
Abstract
A novel electrolytic process that uses a liquid–metal cathode was investigated to produce high-purity magnesium (Mg) metal using magnesium oxide (MgO). Electrolysis of MgO in a magnesium fluoride (MgF2)-lithium fluoride (LiF) molten salt was carried out with applied voltages of 2.5 V to 3.0 V at 1053 K to 1083 K using tin (Sn), silver (Ag), or copper (Cu) as the cathode and graphite or platinum (Pt) as the anode. After electrolysis, Mg alloys with Mg2Sn, AgMg, and Cu2Mg phases were produced with current efficiencies of 77.2 to 83.8 pct when the concentration of Mg in the Mg alloys was 11.9 to 12.9 mass pct. For the production of high-purity Mg metal directly from the Mg alloys, vacuum distillation was performed. When vacuum distillation was conducted at 1200 K to 1300 K, the concentration of Mg in the Mg alloys feed decreased from 30.2 to 34.1 mass pct to 0.32 to 1.75 mass pct, and Mg metal with a purity of 99.975 to 99.999 pct was obtained. Therefore, this study demonstrates that the production of high-purity Mg metal through an efficient and environmentally sound method using the electrolysis of MgO is feasible.

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Magnesium (Mg) is the lightest structural metal and has excellent physical properties, such as high specific strength, castability, and machinability.[1] Owing to these superior properties, one of the major uses of Mg is vehicle applications for the reduction of weight to improve fuel efficiency. For example, 41.2 pct of the global production of Mg was consumed for transportation application in 2015.[2] In addition, the demand for Mg will increase in the future, especially for use in automobiles, because of the need to reduce carbon dioxide (CO2) gas emissions.[3]
Mg metal is commercially produced through thermal reduction or electrolytic methods. At present, the Pidgeon process, which uses the thermal reduction method, is predominantly used for Mg metal production.[4] In the Pidgeon process, dolime (CaO·MgO), which is prepared by the calcination of dolomite (CaCO3·MgCO3) at 1273 K to 1573 K, reacts with ferro-silicon (Fe-Si) at 1373 K to 1473 K under vacuum.[5,6,7] The reduced Mg metal is then evaporated, and Mg crowns are obtained by the condensation of gaseous Mg metal. However, this process has disadvantages, such as high energy consumption, low labor productivity, and environmental concerns about the generation of large amounts of sulfur oxide (SOx) gas by the burning of coal.[7,8]
Several processes, such as the MagCorp, Hydro Magnesium, VAMI, DSM, IG Farben, and Dow processes, were developed utilizing electrolytic methods.[2,7,9,10,11,12] In the electrolytic processes, the preparation of anhydrous magnesium chloride (MgCl2), carnallite (MgCl2·KCl), or partially dehydrated MgCl2 (MgCl2·1.25 H2O) from brine, sea water, magnesite (MgCO3), or serpentine (3 MgO·2 SiO2·2 H2O) is necessary before the electrolysis is conducted.[2,7,11] Afterward, the molten salt electrolysis of MgCl2 is then performed at 928 K to 993 K.[7] As a result of the electrolysis, liquid Mg metal and chlorine (Cl2) gas are produced at the cathode and anode, respectively. However, these electrolytic processes have several disadvantages, such as the intense energy required for anhydrous MgCl2 preparation, generation of Cl2 gas, and contamination of iron (Fe) in the Mg metal produced.[7]
In order to resolve the drawbacks of current commercial electrolytic processes, a few studies have been carried out for the development of a novel Mg metal production process using magnesium oxide (MgO) as a feedstock.[13,14,15,16,17,18,19] Among the Mg production processes that were investigated, the solid oxide membrane (SOM) process has drawn considerable attention because of its promising features.[15,16,17,18] One important feature of this process is the oxygen ions that migrate inside the yttria-stabilized zirconia (YSZ) membrane are oxidized at the liquid silver (Ag) anode during the electrolysis. As a result, Mg metal and oxygen (O2) gas are produced. The advantages of the SOM process are that the preparation of MgCl2 is not necessary and that O2 gas evolves at the anode. However, the disadvantages of the process are the decrease of the current efficiency from 90 pct to 40 to 50 pct when the electrolysis continues without argon (Ar) gas bubbling and degradation of the YSZ membrane.[17]
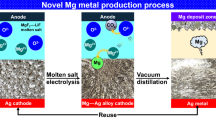
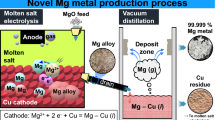
In order to produce high-purity Mg metal directly from MgO using a simple electrolysis cell even with O2 gas evolution, molten salt electrolysis of MgO using a liquid–metal cathode to produce high-density Mg alloys and vacuum distillation of the Mg alloys to produce high-purity Mg metal were investigated. Figures 1 and 2 show the flowchart and schematic of the novel Mg production process investigated, respectively. As shown in Figure 1, MgO is used as the feedstock. Moreover, toxic gases such as Cl2 gas do not evolve in the process. When an inert electrode such as platinum (Pt) is used, O2 gas is generated at the anode. Furthermore, high current efficiencies can be obtained because the reaction between the Mg metal in the Mg alloy and a reactive gas such as the O2 gas generated at the anode can be hindered. High-purity Mg metal can be obtained with the use of vacuum distillation. Finally, scale-up of the process should be feasible because an electrolysis cell similar to the type of the electrolysis cell used in the Hall–Heroult process can be used in the process.

