
Abstract
Electrolytic production of light rare earth elements and rare earth alloys with transition elements takes place in a fluoride-based electrolyte using rare earth oxides as raw material. The optimization of this method, mainly in terms of the energy efficiency and environmental impact control, is rather challenging. Anode effects, evolution of fluorine-containing compounds and side cathode reactions could largely be minimized by good control of the amount of rare earth oxide species dissolved in the fluoride-based electrolyte and their dissolution rate. The Dy2O3 feed rate needed for stable cell operation was studied by following up the anode voltage and gas analysis. On-line analysis of the cell off-gases by FTIR showed that the electrochemical reaction for the formation of Dy-Fe alloy gives mainly CO gas and that CF4 is starting to evolve gradually at anode voltages of ca. 3.25 V. The limiting current density for the discharge of the oxide ions at the graphite anode was in the range of 0.1 to 0.18 A cm−2 at dissolved Dy2O3 contents of ca. 1 wt pct. Modeling of the laboratory cell reactor was also carried out by implementing two models, i.e., an electrical model simulating the current density distribution at the electrodes and a laminal bubbly flow model that explains the electrolyte velocity induced by gas bubble production at the anode.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The biggest challenge to obtaining an environmentally friendly rare earth (RE) electrolytic production is avoiding the anode effects as well as the evolution of fluorine-containing compounds. These hurdles are mostly controlled by the amount of RE oxide (REO) species dissolved in the fluoride-based electrolyte and their dissolution rate. Accordingly, the amount of dissolved oxides needs to match the electrolytic current supply and at the same time avoid the formation of sludge, i.e., non-dissolved REO, which could accumulate at the bottom of the electrolysis reactor. Apart from good and exact knowledge of the oxide saturation levels in the electrolyte at the working temperature, it is necessary to know the optimal electrolysis parameters, including the limiting current density values for the discharge of the oxide ions at the anode, at a given concentration of dissolved oxygen-containing electroactive species, which gives the threshold for releasing detrimental environmental hazardous perfluorocarbon (PFC) gases, mainly CF4 and C2F6.
Dy is a heavy RE element with a high melting point, i.e., 1680 K (1407 °C), which makes it challenging to obtain in the pure form by electrolysis from molten fluoride-based melts. However, it is possible to obtain a liquid Dy-alloy electrolytic product when using a suitable transition metal forming low-melting-point alloys. Using Fe as a consumable cathode in the electrolytic process allows obtaining a liquid Dy-Fe cathode product, free of salt inclusions. Moreover, the Dy-Fe electrolytic product can be used as a master alloy in the manufacturing of Nd-based permanent magnets.
It is believed that Dy-Fe alloys are currently being produced industrially by electrolysis in a DyF3-LiF electrolyte in equimolar composition at temperatures of ca. 1323 K (1050 C) using a vertical electrode set-up cell with graphite anodes and a Fe consumable cathode.[1]
The electrochemical stability of Dy ions in fluoride-based melts has been studied by Saïla et al.[2] Using an Mo inert electrode, the authors found that only Dy(III) ions are stable in the CaF2-LiF eutectic mixture at temperatures ranging from 1113 K to 1203 K (840 °C to 930 °C). The Dy(III) ions are reduced to Dy metal in a one-step diffusion-controlled process exchanging three electrons. The authors also studied the electrochemical reduction of Dy(III) ions using reactive electrodes, i.e., Ni and Cu, showing the evidence of formation of intermetallic compounds.
Castrillejo et al. studied the electrochemical behavior of Dy in an eutectic LiCl-KCl mixture on both inert (W) and reactive (Al) electrodes.[3] The authors demonstrated that the electrodeposition of metallic Dy from Dy(III) ions takes place in two very close electrochemical steps via the intermediate formation of Dy(II). Moreover, the electroreduction of Dy(III) on Al was shown to proceed via a one-step mechanism leading to the formation of DyAl3 alloy.
