
Abstract
In the present work, TiO2 nanotubes (TNT) and carbon-doped TiO2 nanotubes (C-TNT) were produced via the anodization method. Carbon doping was performed on TNT in a tubular oven employing two different 15 cm3/min total flow rates with varying compositions of acetylene (C2H2) and argon (Ar) as VC2H2/Ar = 7/93 (1 cm3/min C2H2 + 14 cm3/min Ar) for C-TNT (7:93) and VC2H2/Ar = 33/67 (5 cm3/min C2H2 + 10 cm3/min Ar) for C-TNT (33:67). The synthesized C-doped TNT was characterized by x-ray diffraction (XRD), Raman spectroscopy, and scanning electron microscopy (SEM). XRD, Raman spectra, and SEM results reveal that a carbon film structure was formed on the TNT surface. In addition, the electronic structure of TNT changed with doping of carbon on the TNT surface. These carbon-doped TNTs were employed as catalysts for the photocatalytic oxidation of glucose (GA). Cyclic voltammetry (CV), chronoamperometry (CA), and electrochemical impedance spectroscopy (EIS) measurements were carried out to investigate the glucose electro-oxidation activity of the carbon-doped TNTs in the dark and under UV illumination (λ = 354 nm). C-TNT (7:93) exhibited the highest glucose electro-oxidation activity under the dark and UV illumination compared to C-TNT (33:67) and TNT. The glucose electro-oxidation (GAEO) current density on C-TNT (7:93) improved significantly under UV illumination compared to glucose electro-oxidation activity obtained in the dark. C-TNT (7:93) enhanced glucose electro-oxidation activity and stability under UV illumination. This electrode production method is promising for the design of photocatalytic glucose fuel cells.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Energy is a vital and persistent need for humankind, and it is supplied from fossil fuels such as petroleum, coal, and natural gas. Fossil fuel exhaust gases such as nitric oxides (NOx), sulfur oxides (SOx), and carbon dioxide (CO2) are hazardous gases, leading to global warming and acid rain. Fossil fuel resources are being depleted due to the increase in population and industrialization. To eliminate these disadvantages of fossil fuel consumption in energy generating systems, a vast number of studies are dedicated to alternative energy sources such as fuel cells,1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28 batteries,29,30,31,32 solar energy,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47 and wind energy.41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60 Fuel cells are environmentally friendly devices that convert chemical energy into electrical energy.4,11 Hydrogen is a clean and sustainable energy source, with high energy density. Hydrogen fuel cells are one of the potential devices for dealing with the problems caused by fossil fuel employment.61 However, the transportation, storage, and flammability of hydrogen gas are important drawbacks to the use of hydrogen as a fuel in hydrogen fuel cells. Direct liquid fuel cells (DLFCs) are types of fuel cells where liquid fuels such as alcohols,62 formic acid,63 formate,64 borohydride,65 or glucose (GA)66 are employed as fuel.67 In DLFCs, electro-oxidation reactions occur in either alkaline or acidic media. DLFCs coupled with anion exchange membranes are more advantageous than acidic DLFCs because alkaline media is better for the kinetics and oxygen reduction ability in terms of electro-oxidation.68
Among these fuels, GA is non-toxic, non-flammable, non-volatile, and the most abundant compound in nature.69,70 GA, with an energy density of 2.87 MJ/mol, is fed to the anode side of a GA fuel cell (DGFC).71,72 As a result, 24 electrons are generated by GAEO to give CO2 as follows:
However, this reaction is uncommon and unlikely to occur, and to date, partial oxidation of GA has been achieved. GA partial oxidation involves breaking a C–H bond to produce gluconic acid.73,74,75,76,77,78,79,80,81
Numerous research studies have been dedicated to improving the efficiency and stability of electro-catalysts for GAEO. The GAEO current density values compiled from the literature are given in Table I. It is notable that metal alloy catalysts such as Ni, Pt, Pd, and Au were used in these studies, and it is clear that their current density values varied in a wide range of 0.44–23.43 mA/cm2.82,83,84,85,86,87 In the present work, carbon-doped TiO2 nanotubes (C-TNT) were produced and their GAEO activity was examined to assess their employment as an anode material in a DGFC.