Flowchart of the novel Mg metal production process

Schematic of the novel Mg metal production process
Mechanism of the Novel Magnesium Metal Production Process
Figure 3 shows the theoretical decomposition voltages of some selected fluorides and MgO at elevated temperatures.[20] The decomposition voltage was obtained from the Nernst equation using the Gibbs free energy change (∆G°) and the number of electrons transferred (n) in a reaction. The theoretical decomposition voltages under the standard state, i.e., MgO in form of the solid phase, were calculated at the elevated temperatures because it is an appropriate method for the evaluation of the stability of species in an electrolyte.

Theoretical decomposition voltages of selected fluorides and magnesium oxide at elevated temperatures
In this study, the eutectic composition of 54 mass pct magnesium fluoride (MgF2)-46 mass pct lithium fluoride (LiF) was selected for the electrolyte after considering the dissolution of MgO, the wide potential windows of MgF2 and LiF, and the low melting temperature of the electrolyte (refer to Figure S-1[21] in the electronic supplementary material). The eutectic point of the electrolyte is 1014 K, therefore, the temperature for the electrolysis of MgO in the MgF2-LiF molten salt was determined to be 1053 K to 1083 K.
In addition, tin (Sn), Ag, and copper (Cu) were selected as the cathode metals. Figures 4(a) through (c) show the binary phase diagrams of the Mg-Sn, Mg-Ag, and Mg-Cu systems, respectively, with the isobaric line of the vapor pressure of Mg (pMg) as a function of the temperature and the concentration of Mg.[21] As shown in Figure 4(a), when Sn metal is used as the cathode, the maximum concentration of Mg electrodeposited is expected to be 27 pct at 1083 K and 30 pct at 1053 K when the pMg is considered. This is because when pMg exceeds 0.01 atm, the Mg metal produced will evaporate during electrolysis,[22] resulting in a decrease in the current efficiency of the process.

Binary phase diagrams of the (a) Mg-Sn, (b) Mg-Ag, and (c) Mg-Cu systems with the isobaric line of vapor pressure of Mg as a function of temperature and Mg concentration
Furthermore, the temperature required for the vacuum distillation of the Mg-Sn alloy can also be assessed from Figure 4(a). When the pressure of the system is lower than 1 atm, the metal begins to evaporate at vapor pressures lower than 0.01 atm.[22] As a result, even though pMg is in the range of 0.0001 atm to 0.001 atm, the Mg in Mg alloys is expected to evaporate during vacuum distillation. Therefore, when the recovery of a large amount of Mg from the Mg-Sn alloy is considered, the appropriate temperature for vacuum distillation is in the range of 1200 K to 1300 K.
As shown in Figure 4(b), when Ag metal is used as the cathode, the maximum concentration of Mg electrodeposited is expected to be 21 pct at 1083 K and 27 pct at 1053 K. In addition, 1300 K is the appropriate temperature for the vacuum distillation of Mg-Ag alloy. Figure 4(c) shows that the maximum concentration of Mg electrodeposited is expected to be 25 pct at 1083 K and 31 pct at 1053 K when Cu metal is used as the cathode. In addition, Mg metal will be recovered from Mg-Cu alloy at 1200 K to 1300 K through vacuum distillation.
Consideration of the densities of the Mg alloys and the electrolyte is also necessary. The theoretical density of 4.8 mass pct MgO-MgF2-LiF is 2.91 g/cm3, while the theoretical densities of 30 mass pct Mg in Sn, Ag, and Cu are 3.45 g/cm3, 3.78 g/cm3, and 3.61 g/cm3, respectively. Therefore, until the maximum concentration of Mg metal of the aforementioned Mg alloys is reached, liquid Mg alloys will be produced at the bottom of the electrochemical reduction cell.
Experimental
Preparation of the Electrodes and Salts
Before the experiments, MgF2 (anhydrous powder, purity > 99.9 pct; Kojundo Chemicals Inc.) and LiF (anhydrous powder, purity > 99.9 pct; Kojundo Chemicals Inc.) were dried at 453 K for more than 72 hours using a vacuum oven (Model no.: VOS-601SD, EYELA).
Each electrode was physically connected to a nickel (Ni) wire (purity > 99.5 pct; ϕ 1 mm; Alfa Aesar Chemical Co. Ltd.) and was inserted into an alumina (Al2O3) tube (purity > 99.8 pct; ϕ = 6 mm, O.D., t = 1 mm, thickness). The gap between the electrode and the Al2O3 tube was filled with an Al2O3-based paste (Aron Ceramic D; Toagosei Co., Ltd.) and dried at room temperature for 24 hours (see Figure 5(b)). Subsequently, the prepared electrodes were dried at 363 K for 2 hours, 423 K for 2 hours, and 773 K for 5 hours under vacuum.