Nohira et al. investigated the formation of Dy-Fe alloy films using an electrochemical process from the eutectic LiCl-KCl mixture containing DyCl3.[4] The authors demonstrated that it was possible to obtain an adhesive DyFe2 film when activating the Fe electrode by first polarizing the working electrode at 0.40 V vs the Li(I)/Li system. The formation potential of the different Dy-Fe alloys was studied by open-circuit potentiometry at the activated Fe electrode.
Scientific papers on the Dy-Fe electrolytic process from a fluoride melt using oxidic raw material are, to the authors’ knowledge, scarce, if not non-existent; there are not even fundamental studies on laboratory-scale electrolysis cells. A few studies have been published in the English language on Nd and Nd-Fe electrolysis, most of them from the US Bureau of Mines.[5,6]
Although there is limited reliable information on the actual electrolysis technology used in China for Nd production, it seems that the typical operational current densities (anodic) are around 1 A cm−2 with CO reported as the main anode off-gas product and that the process releases large amounts of PFC gases almost continuously.[7]
It is believed that conventional electrolytic production of Nd contaminates the atmosphere with PFC as much as the entire aluminum industry if the off-gases are not treated. PFCs are one of the gases with the highest global warming potentials (GWPs) for ozone-depleting substances. The tabulated data for the 100-year GWP relative to CO2 are 6500 and 9200 for CF4 and C2F6, respectively (values refer to the GWP of CO2). The lifetime is established to be 10,000 and 50,000 years for CF4 and C2F6, respectively.[8]
Most of the work published on RE electrolysis is devoted to the study of PFC evolution due to the anode effect and the ways to circumvent this, for example.[9,10] The authors found that during neodymium electrolysis, two cell voltage regimes could be reached before a significant anode effect occurred. One voltage region where only oxide compounds were oxidized at the graphite anode and a high-voltage region where the reaction of oxide and fluoride components resulted in a substantial perfluorocarbon compound content in the off-gases. The authors claimed that during the anode effect C2F6 is the predominant species and that it is difficult to avoid PFC evolution during the operation of the electrolysis cell.
More recently, Vogel et al. analyzed the gas evolved during neodymium electrolysis from fluoride-based melts.[11] Using linear voltammetry and simultaneous off-gas measurements, the authors concluded that the PFC emissions start with a partial anode effect, where CF4 is first detected and the anode is partially passivated. At higher voltages, C2F6 evolution was also detected. The authors determined the critical anode current density at different oxygen contents in the electrolyte.
This article is the second of a series, the first one dealing with determining the oxide saturation limit in the electrolysis melt,[12] the present one devoted to the investigation of the electrolytic production of the Dy-Fe alloy at a laboratory scale. On-line gas analysis and the recorded anode voltages during electrolysis allowed establishing the relation between a batch-wise Dy2O3 feeding rate and stable cell operation, thus helping to assure stable and environmentally friendly electrolytic cell operation for Dy-Fe alloy production using an iron consumable cathode. The last part of the article is devoted to modeling of the laboratory cell reactor using both an electrical and flow model.
Experimental
LiF (98 + pct, Alfa Aesar) and DyF3 [anhydrous 99.9 pct (REO), Strem Chemicals Inc.] salts were carefully mixed in a graphite crucible at the corresponding amounts according to a composition of 50 to 50 mol pct and placed in a gas-tight furnace under inert atmosphere (Ar 5.0). The salt mixture was heated up gradually to ca. 473 K (200 °C), kept there for about 2 hours and then heated up to 773 K (500 °C), where it was kept for another 2 hours and then heated up further to a sufficient temperature to reach the molten state. The furnace was then cooled down under inert atmosphere and the salt mixture stored in a desiccator box until further use.
The pre-melted electrolyte mixture (ca. 580 g) was placed inside the electrolysis reactor together with a sufficient amount of Dy2O3 [anhydrous 99.9 pct (REO), Strem Chemicals Inc.] to reach saturation. A glassy carbon crucible (Ø = 70 mm, Mersen) was used as the reactor container, which was placed in a gas-tight high-temperature tubular furnace with Kanthal heating elements under Ar 5.0. The inert atmosphere was kept during the whole electrolysis experiment and until the electrolysis cell had cooled down to room temperature.