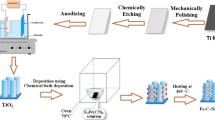
TNT has superior properties including chemical inertness, non-toxicity, high surface area, thermal stability, directional charge transfer, and excellent biocompatibility. TNT can be produced via anodization, organic degradation, and chemical vapor deposition techniques.88,89,90 Among these methods, electrochemical anodization of titanium foil is a promising method to produce one-dimensional self-organized, highly ordered, and vertically oriented TNT. Moreover, carbon doping can be performed as a uniform film on TNT in a tubular oven.88,89,90
In the present work, TNT was firstly synthesized via anodization. Carbon was then doped onto TNT in a tubular oven using two different compositions of both C2H2 and Ar with 15 cm3/min total flow rate as VC2H2/Ar = 7/93 (1 cm3/min C2H2 + 14 cm3/min Ar) for C-TNT (7:93) and VC2H2/Ar = 33/67 (5 cm3/min C2H2 + 10 cm3/min Ar) for C-TNT (33:67). These C-TNT materials were characterized using x-ray diffraction (XRD), Raman spectroscopy, and scanning electron microscopy (SEM). To examine the GAEO activity of C-TNT, Cyclic voltammetry (CV), chronoamperometry (CA), and electrochemical impedance spectroscopy (EIS) electrochemical measurements were utilized.
Experimental
Preparation of TNT and C-TNT
Synthesis of TNT
The anodization of titanium foil (Sigma-Aldrich), employed as substrate, was conducted in two steps as described elsewhere.91 The anodization of titanium foil was performed in NH4F/C2H6O2 electrolyte (3% water and 0.3% NH4F) at 100 V, with voltage kept constant for 2 h. After anodization, to remove the oxide layer, the Ti foils were washed with deionized water and then dried with argon gas. This prepared TNT was placed in the tubular reactor and the reactor temperature was increased to 500°C. After 20 min, the oven was shut down and allowed to cool. This TNT was named TNT-heated.
Synthesis of C-TNT
C-TNT was produced in a tubular oven under C2H2-Ar gas mixtures. First, TNT was cleaned by washing with deionized water before carbon doping. The reactor medium was fixed at varying C2H2-Ar flow rates to define the effect of the flow rate of carbon doping on TNT. Carbon doping was performed on TNT in a tubular oven employing two different 15 cm3/min total flow rates with varying compositions of C2H2 and Ar as VC2H2/Ar = 7/93 (1 cm3/min C2H2 + 14 cm3/min Ar) for C-TNT (7:93) and VC2H2/Ar = 33/67 (5 cm3/min C2H2 + 10 cm3/min Ar) for C-TNT (33:67). The reactor temperature was increased to 500°C. After 20 min, the oven was shut down and allowed to cool.
Characterization of Materials
TNT and C-TNT electrodes were characterized by XRD, Raman spectroscopy, and SEM. The crystallinity of these samples was examined with XRD at 2θ = 20–100o with 0.1o min−1 intervals via Analytical Empyrean (Cu-Kα, λ = 1.5405 Å). Raman spectroscopic measurements were performed using a WITec alpha300 R device. SEM images were obtained using a ZEISS SIGMA 300 instrument to analyze the surface of TNT and C-TNT materials.
Electrochemical Measurements
All electrochemical measurements were performed using a CHI 660 E potentiostat. TNT, C-TNT (7:93), and C-TNT (33:67) were employed as working electrodes with 0.5 cm2 electrode surface area. Pt wire and Ag/AgCl(sat) electrode were used as counter electrode and reference electrode, respectively. All electro-oxidation measurements were investigated to determine the GAEO activity of TNT, TNT-heated, C-TNT (33:67), and C-TNT (7:93) with CV, CA, and EIS in 1 M KOH + 0.5 M GA solution at 100 mV s−1 between −0.8 V and +0.8 V potential range in the dark and under UV illumination (λ = 354 nm). Under UV illumination, the stability of the TNT, C-TNT (33:67), and C-TNT (7:93) electrodes for 1000 s and 0.6 V was examined in CA. In addition, the charge transfer resistance for the electro-oxidation of GA at TNT, C-TNT (33:67), and C-TNT (7:93) electrodes was investigated with EIS at about 320 kHz and 0.05 Hz to 0.005 V amplitude and varying potentials.
Results and Discussion
Physical Characterization
The morphology of TNT, C-TNT (7:93), and C-TNT (33:67) was analyzed using SEM, and SEM images are given in Figs. 1 and 2. Figure 1 shows the morphology of the unmodified TNT. The amorphous nature of this material is observed by SEM. After exposure to C2H2 for carbon doping at high temperature, some crystallites appear on the outer surface of the tubes in C-TNT (7:93) and C-TNT (33:67), visible on 500 nm images in Fig. 2c and f. Carbon layers also formed on these structures. As shown in Fig. 1c, the carbon structure on the TNT surface changes with the ratio of C2H2 (Fig. 2a–b and d–e). The carbon layer on the outer surface of the nanotubes and on the surface of the electrode is clearly observed on the images of C-TNT (33:67).

SEM images of TNT material at 5 μM, 2 μM, and 0.5 μM resolution.

SEM images of (a–c) C-TNT (7:93), (d–f) C-TNT (33:67).