Experimental apparatus used for cyclic voltammetry: (a) schematic diagram, (b) photographs of the electrodes assembled with the top flange, and (c) stainless steel reactor
Cyclic Voltammetry
Figure 5 shows the schematic and photographs of the experimental apparatus used for the cyclic voltammetry (CV). As shown in Figure 5(a), a mixture of MgF2-LiF or MgF2-LiF-MgO (powder, purity > 99.99 pct; Kojundo Chemicals Inc.) was placed in a carbon (C) crucible. In addition, the Ag shot (purity: 99.99 pct, Sungeel Himetal) was placed in the C crucible to absorb the generated metals during the pre-electrolysis of the mixtures of MgF2-LiF and MgF2-LiF-MgO. The crucible was positioned in a stainless steel reactor, and the electrodes assembled with the top flange were set up with the reactor, as shown in Figures 5(b) and (c).
When the cathodic sweep was conducted, a molybdenum (Mo: purity > 99.95 pct; ϕ 0.5 mm; Alfa Aesar Chemical Co. Ltd.) wire, Pt (purity > 99.95 pct; ϕ 1 mm; Alfa Aesar Chemical Co. Ltd.) wire, and graphite (ϕ 3 mm; SGL Carbon Group) or glassy C (ϕ 3 mm; Tokai Fine Carbon Co., Ltd.) rod were used as the working, quasi-reference, and counter electrodes, respectively. In addition, when the anodic sweep was performed, a graphite or glassy C rod, Pt wire, and Mo wire were employed as the working, quasi-reference, and counter electrodes, respectively.[23] The reactor was evacuated for 10 min, and Ar gas (purity: 99.9999 pct) was flowed into the reactor until the internal pressure reached 1 atm. During the experiments, Ar gas was flowed maintaining 1 atm of internal pressure.
After the reactor was positioned in an electric furnace, the temperature was increased and kept at 773 K for 24 hours to remove residual H2O from the system. Subsequently, the CV measurements were conducted at 1083 K using a potentiostat (Model no.: VMP3, booster: VMP3B, 2 A to 20 V; Biologic Science Instruments).
Electrolysis of MgO Using a Liquid–Metal Cathode
Before the electrolysis of MgO, pre-electrolysis was conducted at 1083 K to remove the impurities in the molten salt used. The mixture of MgF2-LiF-MgO was placed in the C crucible, and the crucible was positioned inside the stainless steel reactor. The Ni wire cathode and the graphite rod anode were set up with the top flange. The reactor was then evacuated for 10 minutes, and Ar gas was flowed into the reactor until the internal pressure reached 1 atm. During the experiments, Ar gas was flowed maintaining 1 atm of internal pressure. After the atmosphere control was finished, the temperature of the reactor was increased and kept at 1083 K. In the pre-electrolysis process, 0.5 A was applied for 4 hours. When the pre-electrolysis was finished, the electrodes were removed from the molten salt, and the reactor was cooled to room temperature.
After the pre-electrolysis was completed, an Al2O3 crucible (purity > 99.5 pct, ϕ = 18 mm, O.D., t = 1.5 mm, thickness, h = 25 mm, height) containing Cu, Ag, or Sn was assembled with a graphite rod shielded by an Al2O3 tube and placed at the bottom of the new empty C crucible, as shown in Figure 6. In addition, the Ag shot and pieces of MgF2-LiF-MgO mixture obtained after pre-electrolysis were placed in the crucible. A graphite rod or Pt plate for the anode was assembled with a stainless steel flange in Figure 6 before the experiments. The anode was lowered into the electrolyte during the experiments. When the graphite anode was used, two graphite rods connected externally via Ni wiring were used to control the current density.

Schematic of the experimental apparatus used for the electrolysis of MgO
The reactor was evacuated for 10 minutes, and Ar gas flowed into the reactor until the internal pressure reached 1 atm. During the experiments, Ar gas was flowed maintaining 1 atm of internal pressure. After a controlled atmosphere was established, the temperature of the reactor was increased and kept at 1053 K to 1083 K. For the electrolysis of MgO, 2.5 V to 3.0 V was applied for 10.4 to 15.5 ks using the potentiostat. After the electrolysis, the reactor was cooled to room temperature and the Mg alloys obtained in the Al2O3 crucible were recovered. After the Mg alloys were separated from the Al2O3 crucible, the salt on the surface of the Mg alloys was completely removed using abrasive paper. Thereafter, the Mg alloys were pulverized using a mortar for the analysis.
Vacuum Distillation of Mg Alloys
In order to demonstrate the separation of Mg metal from the Mg alloys by utilizing the difference of vapor pressure between Mg and the cathode metals such as Cu, Ag, or Sn, vacuum distillation of the Mg alloys prepared by melting of Mg and Cu, Ag, or Sn was conducted. Before the vacuum distillation, melting of Mg and Cu, Mg and Ag, and Mg and Sn mixtures in the C crucible were conducted under vacuum at 1023 K to 1073 K for 4 to 5 hours, at 1023 K to 1073 K for 5 to 7 hours, and at 1013 K for 15 hours, respectively, using an electric or high frequency induction furnace. The melting was conducted to use the sufficient amount of Mg alloy feed for the vacuum distillation.
