–7118. https://doi.org/10.1021/jm060814b
Thomae D, Perspicace E, Henryon D, Xu Z, Schneider S, Hesse S, Seck P (2009) One-pot synthesis of new tetrasubstituted thiophenes and selenophenes. Tetrahedron 65(50):10453–10458. https://doi.org/10.1016/j.tet.2009.10.021
Varvounis G, Giannopoulos T (1996) Synthesis, Chemistry, and Biological Properties of Thienopyrimidines. Academic Press, London. https://doi.org/10.1016/S0065-2725(08)60307-4
Warshakoon NC, Sheville J, Bhatt RT, Ji W, Mendez-Andino JL, Meyers KM, Hu XE (2006) Design and synthesis of substituted quinolines as novel and selective melanin concentrating hormone antagonists as anti-obesity agents. Bioorg Med Chem Lett 16(19):5207–5211. https://doi.org/10.1016/j.bmcl.2006.07.006
Washburn WN, Manfredi M, Devasthale P, Zhao G, Ahmad S, Hernandez A, Murphy BJ (2014) Identification of a nonbasic melanin hormone receptor 1 antagonist as an antiobesity clinical candidate. J Med Chem 57(18):7509–7522. https://doi.org/10.1021/jm500026w
Wei Y, Liu J, Lin S, Ding H, Liang F, Zhao B (2010) Acetoacetanilides as masked isocyanates: facile and efficient synthesis of unsymmetrically substituted ureas. Org Lett 12(19):4220–4223. https://doi.org/10.1021/ol101474f
Wiley PF (1949a) The reaction of amides with isocyanates. II. N-substituted amides. J Am Chem Soc 71(11):3746–3748. https://doi.org/10.1021/ja01179a045
Wiley PF (1949b) The reaction of amides with isocyanates. J Am Chem Soc 71(4):1310–1311. https://doi.org/10.1021/ja01172a047
Zhang M, Tamiya J, Nguyen L, Rowbottom MW, Dyck B, Vickers TD, Goodfellow VS (2007) Thienopyrimidinone bis-aminopyrrolidine ureas as potent melanin-concentrating hormone receptor-1 (MCH-R1) antagonists. Bioorg Med Chem Lett 17(9):2535–2539. https://doi.org/10.1016/j.bmcl.2007.02.012
Zheng GZ, Bhatia P, Daanen J, Kolasa T, Patel M, Latshaw S, Stewart AO (2005) Structure–activity relationship of triazafluorenone derivatives as potent and selective mGluR1 Antagonists. J Med Chem 48(23):7374–7388. https://doi.org/10.1021/jm0504407
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
El-Meligie, S.E.M., Khalil, N.A., El-Nassan, H.B. et al. New synthetic approaches to thieno[3,2-d]pyrimidine and thieno[3,4-b]pyridine derivatives. Chem. Pap. 74, 2501–2514 (2020). https://doi.org/10.1007/s11696-020-01089-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-020-01089-2