Raman spectroscopy was performed on TNT, C-TNT (7:93), and C-TNT (33:67) films. Figure 3 illustrates the Raman spectra for TNT, C-TNT (7:93), and C-TNT (33:67) prepared films. TNT, the unmodified sample, exhibits three very broad bands confirming the presence of the amorphous structure of TiO2. It is notable that the Raman spectra depict broad peaks for the TNT sample, revealing that TNT is amorphous.92,93 Such broad features can be assigned to Ti–O bending (180 cm−1 and 400 cm−1) and Ti–O stretching (600 cm−1) vibrations. For C-TNT (7:93) and C-TNT (33:67) samples, TNT was inserted in a furnace and subjected to heat, and thus the structure of TNT was altered during the solid/liquid/vapor interaction through crystallization.92,93 It is clear that the relatively strong intensity of these peaks indicates the good crystallinity of C-TNT. According to the literature, anatase shows six Raman active modes: A1g (519 cm−1), 2B1g (399 cm−1 and 519 cm−1) and 3Eg (144 cm−1, 197 cm−1, and 639 cm−1). Sharp peaks obtained at A1g (510.6 cm−1), 2B1g (393.6 cm−1 and 510.6 cm−1), and 3 Eg (158.1 cm−1, 206 cm−1, and 632.11 cm−1) for CNT (7:93) and peaks obtained at A1g (512.10 cm−1), 2B1g (393.59 cm−1 and 512.10 cm−1), and 3 Eg (157.6 cm−1, 194.9 cm−1, and 632.11 cm−1) for C-TNT (33:67) correspond to the anatase structure Raman active modes as reported in the literature. These obtained Raman active modes are related to the tetragonal structure of anatase TiO2 with a D4h space group.92,93 All peaks could be observed in the Raman spectrum and confirmed that the prior amorphous TNT layer was successfully transformed into anatase phase via C-doping due to heating. Compared with the values reported in the literature and the broad peaks of amorphous TNT, significant peak shifting was observed after doping with carbon black. The peak shifting became more obvious with the increase in C-doping content (Fig. 3). In detail, for the peak at 144 cm−1, after C-doping, the peaks shifted to the right at 158.1 cm−1 for C-TNT (7:93) and 157.6 cm−1 for C-TNT (33:67). At the peak of 197 cm−1, after C-doping the peaks shifted to the right at 206 cm−1 for C-TNT (7:93) and shifted to the left at 194.6 cm−1 for C-TNT (33:67). The peak at 639 cm−1 shifted to the left for both of the samples to 632.11 cm−1. One can note that the TNT structure was altered and peak shifts were observed after carbon doping. This phenomenon can be attributed to the difference in the stoichiometry of TiO2, difference in the crystallite size, and the existence of carbon in the crystalline structure affecting peak positions due to electronic interactions.92 For C-TNT (7:93) and C-TNT (33:67), Raman peaks corresponding to the pure anatase TiO2 phase were found. For the amorphous carbon structure, the Raman spectrum is generally composed of two peaks: the first in the range of 1320 to 1360 cm−1, called the D-band, and the second peak ranging from 1500 to 1600 cm−1, called the G-band. The D line appears due to the sp2 hybridized carbon structure. The physical properties of amorphous carbon films strongly depend on the ratio of these two types of C–C bonds.87 The inset graph in Fig. 3 illustrates the magnified graph in the 900-100 cm−1 range. For C-TNT (7:93) and C-TNT (33:67), the D line of amorphous carbon was observed at 1374 and 1352, respectively. The G-band of C-TNT (7:93) and the G-band of C-TNT (33:67) were detected at 1586 and 1589, respectively. For TNT, D and G peaks corresponding to the amorphous carbon were not detected. However, the intensity of amorphous carbon peaks increased with the increasing amount of carbon. The D and G peaks obtained for C (7:93) were broad and the intensity was low due to the low amount of carbon doping.87,92,93

Raman spectra of (a) TNT, (b) C-TNT (7:93), and C-TNT (33:67).