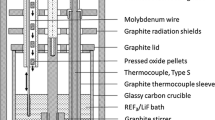
Vertical electrode arrangement was always used, with a 5-mm-diameter Fe rod (99.99 pct metal basis, Alfa Aesar) as consumable cathode, which was surrounded by a cylindrical graphite anode (G330 grade, Schunk Tokai Scandinavia AB). This ensured an even current distribution and allowed a large anode area compared with the cathode area. The anode-to-cathode distance (ACD) was ca. 2 cm. The carbon container was used as a quasi-reference electrode, so the anode and cathode voltages could be followed during the electrolysis.
The temperature was controlled continuously during electrolysis using a thermocouple type S (Pt-Pt 10 pct Rh) situated right above the electrolyte. The working temperature was kept at ca. 1323 K (1050 °C).
Electrolysis experiments were performed by applying constant current using an HP 6031A System Power Supply. The cell voltages and temperature variations were logged during the course of the electrolysis by means of a multichannel Keithley 2000 Multimeter. A Fourier transform infrared spectrometer (FTIR) apparatus (Protea ProtIR 204m) was used to analyze the off-gases of the cell during electrolysis. A 500 mL min−1 Ar flow was used as carrier gas and kept constant during the experiment to obtain the best dynamics in the FTIR analyzer. Existing models were used to transform the FTIR readings to actual gas compositions upon calibration of the apparatus. Two alumina tubes placed above the melt were used for feeding the Dy2O3 material and gas sampling, respectively.
Melt samples were analyzed by the carbothermal method using LECO TC-436 DR (Leco Corp.). To avoid disruptions in the cell flow, samples were taken before and after electrolysis. Otherwise, it was difficult to correlate gas analysis readings with the dynamics of the electrolysis process.
The modeling calculations were made using COMSOL Multiphysics 5.1.
Results and Discussion
Electrolysis and Gas Analysis Results
The initial amount of oxide dissolved in the electrolyte was always determined by LECO analysis prior the electrolysis experiments. In all the cases, the amount obtained was ca. 2 wt pct Dy2O3, which corresponded to a value close to the previously determined oxide saturation levels.[12]
Due to the low REO solubility in the molten fluoride-based melt and the slow dissolution kinetics of the feed material,[12] sludge formation is a big issue in RE electrolysis. Thus, special care was taken to try to avoid over-feeding. Subsequent batch-wise additions of Dy2O3 were fed continuously during the course of the electrolysis in quantities corresponding to 60 pct of the amount of charge passed, which was assumed to be the Faradaic efficiency, for completion of the assumed cell reaction shown in Eq. [1]:
The experiments were run for about 5 to 6 hours and in galvanostatic mode, although the applied current was sometimes adjusted, depending on the anode voltages obtained. Typical current densities applied were ca. 0.1 and 1.5 A cm−2 at the anode and cathode, respectively, in the same range as those reported for the industrial RE electrolysis cell.[13]
The electrolysis experiments were stopped at a point where CF4 started to evolve. Then, an electrolyte sample was taken and the oxygen content analyzed, being typically ca. 1.2 wt pct, thus showing that the limiting anode current density for the discharge of the oxide ions at the graphite anode was in the range of 0.1 to 0.18 A cm−2 at dissolved oxide Dy2O3 contents of ca. 1 wt pct.
Figure 1 shows typical values of different voltages obtained during electrolysis.

Electrolysis data obtained with control of the oxide feed to avoid the anode effect (after gas analysis results). Anode voltage, in volts: continuous gray line; cathode voltage, absolute value in volts: discontinuous black line; amount of CO, in vol pct: continuous black line; amount of CO2, in ppm: discontinuous gray line; amount of CF4, in ppm: dotted black line. The dots indicate the point in time for the Dy2O3 feeding
Sudden and uncontrolled increases in the anode voltage at values of ca. 3.25 V were obtained, whereas the cathode voltage (measured against the carbon container) was kept relatively constant (see Figure 1). Despite this anode voltage increase not being a “full anode effect,” the gas analysis results showed the generation of CF4, which is a harmful greenhouse gas (see Figure 1). This indicated the onset of fluoride decomposition at the graphite anode as the depletion of dissolved oxides at the anode interface led to a potential increase; this cell reaction is shown in Eq. [2]:
Addition of oxide at the voltage increase values led to a decline in the potential and the evolution of PFC gas ceased, thus establishing the stable cell operation again. In this way, the Dy2O3 feed rate, in terms of the batch addition frequency needed for stable cell operation, could be controlled by following the anode voltage and gas analysis.