Figure 7 shows the schematic and a photograph of the experimental apparatus used for the vacuum distillation. The Mg alloys were placed at the bottom of the steel reactor. The reactor was plugged using a silicone plug and evacuated during the experiments using a rotary pump (Model no.: GLD-201B, ULVAC KIKO, Inc.). The steel reactor was positioned in the electric furnace, which was preheated to 1200 K to 1300 K for 5 to 10 hours. After the vacuum distillation experiments were finished, the reactor was immediately removed from the furnace and cooled to room temperature. The reactor was then cut and the samples were collected.

Experimental apparatus used for vacuum distillation: (a) schematic diagram and (b) photograph
Analysis
The concentration of elements in the samples was analyzed using inductively coupled plasma optical emission spectroscopy (ICP-OES: Perkin Elmer, Optima 5300DV and Thermo Fisher Scientific, iCAP 6500) or glow discharge mass spectroscopy (GD-MS: MSI, GD90RF). The crystalline phases of the samples obtained were identified using X-ray diffraction (XRD: Rigaku, SmartLab, Cu-Kα radiation). Additionally, the compositions and surfaces of the samples were analyzed using field emission scanning electron microscopy/energy dispersive X-ray spectroscopy (FE-SEM/EDS, Merlin Compact, Carl Zeiss AG/AZTEC, Oxford Instruments).
Results and Discussion
Cyclic Voltammetry
Figures 8(a) through (c) show the results of the CV measurement of the MgF2-LiF molten salt before the pre-electrolysis, the MgF2-LiF molten salt after the pre-electrolysis, and the addition of MgO in the MgF2-LiF molten salt obtained after the pre-electrolysis using a graphite anode with respect to the Pt quasi-reference electrode at 1083 K, respectively. The potentials indicated in the figures were not corrected for the ohmic drop.

Results of CV measurement of the (a) MgF2-LiF molten salt before pre-electrolysis, (b) MgF2-LiF molten salt after pre-electrolysis, and (c) addition of MgO in the MgF2-LiF molten salt after pre-electrolysis using a graphite anode with respect to the Pt quasi-reference electrode at 1083 K
As shown in Figure 8(a), a small cathodic current was observed at − 0.68 V (vs Pt quasi-reference electrode) owing to the reduction of cations of impurities in the molten salt. In addition, a large cathodic current was observed at − 1.70 V (vs Pt quasi-reference electrode) owing to the reduction of Mg2+ to Mg in the molten salt, as shown in Eq. [1]. When the anodic sweep was conducted, a small anodic current was observed at 0.90 V (vs Pt quasi-reference electrode) and a large anodic current was observed at 3.01 V (vs Pt quasi-reference electrode). The result of the anodic current at 0.90 V (vs Pt quasi-reference electrode) indicates that the anions of the impurities contained in the MgF2-LiF molten salt were oxidized. As shown in Figure 8(a), the span of the potential window of impurities contained in MgF2-LiF molten salt (vs Pt quasi-reference electrode) was 1.58 V. Therefore, it is expected that impurities in the form of oxides were contained in the MgF2-LiF molten salt because the theoretical decomposition voltages of MgF2 and LiF are 4.84 V to 5.31 V.[20] In addition, a large anodic current was observed at 3.01 V (vs Pt quasi-reference electrode) owing to the generation of fluorine (F2) gas by the oxidation of F− in the MgF2-LiF molten salt, as shown in Eq. [2].
However, when the CV measurements were conducted after pre-electrolysis, even a small anodic current was not observed until the generation of the F2 gas. The results shown in Figure 8(b) indicate that the estimated decomposition voltage of MgF2 is 4.62 V.
When the CV measurements were conducted at 1083 K for the 4.8 mass pct MgO-MgF2-LiF molten salt obtained after the removal of impurities, the first large anodic current was analyzed at 0.36 V (vs Pt quasi-reference electrode), as shown in Figure 8(c). This is because the O2− was oxidized at the graphite anode, as shown in Eq. [3]. These results indicate that the estimated decomposition voltage of MgO is 2.01 V at 1083 K when a graphite anode is used under the experimental conditions. In addition, a second large anodic current started at 3.03 V (vs Pt quasi-reference electrode) due to the oxidation of F− to F2 gas. Therefore, the estimated decomposition voltage of MgF2 is 4.68 V.
Figures 9(a) through (c) show the results of the CV measurements of the MgF2-LiF molten salt before the pre-electrolysis, the MgF2-LiF molten salt after the pre-electrolysis, and the addition of MgO in the MgF2-LiF molten salt obtained after pre-electrolysis, using the glassy C anode with respect to the Pt quasi-reference electrode at 1083 K, respectively.

Results of CV measurement of the (a) MgF2-LiF molten salt before pre-electrolysis, (b) MgF2-LiF molten salt after pre-electrolysis, and (c) addition of MgO in the MgF2-LiF molten salt after pre-electrolysis using a glassy C anode with respect to the Pt quasi-reference electrode at 1083 K
As shown in Figure 9(a), the redox potential for Mg2+/Mg of MgF2-LiF molten salt before the pre-electrolysis was − 1.75 V (vs Pt quasi-reference electrode). In addition, a large anodic current was observed at 1.11 V (vs Pt quasi-reference electrode). As a result, it is expected that impurities in the form of oxides in the MgF2-LiF molten salt were decomposed at 1083 K when the cell voltage exceeded 2.86 V.