XRD patterns of TNT and C-TNT (7:93) electrodes are illustrated in Fig. 4a and b. Crystallographic data revealed that TNT was amorphous just after anodization, and thus there were no peaks confirming the structure in agreement with the Raman spectra of TNT [35]. XRD and Raman results revealed that TNT is amorphous. The XRD pattern of the TNT sample exhibited a pattern with a broad peak at nearly 25.31° related to the TNT anatase (101) plane. For the C-doping of TNT, TNT was inserted in a furnace and subjected to heat. Following this, rapid structural evolution occurred during the solid/liquid/vapor interaction.92,93 As observed in the XRD spectra given in Fig. 4a, the crystallization of the material occurred in anatase form and a sharp peak was observed at 25.31°, related to the (101) crystal face. From the XRD pattern of C-TNT (7:93), peaks at 2θ values of 25.31°, 37.84°, 48.08°, 53.96°, 54.98°, 62.64°, 68.75°, 70.18°, and 75.09° can be ascribed to (101), (004), (200), (105), (211), (204), (116), (200), and (215) planes, respectively, which are characteristic of the anatase phase. The relatively strong intensity of these peaks indicates good crystallinity of C-TNT. For C-TNT, as was mentioned above, the diffraction peaks corresponding to the pure anatase TiO2 phase were found, but other phases assigned to carbon were not observed. This phenomenon was attributed to the fact that carbon content was low and had an amorphous structure. As was mentioned previously, for C-TNT (7:93), Raman spectra illustrated that D and G peaks obtained for C (7:93) were broad and intensity was low due to the low amount of carbon doping, in agreement with the XRD results.92,93

(a) Evolution of the main anatase x-ray diffraction peak ((101) crystal face) during C-doping for TNT and C-TNT (7:93), (b) XRD pattern of C-TNT (7:93).
Electrochemical Measurements
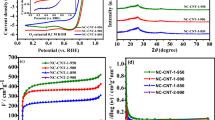
The photocatalytic activity of TNT, C-TNT (33:67), and C-TNT (7:93) was examined by CV in 1 M KOH + 0.5 M GA solution in the dark and under UV illumination. Figure 5a shows the cyclic voltammograms of TNT, C-TNT (33:67), and C-TNTs (7:93) in 1 M KOH solution under dark conditions. According to these measurements, it is clear that C-TNT (7:93) is more active at −0.8 V in the hydrogen adsorption–desorption region. Figure 5b shows the cyclic voltammograms of TNT, C-TNT (33:67), and C-TNT (7:93) in 1 M KOH solution under UV illumination. C-TNT (7:93) exhibits similar behavior under UV illumination as the voltammograms taken in the dark. Figure 5c displays the comparison of the voltammograms taken for C-TNT (7:93) in 1 M KOH solution in dark conditions and under UV illumination. It is clear that electro-oxidation increases under UV illumination.

Cyclic voltammograms of TNT, C-TNT (33:67), and C-TNT (7:93) in 1 M KOH solution at 100 mV s−1 (a) in the dark and (b) under UV illumination, and (c) comparison of cyclic voltammograms on C-TNT (7:93) taken in the dark and under UV illumination.
The photocatalytic activity of TNT, TNT-heated, C-TNT (33:67), and C-TNT (7:93) was measured by CV in 1 M KOH + 0.5 M GA solution in the dark and under UV illumination. GAEO measurements on TNT, C-TNT (33:67), and C-TNT (7:93) in the dark and under UV illumination conditions at the potential range of −0.8–0.8 V and at 100 mV s−1 scan rate are given in Fig. 6a and b, respectively. Figure 6c displays the comparison of the voltammograms taken for C-TNT (7:93) in 1 M KOH + 0.5 M GA solution in dark conditions and under UV illumination. C-TNT (7:93) has the highest specific activity under UV illumination compared to TNT, TNT heated, and C-TNT (33:67) 94,95,96,97,98,99,100,101 This result shows that GAEO is enhanced by altering the surface electronic structure and crystal properties of TNT, called structure sensitivity. By carbon doping on TNT, the surface electronic properties and crystal structure of C-TNT (7:93) and (33:67) were altered, as noted from XRD, Raman spectra, and SEM results. On the other hand, GAEO reaction activity is strongly enhanced under UV illumination on C-TNT (7:93), revealing that the GAEO reaction is a UV-light-driven reaction.

Cyclic voltammograms taken for TNT, TNT-heated, C-TNT (33:67), and C-TNT (7:93) in 1 M KOH + 0.5 M GA solution at 100 mV s−1 (a) in the dark and (b) under UV illumination, and (c) comparison of cyclic voltammograms for C-TNT (7:93) taken in the dark and under UV illumination.
Chronoamperometric measurements were performed on TNT, C-TNT (33:67), and C-TNT (7:93) in 1 M KOH and 0.5 M GA solution at 0.6 V under UV illumination. Figure 7 shows the chronoamperomograms of TNT, C-TNT (33:67), and C-TNT (7:93) under UV illumination. Among these TNT-based electrodes, C-TNT (7:93) exhibits better activity and stability than the other electrodes. Furthermore, the C-TNT (7:93) electrode reveals better activity under UV illumination than that obtained in dark conditions.

Chronoamperomograms taken on TNT, C-TNT (33:67), and C-TNT (7:93) at 0.6 V in 1 M KOH + 0.5 M GA solution under UV illumination.