Analysis of the off-gases showed that CO is the main component in the gas produced, thus making probable that the reaction in Eq. [1] is the overall cell reaction. Moreover, CO2 was present in significant amounts (ca. 2000 ppm, or 0.2 pct; see Figure 1) in the first 30 minutes after the electrolysis started. The amount decreased slowly until after 60 minutes of electrolysis time and onwards where the value was ca. 100 to 200 ppm. This could be because some CO2 was produced in the primary anode reaction but was consumed afterwards, either by a reaction with the dissolved metal, as shown in the general Eq. [3], or through the Boudouard reaction, shown in Eq. [4]:
Moreover, it is interesting to observe that the amounts of CF4 gas detected after a depletion of oxide compounds at the vicinity of the anode were accompanied by small amounts of CO2. The amounts of CO2 and CO were again re-established when the release of CF4 disappeared after more Dy2O3 was fed to the electrolysis cell.
The gas analysis results showed that significant amounts of PFC gas (mainly CF4) also could be released during the production of the Dy-Fe alloy without noticing any full anode effect, i.e., while there was no current blockage. This occurrence was also demonstrated in Al-electrolysis cells and the effect named the non-detected anode effect.[14] The results obtained showed that a similar reaction takes place during RE electrolysis; thus, it is very important to have good control over the cell feed. Although it is difficult to have the cell/anode voltage as a monitoring parameter for the optimal operation of the electrolysis cell since there are just some hundreds of mV difference between the desired anode reaction (Eq. [1]) and the PFC evolution, it might be possible for a given cell where the current/voltage characteristics are well known. Moreover, an environmentally friendly electrolytic process could be realized by a good off-gas treatment system. This can be achieved by using a scrubbing tower with RE2O3, in this case with Dy2O3, giving back RE-fluoride, which can be fed to the electrolysis cell, according to the reaction shown in Eq. [5], as suggested by Keller and Larimer.[5]
Electrolysis Product
After electrolysis, the electrodes were lifted from the electrolyte while still polarized. The current was then turned off and the reactor cooled down, still under Ar atmosphere. After typically 5 hours electrolysis, ca. 19 g of a well-coalesced metal lump was recovered from the bottom of the glassy carbon crucible (see Figure 2).

Frozen electrolyte after electrolysis showing the Dy-Fe cathode product harvested at the bottom of the glassy carbon container as well as the different layer of electrolytes, i.e., ➀ top layer; ➁ middle layer; ➂ bottom layer
XRD analysis showed the presence of both the DyFe2 and Dy phases (see Figure 3), which corresponded to the expected phases after slow cooling of the alloy produced during electrolysis, which, according to the phase diagram[15] and working temperature, corresponded to Dy-Fe contents of 80 and 20 wt pct, respectively. The diffractogram of the cathode product also showed the presence of the remaining salt components, i.e., DyF3, LiDyF4 and Dy1+xO3xF3−3x.

XRD diffractogram of the cathode product. (Open triangle) Dy; (asterisks) DyFe2; (open square) LiDyF4; (open circle) DyF3; (filled circle) Dy1+xO3xF3−3x
The weighed amounts of metal recovered gave current efficiencies of ca. 20 pct, which is low but comparable to short-term small-scale laboratory experiments where the back reaction between the cathode metal product and anode gas is more extensive.