Figure 9(b) shows the results of the CV measurement of MgF2-LiF molten salt after the pre-electrolysis at 1083 K. When the anodic sweep was conducted, a large anodic current was observed at 2.45 V (vs Pt quasi-reference electrode) owing to the oxidation of F− to F2 gas, as shown in Eq. [2]. As a result, the estimated decomposition voltage of MgF2 at 1083 K using the glassy C anode was 4.08 V, when the redox potentials for Mg2+/Mg and F−/F2 were considered.
When the CV measurements were conducted at 1083 K for the 4.8 mass pct MgO-MgF2-LiF molten salt obtained after the removal of impurities, the first large anodic current was observed at 1.37 V (vs Pt quasi-reference electrode), as shown in Figure 9(c), which was attributed to the oxidation of O2− in the MgO-MgF2-LiF molten salt. Therefore, the estimated decomposition voltage of MgO using the glassy C anode at 1083 K was 3.02 V under the experimental conditions when the redox potential for Mg2+/Mg, − 1.65 V (vs Pt quasi-reference electrode), was considered. These results show that the estimated decomposition voltage of MgO using the glassy C anode is larger than that of MgO using the graphite anode by approximately 1.0 V. From these results, O2 gas can be expected to have evolved when a glassy C anode was used. In addition, a second large anodic current was observed at 2.51 V (vs Pt quasi-reference electrode), as shown in Figure 9(c), due to the oxidation of F− to F2 gas. As a result, the estimated decomposition voltage of MgF2 at 1083 K using the glassy C anode was 4.16 V.
Electrolysis of MgO using a Liquid–Metal Cathode
Table I shows the experimental conditions of the electrolysis of MgO using a Sn, Ag, or Cu cathode. Table II shows the results of the electrolysis of MgO using a liquid–metal cathode and the analytical results of the Mg alloys obtained.
As shown in Tables I and II, when the electrolysis of MgO was conducted using a Sn cathode and a graphite anode with an applied cell voltage of 2.5 V at 1083 K and 1053 K for 10.4 to 14.2 ks, the current efficiencies were 82.2 pct and 80.1 pct, respectively. These results indicate that the influence of the difference in the reaction temperatures on the current efficiency is not large until the concentration of Mg in the Mg-Sn alloy reached 12.6 to 12.7 mass pct. However, as expected in Section II, the decrease of the temperature required for the electrolysis, from 1083 K to 1053 K, will increase the current efficiency when the concentration of electrodeposited Mg increases beyond 12.6 to 12.7 mass pct owing to the decrease of the pMg of the Mg-Sn alloy.
Figure 10 shows the results of the XRD analysis of Mg alloys obtained after the electrolysis of MgO at 1053 K to 1083 K using Sn, Ag, or Cu as the cathode and graphite or Pt as the anode. Figure 11 shows the results of the SEM and EDS analysis of the Mg alloys obtained after the electrolysis at 1053 K to 1083 K using Sn, Ag, or Cu as the cathode and graphite as the anode.

Results of the XRD analysis of Mg-Sn alloy obtained at (a) 1083 K and (b) 1053 K, those of (c) Mg-Ag and (d) Mg-Cu alloys obtained at 1053 K using a graphite anode, and that of (e) Mg-Sn alloy obtained at 1053 K using a Pt anode

Results of SEM and EDS analysis of the cross-sections of the obtained (a) Mg-Sn (Exp. no. 190930), (b) Mg-Ag (Exp. no. 191209), and (c) Mg-Cu (Exp. no. 200106) alloys
As shown in Figures 10(a) and (b), Mg2Sn and Sn (Mg) were produced at the cathode after the electrolysis of MgO, using a Sn cathode and a graphite anode at 1053 K to 1083 K, as expected from the binary phase diagram of the Mg-Sn system when the concentration of Mg is 12.6 to 12.7 mass pct.[24] Furthermore, as shown in Figure 11(a), the microstructure of the Mg-Sn alloy is a mixture of Sn-rich and Mg-rich phases. For example, the results of the EDS analysis indicated that the concentrations of Mg in the Sn- and Mg-rich phases were 0.4 mass pct and 27.2 mass pct, respectively.
In addition, the electrolysis of MgO was conducted using an Ag or Cu cathode and a graphite anode with an applied cell voltage of 2.5 V at 1053 K for 12.1 to 14.3 ks. It is worth noting that Ag or Cu was solid before the electrolysis because the melting temperature of Ag and Cu is higher than 1053 K. Figures 12(b), (c), (e), and (f) show the photographs of Ag and Cu metals before the electrolysis and those of Mg alloys obtained after the electrolysis of MgO using Ag and Cu as the cathodes. As shown in Figures 12(e) and (f), lumps of Mg alloy were obtained after the electrolysis because the liquid cathodes were formed at 1053 K as the electrolysis proceeded, owing to Mg metal that was electrodeposited, as expected from the binary phase diagrams of the Mg-Ag and Mg-Cu systems.[24]

Photographs of (a) Sn, (b) Ag, and (c) Cu before the electrolysis and those of (d) Mg-Sn (Exp. no. 190930), (e) Mg-Ag (Exp. no. 191209), and (f) Mg-Cu (Exp. no. 200106) alloys after the electrolysis
Table II shows that the current efficiencies of the electrolysis of MgO at 1053 K using Ag and Cu cathodes were 83.8 and 77.2 pct, respectively. In these experiments, the concentrations of Mg in Mg-Ag alloy and Mg-Cu alloy were 12.9 and 11.9 mass pct, respectively. When the pMg of the Mg-Cu alloy is considered, as shown in Figure 4(c), it is expected that the current efficiency would be similar to the current efficiencies obtained by the electrolysis using the Sn or Ag cathode. However, the current efficiency of the electrolysis of MgO using the Cu cathode was found to be slightly smaller than that obtained using the Ag or Sn cathode. However, the exact reason for this reduction in the current efficiency is still under investigation.