The EIS technique was used to investigate the chemical and physical processes at the electrode surface that control the activity of the catalyst. Figure 8a shows the electrochemical impedance behavior of C-TNT (7:93) recorded at different potentials under UV illumination. The EIS results indicate that the GA electro-oxidation on C-TNT (7:93) at various potentials has different impedance behaviors. The semicircle diameter of the impedance spectra is equal to the electron transfer resistance (Rct). The smallest Rct was obtained at 0.6 V for C-TNT (7:93). The large arc shown in Fig. 8 exhibits the slow reaction rate of GAEO. This slow kinetics are caused by COads blocking further adsorption and dehydrogenation of GA. The decrease in the arc diameter with increasing potential indicates that the charge transfer resistance of the GA electro-oxidation reaction decreases. There are only capacitive behaviors from the double layer charge in the impedance plots at low potentials.3,4 EIS measurements of TNT, C-TNT (33:67), and C-TNT (7:93) electrodes and Nyquist plots for GAEO under UV illumination are given in Fig. 8b, which shows that the resistance values for of TNT, C-TNT (33:67), and C-TNT (7:93) electrodes at 0.6 V Rct were obtained in the following order: C-TNT (7:93) < C-TNT (33:67) < TNT. As mentioned above, the smallest arc is related to the fast oxidation kinetics of GAEO.94,95,96,97,98,99,100,101 Thus, it is noted that the C-TNT (7:93) electrode exhibits the best GAEO performance in the dark and under UV illumination. In this context, these EIS results are in agreement with results obtained from CV and CA both in the dark and under UV illumination.

Nyquist plots of (a) C-TNT (7:93) at different voltages, (b) TNT, C-TNT (33:67), and C-TNT (7:93) at 0.6 V in 1 M KOH + 0.5 M GA solution under UV illumination.
Conclusion
Herein, TNT was prepared by the anodization method, and carbon doping on TNT was performed in a tubular oven employing two different 15 cm3/min total flow rates with varying compositions of C2H2 and Ar as VC2H2/Ar = 7/93 (1 cm3/min C2H2 + 14 cm3/min Ar) for C-TNT (7:93) and VC2H2/Ar = 33/67 (5 cm3/min C2H2 + 10 cm3/min Ar) for C-TNT (33:67). These TNT, C-TNT (7:93), and C-TNT (33:67) electrode materials were employed as anode materials to investigate their photocatalytic GAEO activity. The materials were characterized by XRD, Raman spectroscopy, and SEM. SEM images revealed that the TNT structure formed as a result of anodization. XRD and Raman results showed broad peaks for the TNT sample assigned to the Ti–O bending and Ti–O stretching vibrations, revealing that TNT was amorphous. For C-TNT (7:93) and C-TNT (33:67) samples, relatively strong intensity of these peaks indicated good crystallinity of C-TNT. Raman peak shifts were also ascribed to the surface electronic change. By carbon doping of TNT, the surface electronic properties and crystal structure of C-TNT (7:93) and C-TNT (33:67) were altered, as noted from XRD, Raman, and SEM results. Photocatalytic GAEO measurements were performed on TNT, C-TNT (7:93), and C-TNT (33:67). GAEO measurements revealed that C-TNT (7:93) had the highest catalytic activity and stability in the dark and under UV illumination. The C-TNT (7:93) electrode exhibited enhanced electro-catalytic activity under UV illumination compared to that measured in dark conditions. This result indicates that GAEO is enhanced for C-TNT (7:93) by altering the surface electronic structure and crystal properties of TNT, called structure sensitivity. The enhanced photocatalytic activity of the electrode could be related to structure sensitivity. This electrode production method is promising for the design of photocatalytic DGFC materials.
References
B. Ulas, A. Kivrak, N. Aktas, and H. Kivrak, Fuller. Nanotub. Carbon Nanostructures 27, 545 (2019).
H. Kivrak, S. Kuliyev, H. Tempel, J. Schneider, and D. Uner, Int. J. Chem. React. Eng. 9, 1 (2011).
O. Sahin and H. Kivrak, Int. J. Hydrog. Energy 38, 901 (2013).
B. Ulas, A. Caglar, O. Sahin, and H. Kivrak, J. Colloid Interface Sci. 532, 47 (2018).
B. Ulas, A. Caglar, and H. Kivrak, Int. J. Energy Res. 43, 3436 (2019).
H. Demir Kivrak, Turk. J. Chem. 39, 563 (2015).
O. Sahin, D. Duzenli, and H. Kivrak, Energ. Source. Part A. 38, 628 (2016).
A. Caglar and H. Kivrak, Int. J. Hydrog. Energy 44, 11734 (2019).
H. Kivrak, M. Can, H. Duru, and O. Sahin, Int. J. Chem. React. Eng. 12, 1 (2014).
H.C. Kazici, F. Yildiz, M.S. Izgi, B. Ulas, and H. Kivrak, Int. J. Hydrog. Energy 44, 10561 (2019).