To avoid extensive back reaction, it could be possible to use diaphragms, membranes or porous separators between the anode and cathode, as done during aqueous electrolysis of metals, i.e., Cu, Co, Ni or water electrolysis. Some studies have been carried out during electrolysis of different reactive metals, i.e., Mg, Ti, Al and RE, in both molten chlorides and fluorides.[16,17,18] However, the results obtained so far do not seem to be encouraging, mainly because of the high reactivity of the molten salts toward any kind of ceramic material at elevated working temperatures, especially in the case of rare earth electrolysis.
Di-valent Dy species are reported to be stable in molten chlorides.[3] However, in molten fluorides the highest valences of elements are stabilized, and for Dy only three valent species are stable in the electrolyte.[2] Therefore, it is unlikely that the loss in the current is due to the presence of shuttled reactions caused by multivalent Dy species.
Metal solubility is considered to be quite significant in this type of melt at working temperature not only for the solubility of the RE metal itself,[19] but also because of the presence of dissolved oxide and thus the formation of sub-valent oxides.[5] All this leads to a considerable electronic conductivity in the electrolyte, thus reducing the Faradaic efficiency. It is believed that long-term electrolysis would allow establishing steady-state saturation conditions of the dissolved metal, thus making it possible to achieve higher efficiencies. Nevertheless, the reported Faradaic efficiencies for industrial RE electrolysis cells are in the order of 75 to 80 pct.[1]
An oxygen balance could also be carried out considering the main reaction (Eq. [1]), amount of metal recovered, amount of Dy2O3 fed during the electrolysis and amount of Dy-oxyfluoride dissolved in the melt before and after the electrolysis, determined by the LECO analysis of electrolyte samples taken from the molten bath. The results showed that a large amount of Dy2O3 was lost, probably as sludge. If the instantaneous feeding rate becomes larger than the REO dissolution rate, sludge will form, which will dissolve more slowly than the REO feed material, as reported by Dysinger and Murphy.[6] The concentration of oxygen-containing electroactive species available at the anode is controlled by the dissolution rate, and no matter how much REO is added to the electrolyte, it still needs to dissolve before being discharged. The formation of sludge might then be minimized by establishing a continuous and uniform feeding of small amounts of Dy2O3 through the progress of the electrolysis instead of batch-wise feeding.
Figure 2 shows the formation of two clear layers of frozen electrolyte together with a darker middle layer. XRD analysis of electrolyte samples taken in both the molten and frozen state showed the presence of LiDyF4 and Dy1+xO3xF3−3x phases.
Moreover, SEM-EDS analysis of the different parts of the frozen electrolyte showed a more extensive presence of Dy-oxyfluoride phases in the bottom layer (see Figure 4). Similar results are reported by Vogel and Friedrich.[20] The middle layer seemed to be rich in carbon and oxygen.

(a) Back-scattered SEM image of the frozen electrolyte layers. (b) SEM-EDS mapping analysis showing bright particles rich in oxygen. (c) SEM-EDS line-scan analysis of the oxygen-rich particles
Despite efforts to avoid over-feeding, these results also confirmed that some of the dysprosium oxyfluoride species precipitated during the electrolysis, and so a considerable amount of oxide feed was lost in the form of sludge lying at the bottom of the cell. Nevertheless, the sludge apparently did not create a problem for the cathode Dy-Fe alloy product as it coalesced well and remained at the bottom of the glassy carbon crucible, where it was harvested at the end of the experiment.
Modeling of the Electrolysis Cell Reactor
An electrical and computational fluid dynamics model was applied to the laboratory-scale electrolysis cell using COMSOL Multiphysics 5.1. The electrical model accounts for the electrochemical reaction, whereas the flow model accounts for the movement in the electrolyte caused by the gas formed during the anode reaction. A unidirectional coupling between the electrical model and the bubbly flow model is assumed, where the electrical behavior of the cell determines the electrolyte motions, as the bubble formation rate is determined by the value of the anodic current density. Contrarily, it is assumed that the electrical model is not influenced by the bubbly flow model, i.e., no bubble overpotential is accounted for in the electrical model.