Figures 10(c) and (d) show the results of the XRD analysis of the Mg-Ag and Mg-Cu alloys obtained after the electrolysis using a graphite anode at 1053 K, respectively. As shown in Figure 10(c), an AgMg phase was identified after the electrolysis of MgO using the Ag cathode. When the binary phase diagram of the Mg-Ag system and the concentration of Mg in the Mg-Ag alloy are considered, the production of AgMg and Ag3Mg phases is expected under slow cooling.[24] However, the binary phase diagram of the Mg-Ag system is incomplete for compositions that have less than 45 mass pct Mg. Figure 10(d) shows that the Cu2Mg phase and the Cu (Mg) phase were identified, as expected from the binary phase diagram of the Mg-Cu system when the concentration of Mg in Mg-Cu alloy is 11.9 mass pct.
Figures 11(b) and (c) show the microstructure of the Mg-Ag and Mg-Cu alloys obtained after the electrolysis of MgO using Ag and Cu cathodes, respectively. As shown in Figure 11(b), Ag-rich phases were not observed. These results indicate that only intermetallic phases, such as AgMg, were produced. Unlike in the microstructure of the Ag-Mg alloy, a mixture of Cu- and Mg-rich phases was observed in the microstructure of the Mg-Cu alloy. For example, the results of the EDS analysis showed that the concentrations of Mg in the Cu- and Mg-rich phases were 2.6 and 14.3 mass pct, respectively.
In order to demonstrate the feasibility of the novel electrolytic process under the evolution of O2 gas at the anode, the electrolysis of MgO was conducted at 1053 K for 15.5 ks with an applied voltage of 3.0 V using a Sn cathode and a Pt anode. When the concentration of Mg in the Mg-Sn alloy obtained was 12.6 mass pct, the current efficiency was 81.7 pct. As shown in Table II, this result is similar to the current efficiencies obtained when the electrolysis of MgO was conducted using a graphite anode. In addition, it is expected that O2 gas was generated at the anode under these conditions when the results of the CV measurements in Sect. IV–A were considered. Therefore, these results indicate that the novel electrolytic process investigated in this study exhibited 77.2 to 83.8 pct current efficiency when the graphite or inert electrode was used at 1053 K until the concentration of Mg was 11.9 to 12.9 mass pct.
Figure 10(e) shows the results of the XRD analysis of the Mg-Sn alloy obtained after the electrolysis of MgO at 1053 K using a Sn cathode and a Pt anode. As expected from the binary phase diagram of the Mg-Sn system, when the concentration of Mg is 12.6 mass pct, a mixture of Mg2Sn and Sn (Mg) was obtained after the electrolysis.[24]
Table II also shows that the Mg alloys obtained after electrolysis contained aluminum (Al). It is expected that the Al originated from the Al2O3 crucible that was used for holding the liquid–metal cathode in the reaction between Mg in the Mg alloys and the crucible. However, it is worth noting that the Al2O3 crucible maintained its shape even after the electrolysis experiments. These results indicate that the effect of Mg in Mg alloys on the reduction of the Al2O3 crucible was not large owing to the low activity of Mg. However, further investigation on the solubility of Al2O3 in the MgF2-LiF molten salt at 1053 K to 1083 K is necessary.
As shown in Table II, the current efficiency of the electrolysis of MgO in the MgF2-LiF molten salt at 1053 K to 1083 K did not reach 100 pct and was 77.2 to 83.8 pct. The electrodeposition of the impurities, reactions between the Mg alloy and the Al2O3 crucible, and the evaporation loss of Mg from the Mg alloy could be the reasons that reduced the current efficiency below 100 pct. Moreover, under these electrolysis conditions, the current density was 0.372 to 0.554 A/cm2. The current efficiency and current density of a commercial electrolytic process using MgCl2 are approximately 70 to 90 pct and 0.55 to 0.60 A/cm2, respectively.[25, 26] The current efficiency of the new Mg production process developed is close to that of the commercial electrolytic process although the current density is slightly lower than that of the commercial electrolytic process.
The anode system is important for a commercial electrolysis process. When large-scale production of Mg was considered using the developed process in this study, the use of a C anode is preferred from the short-term perspective. In this process, Mg metal is produced at the bottom of the cell. Therefore, C contamination is prevented because C powders can float on the surface of the electrolyte.