A. Caglar, T. Sahan, M.S. Cogenli, A.B. Yurtcan, N. Aktas, and H. Kivrak, Int. J. Hydrog. Energy 43, 11002 (2018).
C. Avci, F. Cicek, H.C. Kazici, A. Kivrak, and H. Kivrak, Int. J. Nano Dimens. 9, 15 (2018).
A. Caglar, B. Ulas, M.S. Cogenli, A.B. Yurtcan, and H. Kivrak, J. Electroanal. Chem. 850, 113402 (2019).
A. Caglar, B. Ulas, O. Sahin, and H. Kivrak, Int. J. Energy Res. 43, 8204 (2019).
B. Ulas, A. Caglar, S. Yilmaz, U. Ecer, Y. Yilmaz, T. Sahan, and H. Kivrak, Int. J. Energy Res. 43, 8985 (2019).
T. Oka, H. Mizuseki, and Y. Kawazoe, J. Jpn. Inst. Metals. 70, 495 (2006).
I.M. Al-Akraa, A.M. Mohammad, M.S. El-Deab, and B.E. El-Anadouli, Int. J. Electrochem. Sci. 10, 3282 (2015).
M. Carmo, V.A. Paganin, J.M. Rosolen, and E.R. Gonzalez, J. Power Sources 142, 169 (2005).
F.G.B. San, I. Isik-Gulsac, and O. Okur, Energy 55, 1067 (2013).
H.D. Du, B.H. Li, F.Y. Kang, R.W. Fu, and Y.Q. Zeng, Carbon 45, 429 (2007).
E. Auer, A. Freund, J. Pietsch, and T. Tacke, Appl. Catal. A-Gen. 173, 259 (1998).
C. Rice, S. Ha, R.I. Masel, and A. Wieckowski, J. Power Sources 115, 229 (2003).
M. Takahashi, T. Mori, A. Vinu, D.R. Ou, H. Kobayashi, and J. Drennan, Adv. Appl. Ceram. 107, 57 (2008).
A.K. Shukla, M.K. Ravikumar, and K.S. Gandhi, J. Solid State Electrochem. 2, 117 (1998).
K.M. McGrath, G.K.S. Prakash, and G.A. Olah, J. Ind. Eng. Chem. 10, 1063 (2004).
A. Bayrakceken, U. Kitkamthorn, M. Aindow, and C. Erkey, Scripta Mater. 56, 101 (2007).
B. Guvenatam, B. Ficicilar, A. Bayrakceken, and I. Eroglu, Int. J. Hydrog. Energy 37, 1865 (2012).
A. Bayrakceken, B. Cangul, L.C. Zhang, M. Aindow, and C. Erkey, Int. J. Hydrog. Energy 35, 11669 (2010).
X. Min, B. Sun, S. Chen, M.H. Fang, X.W. Wu, Y.G. Liu, A. Abdelkader, Z.H. Huang, T. Liu, K. Xi, and R.V. Kumar, Energy Storage Mater. 16, 597 (2019).
A. Kivrak, O.F. Er, H. Kivrak, Y. Topal, M. Kus, and Y. Camlisoy, Opt. Mater. 73, 206 (2017).
A. Kivrak, H. Calis, Y. Topal, H. Kivrak, and M. Kus, Sol. Energy Mater. Sol. Cell. 161, 31 (2017).
A. Kivrak, C. Zobi, Y. Torlak, Y. Camlisoy, M. Kus, and H. Kivrak, Appl. Organomet. Chem. 32, e4512 (2018).
V.M. Aroutiounian, V.M. Arakelyan, and G.E. Shahnazaryan, Sol. Energy 78, 581 (2005).
J.H. Park, S. Kim, and A.J. Bard, Nano Lett. 6, 24 (2006).
M. Gratzel, Inorg. Chem. 44, 6481 (2005).
M. Hambourger, G. Kodis, M.D. Vaughn, G.F. Moore, D. Gust, A.L. Moore, and T.A. Moore, Dalton Trans. 45, 9979 (2009).
Y. Jeyashree, Y. Sukhi, A.V. Juliet, S.L. Jame, and S. Indirani, Mater. Sci. Semicond. Process. 107, 104782 (2020).
R. Mirzanamadi, C.E. Hagentoft, and P. Johansson, Renew. Energy 147, 447 (2020).
K. Rashid, K. Mohammadi, and K. Powell, J. Clean. Prod. 248, 119193 (2020).
M.H. Khanmirzaei, S. Ramesh, and K. Ramesh, J. Nanosci. Nanotechnol. 20, 2423 (2020).
P.B.L. Neto, O.R. Saavedra, and D.Q. Oliveira, Renew. Energy 147, 339 (2020).
J.S. Lacerda and J. van den Bergh, Renew. Sustain. Energy Rev. 118, 2 (2020).