The influence of several parameters such as the mesh refinement, anode-cathode distance or gas bubble diameter on the numerical results was analyzed. The numerical method has been described elsewhere.[21]
Figure 5 shows current density mappings obtained after applying the electrical model for two different ACD values. The results show the current distribution during the electrochemical process. The outer part of the anode has an almost negligible current density; thus, only the inner part could be considered the electroactive surface. Moreover, edge effects are also clear at both the anode and cathode. Increasing ACD does not imply any significant change of the current distribution at the electrodes. The average current density at the cathode is only shifted toward higher values as the anodic current density is kept constant.

Current density simulated at the surface of the electrodes at various ACDs. (a) 2 cm; (b) 3.5 cm. The anodic current density was kept constant in both cases and equal to 0.075 A cm−2. Scale A cm−2
Moreover, the bubbly flow model could predict the convective movement of the electrolyte due to the gas evolution during the anode reaction. Considering an average bubble diameter of 1 mm, the velocity field for two different ACD values, keeping the anodic current density constant, was determined (see Figure 6). At the lowest ACD, a higher velocity in the space in between the electrodes and especially near the cathode is seen. Moreover, the influence of the gas bubble diameter was also studied, and the results are shown in Figure 7.

Results obtained after applying the flow model with COMSOL Multiphysics 5.1 when using two different ACDs, i.e., (a) 2; (b) 3.5 cm. Bubble diameter 1 mm in both cases. Scale m s−1. The size of the arrows gives an idea of the intensity of the flow

Numerical simulations of the electrolyte motions due to bubble gas evolution at the anode. Results obtained at different bubble diameters: (a) 0.5; (b) 1; (c) 1.5; (d) 2 mm. In all cases, ACD and anodic current density values of 2 cm and 0.075 A cm−2, respectively, were applied. Scale m s−1. The size of the arrows gives an idea of the intensity of the flow
To keep a reasonably high anodic current density and at the same time not release significant amounts of PFC gas, the bubble diameter of the CO gas evolving at the anode has to be small. The results showed that smaller gas bubbles implied higher velocities in the electrolyte, which can have both positive and negative effects on the electrolysis process. On one hand, the higher convective movement facilitates the dissolution process of the Dy2O3 feed, which has been shown to be slow.[12] However, it also implies a more extensive back reaction, as it is more likely that the reaction products (metal and CO gas) will be in contact in the turbulent electrolyte flow.
Conclusions
Together with the solubility values of dysprosium oxide species determined in a previous paper, the analysis of the anode gases during electrolysis allowed establishing the optimal anode voltages and oxide feed rates, thus leading to an understanding of the fundamental requirements to run the electrolysis cells for Dy-Fe alloy production in a more efficient and environmentally friendly way.
The results showed that the Dy2O3 feed rate needed for stable cell operation could be established by following up the anode voltage and the on-line reads of the gas analysis. The FTIR analyses of the cell off-gas showed that the electrochemical reaction for the formation of the Dy-Fe alloy using Fe as a consumable cathode gives mainly CO gas and that CF4 is starting to evolve gradually at anode voltages of ca. 3.25 V. The limiting anode current density for the discharge of the oxide ions at the graphite anode was in the range of 0.1 to 0.18 A cm−2 at dissolved oxide contents of 1 wt pct.
XRD analysis of the cathode product recovered at the bottom of the electrolysis cell showed the presence of DyFe2 and Dy, as expected after the formation the Dy-Fe alloy in 80 and 20 wt pct composition, respectively.
Taking into account the amounts of alloy produced and gases analyzed, the current efficiency of the process was determined to be ca. 20 pct. The current losses are mainly attributed to the high metal solubility in the RE-halide system and the extensive “back reaction” between the cathode product and anode gases in the small-sized laboratory cell.
Numerical simulations were focused on the prediction of the current distribution at the surface of the electrodes and on the electrolyte motions due to bubble gas evolution at the anode. The effect of varying the ACD as well as the average bubble diameter was assessed in a parametric study. The results showed that the current at the anode is mainly located of the side facing the cathode, while the opposite side of the anode works with a lower reaction rate. Increasing ACD does not imply any significant change in the current distribution at the electrodes. In the bubbly flow simulations, the electrolyte velocity remains globally the same in the anode vicinity and at the electrolyte surface close to the anode when changing the ACD. Increasing the bubble diameter causes the average electrolyte velocity to decrease significantly in the space between the electrodes. Even if the upward velocity of small bubbles is lower, their “dragging” effect on the surrounding liquid seems to be more efficient, leading to a higher overall velocity of the electrolyte.