Vacuum Distillation of Mg Alloys
In order to demonstrate the feasibility of the production of high-purity Mg metal from Mg alloys, such as Mg-Sn, Mg-Ag, and Mg-Cu, vacuum distillation of the Mg alloys was conducted. Table III shows the experimental conditions and analytical results of the Mg metal obtained at the low-temperature part and the residues obtained at the bottom of the reactor after vacuum distillation.
Vacuum distillation of the Mg-Cu alloy was conducted at 1200 K to 1300 K for 5 to 10 hours. As shown in Table III, when the vacuum distillation was conducted at 1200 K for 5 hours, the concentration of Mg in the Mg-Cu alloy feed decreased from 33.9 to 7.02 mass pct, and Mg metal with a purity of 99.965 pct was obtained at the low-temperature part of the reactor (see Figure S-2 in the electronic supplementary material). To increase the recovery efficiency of Mg from the Mg-Cu alloy, the temperature of the vacuum distillation was increased to 1300 K. As a result, the concentration of Mg in Mg-Cu alloy feed decreased from 30.2 to 0.78 mass pct, and Mg metal with a purity of 99.999 pct was obtained. From the experimental results, it can be concluded that Mg can be efficiently recovered from a Mg-Cu alloy at 1300 K using vacuum distillation.
In addition, the vacuum distillation of the Mg-Ag alloy was conducted at 1200 K to 1300 K for 5 hours. As shown in Table III, when the vacuum distillation was conducted at 1200 K, the concentration of Mg in the Mg-Ag alloy feed decreased from 30.5 to 5.72 mass pct, and Mg metal with a purity of 99.999 pct was obtained at the low-temperature part of the reactor (see Figure S-3 in the electronic supplementary material). The recovery efficiency of Mg from Mg-Ag alloy increased when the vacuum distillation was conducted at 1300 K because the concentration of Mg in the residue obtained at the bottom of the reactor was 1.75 mass pct. In addition, Mg metal with a purity of 99.975 pct was produced. These results indicate that 1300 K is appropriate for the vacuum distillation of a Mg-Ag alloy, as mentioned in Section II.
Vacuum distillation of the Mg-Sn alloy was conducted at 1200 K for 5 hours. As shown in Table III, Mg metal with a purity of 99.998 pct was obtained at the low-temperature part of the reactor (see Figure S-4 in the electronic supplementary material), and the concentration of Mg in the Mg-Sn alloy feed decreased from 34.1 to 0.32 mass pct. Therefore, it can be concluded that Mg can be efficiently recovered from a Mg-Sn alloy at 1200 K, as expected in Section II.
Figure 13 shows the results of XRD analysis of Cu-Mg, Ag-Mg, and Sn-Mg alloys used as a feedstock for the vacuum distillation and the Mg metal obtained. As shown in Figure 13, high-purity Mg metal was obtained by the vacuum distillation of Cu-Mg, Ag-Mg, Sn-Mg alloys at 1200 K to 1300 K.

Results of the XRD analysis of (a) Cu-Mg, (b) Ag-Mg, and (c) Sn-Mg alloys used as feedstock for the vacuum distillation and (d) Mg metal obtained after the vacuum distillation
When the results of the electrolysis of MgO followed by the vacuum distillation are taken into consideration, the use of Sn as the cathode for the developed process is preferred. Table II shows no significant difference in the current efficiencies obtained when Sn, Ag, or Cu was used as the cathode and C or Pt was used as the anode until the concentration of Mg was 11.9 to 12.9 mass pct. However, the vacuum distillation temperature for the recovery of Mg from Mg-Sn alloy was 1200 K, which was the lowest among the recorded temperatures for the vacuum distillation. In addition, 30 mass pct of Mg can be electrodeposited on the Sn cathode, as expected from the discussion in Section II, and Sn has the lowest melting point among Sn, Ag, and Cu.
Conclusions
To develop an efficient and environmentally friendly Mg production process, molten salt electrolysis using a liquid–metal cathode (MSE-LMC) and vacuum distillation to produce high-purity Mg metal using MgO as the feedstock was investigated. The estimated decomposition voltages of MgO in 54 mass pct MgF2-46 mass pct LiF molten salt at 1083 K under the experimental conditions when using graphite and glassy C anodes were 2.01 V and 3.02 V, respectively. Through the electrolysis of MgO in MgF2-LiF molten salt with an applied voltage of 2.5 V to 3.0 V at 1053 K to 1083 K using graphite or Pt as the anode, Mg alloys such as a Mg2Sn and Sn (Mg) mixture, AgMg, and a Cu2Mg and Cu (Mg) mixture were produced when Sn, Ag, and Cu were used as the cathode, respectively. The current efficiency and current density were 77.2 to 83.8 pct and 0.372 to 0.554 A/cm2, respectively, when the concentration of Mg in the Mg alloys was 11.9 to 12.9 mass pct. In addition, Mg metal with a purity of 99.975 to 99.999 pct was obtained by the vacuum distillation of Mg-Cu, Mg-Ag, and Mg-Sn alloys prepared in advance at 1300 K, 1300 K, and 1200 K, respectively.
References
C. Zhong, F. Liu, Y. Wu, J. Le, L. Liu, M. He, J. Zhu, and W. Hu: J. Alloy. Compd, 2012, vol. 520, pp. 11–21.
Roskill: Magnesium Metal: Global Industry, Markets and Outlook to 2020, 12th ed., Roskill Information Services Ltd., London, 2016.