C.Y. Chou, C.P. Lee, R. Vittal, and K.C. Ho, J. Power Sources 196, 6595 (2011).
R. Hahn, T. Stergiooulus, J.M. Macak, D. Tsoukleris, A.G. Kontos, S.P. Albu, D. Kim, A. Ghicov, J. Kunze, P. Falaras, and P. Schmuki, Phys. Status Solidi Rapid Res. Lett. 1, 135 (2007).
Q.L. Liu, Z.Y. Zhao, R.D. Zhao, and J.H. Yi, J. Alloys Compd. 819, 153032 (2020).
A. Kudo and Y. Miseki, Chem. Soc. Rev. 38, 253 (2009).
M.Y. Abdelsalam, H.M. Teamah, M.F. Lightstone, and J.S. Cotton, Renew. Energy 147, 77 (2020).
M.J. Cao, Q.S. Xu, X.Y. Qin, and J.L. Cai, Int. J. Electr. Power Energy Syst. 115, 105471 (2020).
S.Q. Hao, A.T.H. Kuah, C.D. Rudd, K.H. Wong, N.Y.G. Lai, J.N. Mao, and X.L. Liu, Sci. Total Environ. 702, 135054 (2020).
K.A. Naik, C.P. Gupta, and E. Fernandez, Int. J. Electr. Power Energy Syst. 115, 105468 (2020).
K. Suleimenov, B. Sarsembayev, B. DucHongPhuc, and T.D. Do, Wind Energy 23, 1026 (2020).
T.T. Guo, Y.B. Liu, J.B. Zhao, Y.W. Zhu, and J.Y. Liu, Int. J. Electr. Power Energy Syst. 116, 105579 (2020).
M. Elkazaz, M. Sumner, and D. Thomas, Int. J. Electr. Power Energy Syst. 115, 105483 (2020).
Z.N. Wang, H. Wang, S. Ji, X.Y. Wang, B.G. Pollet, and R.F. Wang, J. Power Sources 446, 227348 (2020).
K. Anwar and S. Deshmukh, J. Power Energy 234, 96 (2020).
R. Cisneros, R. Gao, R. Ortega, and I. Husain, Int. J. Control Article in press, 1 (2020)
A.A. Candade, M. Ranneberg, and R. Schmehl, Wind Energy 23, 1006 (2020).
M. Rezaei, N. Naghdi-Khozani, and N. Jafari, Renew. Energy 147, 1044 (2020).
S. Kuskaya and F. Bilgili, J. Clean. Prod. 245, 118872 (2020).
F. Fouladi, P. Henshaw, D.S.K. Ting, and S. Ray, Heat Tran. Eng. 41, 407 (2020).
B. Bozkaya and W. Zeiler, Renew Energy 146, 1932 (2020).
A. Çağlar, A. Aldemir, and H. Kivrak, Fullerenes. Nanotub. Carbon Nanostructures 26, 863 (2018).
R. Li, Z. Wei, T. Huang, and A. Yu, Electrochim. Acta 56, 6860 (2011).
S. Sankar, G.M. Anilkumar, T. Tamaki, and T. Yamaguchi, ChemCatChem 11, 4731 (2019).
T.A. Hansu, A. Caglar, O. Sahin, and H. Kivrak, Mater. Chem. Phys. 239, 12201 (2020).
C.-T. Hsieh, W.-H. Lin, Y.-F. Chen, D.-Y. Tzou, P.-Q. Chen, and R.-S. Juang, J Taiwan Inst Chem Eng. 71, 77 (2017).
K.K. Maniam, V. Muthukumar, and R. Chetty, Int. J. Hydrog. Energy 41, 18602 (2016).
B. Ong, S. Kamarudin, and S. Basri, Int. J. Hydrog. Energy 42, 10142 (2017).
D. Chai, X. Zhang, S.H. Chan, and G. Li, J. Taiwan Inst. Chem. Eng. 95, 139 (2019).
D. Basu and S. Basu, Int. J. Hydrog Energy 36, 14923 (2011).
L. Li, K. Scott, and E.H. Yu, J. Power Sources 221, 5 (2013).
A. Brouzgou, L.L. Yan, S.Q. Song, and P. Tsiakaras, Appl. Catal. B Environ. 147, 481 (2014).
A. Brouzgou and P. Tsiakaras, Topics in Catal. 58, 1311 (2015).
Q. Chen, Z. Xia, Y. Zhang, and S. Wang, J. Solid State Electrochem. 23, 3399 (2019).
A. Dokouzis, F. Bella, K. Theodosiou, C. Gerbaldi, and G. Leftheriotis, Mater. Today. Energy 15, 100365 (2020).
F. Bella, A.B. Muñoz-García, F. Colò, G. Meligrana, A. Lamberti, M. Destro, M. Pavone, and C. Gerbaldi, ACS Omega 3, 8440 (2018).