References
J. Lucas et al., eds.: Rare Earths. Science, Technology, Production and Use, Elsevier B.V., Amsterdam, Netherlands, 2015.
A. Shaïa, M. Gibilaro, L. Massot, P. Chamelot and P. Taxil, J. Electroanal. Chem., 2010, vol 642, pp. 150-156.
Y. Castrillejo, M.R. Bermejo, A.I. Barrado, R. Pardo, E. Barrado and A.M. Martinez, Electrochim. Acta, 2005, vol 50, pp. 2047-2057.
H. Konishi, T. Nohira and Y. Ito, Electrochim. Acta, 2002, vol 47, pp. 3533-3539.
R. Keller and K.T. Larimer: Final Report for US Department of Energy, DOE/ID/13104, 1997.
D.K. Dysinger and J.E. Murphy: Report of Investigations No. 9504, US Department of Interior, Bureau of Mines, Washington, 1994.
R. Keller: Proceedings of Joint ECS/ISE Meeting, 1997.
Climate Change Connection website, http://www.climatechangeconnection.org/, Accessed 18 Aug 2017.
US Patent 5,810,993.
R. Keller and K.T. Larimer: in Rare Earth Science, Technology and Applications III, R.G. Bautista, ed., The Mineral, Metals and Materials Society, 1997, pp. 175–80.
H. Vogel, B. Flerus, F. Stoffner and B. Friedrich, J. Sustain. Metall., 2017, vol 3, pp. 99-107.
A.M. Martinez, A. Støre and K.S. Osen, Mat.Met.Trans. B, 2018, vol. 49, pp. 783-89.
S. Pang, Y. Shihong, L. Zongan, C. Dehong, X. Lihai and Z. Bin, Chin. J. Rare Met., 2011, vol 34, pp. 440–50 (only abstract in English).
D.S. Wong, P. Fraser, P. Lavoie and J. Kim, JOM, 2015, vol 67, pp. 342-353.
B. Massalski ed.: Binary Alloy Phase Diagrams, vol. 2, ASM International, Ohio, 1990.
X. Guan, U.B. Pal, Y. Jiang, S. Su, J. Sustain.Metall., 2016, vol 2, pp. 152-166.
A. Martin, J.C. Poignet, J. Fouletier, M. Allibert, D. Lambertin and G. Bourges, J. Appl Electrochem., 2010, vol 40, pp. 533-542.
M.V. Ginatta: Proceeding of the International Symposium on Ionic Liquids in Honour of Marcelle Gaune-Escard, H.A, Oye and A. Jagtoyen, eds., 2003.
A.S. Dworkin and M.A. Bredig, J. Phys. Chem., 1971, vol 75, pp. 2340-2344.
H. Vogel and B. Friedrich, Int. J. Nonferrous Metall., 2017, vol 6, pp. 27-46.
A. Oury, P. Namy, A.M. Martinez, K.S. Osen and A. Solheim: Proceedings of COMSOL Conference, 2016. https://www.comsol.fr/paper/electrical-and-bubbly-flow-modeling-of-a-molten-salt-electrolysis-cell-40791.
Acknowledgment
This work received funding from the European Union’s Seventh Framework Programme (FP7/2007-2013) under Grant Agreement No. 603564.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted September 29, 2017.
Rights and permissions
About this article

Cite this article
Martinez, A.M., Osen, K.S., Støre, A. et al. Manufacturing of Dysprosium-Iron Alloys by Electrolysis in Fluoride-Based Electrolytes. Electrolysis in a Laboratory-Scale Cell. Metall Mater Trans B 49, 2063–2070 (2018). https://doi.org/10.1007/s11663-018-1270-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-018-1270-3