U.S. Department of Energy: Materials Technologies: Goals, Strategies, and Top Accomplishments, 2010, https://www.enegy.gov/sites/prod/files/2014/03/f13/materials_tech_goals.pdf.
M. Tauber: Proceedings of 76th International Magnesium Association World Magnesium Conference, Int. Magnesium Ass., Budapest, Hungary, 2019, pp. 1–16.
Y. Wada, S. Fujii, E. Suzuki, M. M. Maitani, S. Tsubaki, S. Chonan, M. Fukui, and N. Inazu: Sci. Rep., 2017, vol. 7, pp. 1-7.
L.M. Pidgeon and W.A. Alexander: T. Am. I. Min. Met. Eng., 1944, vol. 159, pp. 315-52.
H.E. Friedrich and B.L. Mordike: Magnesium Technology: Metallurgy, Design Data, Applications, Springer-Verlag Berin Heidelberg, Berin, 2006.
G. Holcroft: Proceedings of 75th International Magnesium Association World Magnesium Conference, Int. Magnesium Ass., New Orleans, USA, 2018, pp. 239-47.
R.L. Thayer and R. Neelameggham: JOM, 2001, vol. 53, pp. 15-17.
G.T. Mejdell, H.M. Baumann, and K.W. Tveten: Method for Production of Magnesium Chloride, US 5112584, 1992.
G.J. Kipouros and D.R. Sadoway: Advances in Molten Salt Chemistry 6, Elsevier Science, Amsterdam, 1987, pp. 127-209.
D. Chunming: Proceedings of 76th International Magnesium Association World Magnesium Conference, Int. Magnesium Ass., Budapest, Hungary, 2019, pp. 86–113.
R.A. Sharma: JOM, 1996, vol. 43, pp. 39-43.
N. Leonard, M. Korenko, C. Larson, K. Blood, L.J. Venstrom, S. Nudehi, S. Duncan, R. Diver, F. Simko, J. Priscak, J. Schoer, P.T. Kissinger, and R. Palumbo: Chem. Eng. Sci., 2016, vol. 148, pp. 155-69.
A. Krishnan, X.G. Lu, and U.B. Pal: Metall. Mater. Trans. B, 2005, vol. 36, pp. 463-73.
A.C. Powell IV and S.J. Derezinski III: Primary Production of Elements, US 8460535 B2, 2013.
X. Guan, U.B. Pal, and A.C. Powell: Metall. Mater. Trans. E, 2014, vol. 1A, pp. 132-44.
A.C. Powell IV: Final Scientific/Technical Report of Industrial Scale-Up of Low-Cost Zero-Emission Magnesium by INFINIUM Electrolysis, 2018. https://www.osti.gov/biblio/1431302.
L.A. Yerkes: Electrolytic Method for Producing Magnesium Alloys, US 2431723, 1942.
I. Barin: Thermochemical Data of Pure Substances, 3rd ed., VCH Verlagsgesellschaft mbH, Weinheim, 1995.
C.W. Bale, E. Belisle, P. Chartrand, S.A. Decterov, G. Eriksson, A.E. Gheribi, K. Hack, I.H. Jung, Y.B. Kang, J. Melancon, A.D. Pelton, S. Petersen, C. Robelin, J. Sangster, P. Spencer, and M-A. V. Ende: Calphad, 2016, vol. 54, pp. 35–53.
J. Kang and T.H. Okabe: Metall. Mater. Trans. B., 2014, vol. 45, pp. 1260-71.
L. Kong, T. Ouchi, C. Zheng, and T.H. Okabe: J. Electrochem. Soc., 2019, vol. 166, pp. E429-37.
T.B. Massalski: Bianry Alloy Phase Diagrams, 2nd ed., ASM International, Materials Park, Geauga County, 1990.
The Japan Magnesium Association: Handbook of Advanced Magnesium Technology, Kallos Publishing Co., LTD., Tokyo, 2000.
O. Takeda, T. Uda, and T.H. Okabe: Treatise on Porcess Metallurgy, vol. 3, Elsevier, Amsterdam, 2014, pp. 995-1069.
Acknowledgments
The authors are grateful to Dr. DongEung Kim of the Korea Institute of Industrial Technology for the discussions throughout this study. In addition, the authors also thank Ms. Gyeonghye Moon, Ms. Jiyoung Baek, Dr. Jae-Yeol Yang, and Dr. Jae-Sik Yoon for their technical support. Furthermore, the authors are grateful to all the members of the Geoanalysis Department of KIGAM for their technical assistance. This research was supported by the National Research Council of Science & Technology (NST) Grant by the Korea government (MSIT) (No.CRC-15-06-KIGAM).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted May 10, 2020, Accepted September 11, 2020.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lee, TH., Okabe, T.H., Lee, JY. et al. Molten Salt Electrolysis of Magnesium Oxide Using a Liquid–Metal Cathode for the Production of Magnesium Metal. Metall Mater Trans B 51, 2993–3006 (2020). https://doi.org/10.1007/s11663-020-01976-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-020-01976-9