D. Pugliese, A. Lamberti, F. Bella, A. Sacco, S. Bianco, and E. Tresso, Org. Electron. 15, 3715 (2014).
J. Zhou, M. Guo, L. Wang, Y. Ding, Z. Zhang, Y. Tang, C. Liu, and S. Luo, Chem. Eng. J. 366, 163 (2019).
F. Bella, S. Galliano, G. Piana, G. Giacona, G. Viscardi, M. Grätzel, C. Barolo, and C. Gerbaldi, Electrochim. Acta 302, 31 (2019).
F. Bella, A. Verna, and C. Gerbaldi, Mater. Sci. Semicond. Process. 73, 92 (2018).
F. Bella, A. Lamberti, A. Sacco, S. Bianco, A. Chiodoni, and R. Bongiovanni, J. Membr. Sci. 470, 125 (2014).
Y. Gu, Y. Liu, H. Yang, B. Li, and Y. An, Electrochim. Acta 160, 263 (2015).
Y. Gu, H. Yang, B. Li, J. Mao, and Y. An, Electrochim. Acta 194, 367 (2016).
D. Chu, X.H. Li, D.X. Feng, J.S. Gu, and G. Shen, Acta Chim Sin. 62, 2403 (2004).
T. Rafaïdeen, S. Baranton, and C. Coutanceau, Appl. Catal. B 243, 641 (2019).
R.A. Escalona-Villalpando, M.P. Gurrola, G. Trejo, M. Guerra-Balcázar, J. Ledesma-García, and L.G. Arriaga, J Electroanal Chem. 816, 92 (2018).
A. Caglar, B. Ulas, O. Sahin, and H. Demir Kivrak, Energy Storage 1, e73 (2019).
M. Sheikhzadeh, S. Hejazi, S. Mohajernia, O. Tomanec, M. Mokhtar, A. Alshehri, S. Sanjabi, R. Zboril, and P. Schmuki, ChemCatChem 11, 6258 (2019).
N. Khaliq, M.A. Rasheed, G. Cha, M. Khan, S. Karim, P. Schmuki, and G. Ali, Sens. Actuators B. Chem. 302, 127200 (2020).
Z. Zhang, Z. Xu, Z. Yao, Y. Meng, Q. Xia, D. Li, and Z. Jiang, J. Alloys Compd. 805, 396 (2019).
D. Wang, B. Yu, C. Wang, F. Zhou, and W. Liu, Adv. Mater. 21, 1964 (2009).
S. Santangelo, G. Messina, G. Faggio, A. Donato, L. Luca, N. Donato, A. Bonavita, and G. Neri, J. Solid State Chem. 183, 2451 (2010).
A. Lamberti, A. Chiodoni, N. Shahzad, S. Bianco, M. Quaglio, and C.F. Pirri, Sci. Rep. 5, 7808 (2015).
A. Caglar, D. Düzenli, I. Onal, I. Tezsevin, O. Sahin, and H. Kivrak, Int. J. Hydrog. Energy 45, 490 (2020).
O.F. Er, A. Caglar, B. Ulas, H. Kivrak, and A. Kivrak, Mater. Chem. Phys. 241, 122422 (2020).
A. Caglar, M.S. Cogenli, A. Bayrakçeken Yurtcan, and H. Kivrak, Renew. Energy 150, 78 (2020).
B. Ulas, A. Caglar, A. Kivrak, N. Aktas, and H. Kivrak, Ionics 26, 3109 (2020).
T. Avci Hansu, A. Çağlar, O. Sahin, and H. Kivrak, Int. J. Ecosyst. Ecol. Sci. (IJEES). 10, 38 (2020).
H. Demir Kivrak, A. Caglar, T. Avcı Hansu, and O. Sahin, Manas J. Eng (MJEN) 8, 1 (2020).
O. Ozok, E. Kavak, O.F. Er, H. Kivrak, and A. Kivrak, Int. J. Hydrog. Energy 45, 28706 (2020).
A. Caglar, D. Düzenli, I. Önal, İ. Tezsevin, Ö. Sahin, and H. Kivrak, J. Phys. Chem. Solids Article in press, 109684 (2020).
Acknowledgments
The authors would like to thank Kyrgyz-Turk Manas University Scientific Research Fund for financial support with Project No: KTMU-BAP-2019. FBE. 05
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Caglar, A., Kivrak, H., Aktas, N. et al. Fabrication of Carbon-Doped Titanium Dioxide Nanotubes as Anode Materials for Photocatalytic Glucose Fuel Cells. J. Electron. Mater. 50, 2242–2253 (2021). https://doi.org/10.1007/s11664-020-08671-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11664-020-08671-0