
Abstract
In order to clarify the changes in the composition of oxide inclusions in steel, the effect of the metal and oxide composition on the reaction between solid Fe-based alloys and liquid multi-component oxides was investigated using the diffusion couple method at 1473 K. The measured concentration gradients of Mn and Si in the metal indicated that Mn diffused into the metal from the oxide, while the diffusion of Si occurred in the opposite direction. In addition, the MnO content in the oxide decreased with heat treatment time, while the SiO2 content increased. The compositional changes in both phases indicated that the Mn content in the metal near the interface increased with heat treatment with decreasing MnO content in the oxide. Assuming local equilibrium at the interface, the calculated [Mn]2/[Si] ratio at the interface in equilibrium with the oxide increased with increases in the MnO/SiO2 ratio in the oxide. The difference in the [Mn]2/[Si] ratios between the interface and the metal matrix increased, which caused the diffusion of Mn and Si between the multi-component oxide and metal. By measuring the diffusion lengths of Mn and Si in the metal, the chemical diffusion coefficients of Mn and Si were obtained to calculate the composition changes in Mn and Si in the metal. The calculated changes in Mn and Si in the metal agreed with the experimental results.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Steel-wire-rod products are typically used as steel cords for tire reinforcement and valve springs for intake/exhaust valve controllers. The breakage of steel wire rods during wire drawing to submillimeter sizes is caused by oxide inclusions exceeding 10 μm in diameter.[1,2] The major oxide inclusions causing the breakage of wire have been reported to be alumina (Al2O3) and spinel (MgO·Al2O3), which form during Al deoxidation.[1,3,4] The Si-Mn deoxidizing method is useful in the manufacture of steel wire rod because small and low-melting-point oxide inclusions are produced as MnO-SiO2.[5,6,7] Furthermore, the oxide inclusions can also be softened by reaction with CaO-SiO2-Al2O3 bearing fluxes in the secondary refining process. However, it was recently determined that the composition of softened inclusions varied upon reacting with alloying elements at hot rolling temperatures, and the composition of the oxide inclusions changed as precipitation of the solid phase increased.[8,9] In previous studies,[5,6,7] a given oxide, which becomes the seed for the inclusions, was added into the molten steel, and a metal sample including the small inclusions was reacted at hot rolling temperatures under the coagulation of the metal. Then, the morphology, composition, and size of the oxide inclusions were analyzed. However, their reaction mechanism has not been well studied because the size of the oxide inclusions in steel is very small. Recently, in the 304 stainless steel, the transformation rate of inclusion to change MnO-SiO2 to MnO-Cr2O3 was investigated by heat treatment at 1273 K to 1473 K (1000 °C to 1200 °C).[10] The Cr in the solid steel was diffused to the oxide inclusion, and the Si and Mn in the inclusion were exchanged by Cr. By simulation results using kinetic model, the transformation rate of MnCr2O3/MnSiO2 in the inclusion was increased as the increase in temperature and the decrease in the diameter of inclusions.
Even if the diffusion of elements and change in composition between the oxide inclusions and steel occurred, the varying amounts could not be detected and were insufficient to clarify the mechanism. Therefore, it is difficult to clarify the reaction using real non-metallic inclusions because they are too small to be accurately analyzed. In this study, the effect of the oxide composition on the diffusion behaviors of components between multi-component oxide particles and solid at hot rolling temperature was investigated by the diffusion couple method. Based on the authors’ knowledge, the present study is the first to investigate the reaction between liquid multi-component oxides and solid metal the past several decades. The diffusion couple has sufficient oxide present to determine the reaction and mass transfer at the interface.
Experiments
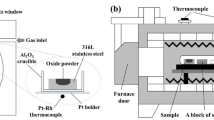
The details of the experimental procedure of the diffusion couple method were described elsewhere.[11,12] Here, the experimental procedure is explained briefly. As illustrated in Figure 1(a), a hole was made at the center of the alloy (1 mm × 1 mm × 1 mm), and the crushed oxides (2 to 3 mg) were inserted into the hole. To prevent external oxidation of the alloy during heat treatment, the alloy and oxide sample were enclosed in a quartz tube with Ar gas at 20 kPa, as illustrated in Figure 1(b). In the quartz tube, Ti foil was used to capture the oxygen in the Ar gas. The entire enclosed sample was heated at 1473 K (1200 °C) for 10 hours before being quenched in water. After quenching, the sample was removed from the quartz tube. The interface between the alloy and oxide was examined using electron probe microanalysis (EPMA).

Schematic image of diffusion couple samples. Figure (a) and (b) represents the images of sample of metal and oxide and preparation of sealed sample, respectively
Table I lists the initial compositions of the oxide and metal. In runs A1, B1, and C1, the Si content in the metal was from 1.44 to 3.34 pct by mass with a constant Mn content. In runs C1, C2, and C3, the CaO/SiO2 ratio in the oxide was maintained between 0.63 and 0.68, while the MnO content was varied from 4.9 to 15.7 pct by mass. The melting points of the oxides were analyzed using thermo-gravimetric analysis and differential thermal analysis (TG–DTA 8120, Rigaku), and the melting temperatures of oxides 1, 2, and 3 were 1340 K, 1358 K, and 1440 K (1067 °C, 1085 °C, and 1167 °C), respectively.
Results
Figure 2 presents a typical image of the interface between the oxide and the metal after the heat treatment at 1473 K (1200 °C) for 10 hours. The oxide phase was homogeneous, and the precipitation of oxide particles in the metal phase near the interface was not observed. The composition change in each phase was analyzed at a distance of 300 μm from the interface at 5 to 50 μm intervals. Especially, since the composition of elements in metal side were assumed to be significantly changed, the 20 μm from interface was analyzed at 5-μm intervals.

Composite image of A1 after heat treatment at 1473 K for 10 h
Figures 3 and 4 represent the composition changes in metal and oxide for runs A1, B1, and C1, investigating the influence of Si content in the metal. Figures 5 and 6 represent the composition changes in the metal and oxide for C1 through C3, investigating the influence of MnO content in the oxide. In Figures 3, 4, 5 and 6, even though the measured points of Mn, Si, MnO, and SiO2 were connected for the tendency of the composition changes, those lines were not the fitting lines and had no physical meaning. The accuracy of the measurement results of Al, Ca, Mg, Mn, Si, and Fe were more than 95 pct, and below 0.2 pct, 0.3, 0.3, 2, 2, and 2 pct of averaged error measurements, respectively.

Chemical composition change Mn (a), Si (b), Ca (c), Fe (d), Mg (e), and Al (f) in the metal of runs A1, B1, and C1 as a function of the distance from the interface after heat treatment at 1473 K for 10 h

Chemical composition changes in MnO (a), SiO2 (b), CaO (c), FeO (d), MgO (e), and Al2O3 (f) in the oxide of runs A1, B1, and C1 as a function of the distance from the interface after heat treatment at 1473 K for 10 h

Chemical composition change Mn (a) and Si (b) in the metals of runs C1, C2, and C3 as a function of the distance from the interface of the metal after heat treatment at 1473 K for 10 h

Chemical composition changes in MnO (a) and SiO2 (b) in the oxides of runs C1, C2, and C3 as a function of the distance from the interface of the oxide after heat treatment at 1473 K for 10 h
Figure 3 shows the composition changes in Mn (a), Si (b), Ca (c), Fe (d), Mg (e), and Al (f) in the metal of runs A1, B1, and C1 as a function of the distance from the interface. In Figure 3(c), the Ca contents were almost constant more than 10 μm from the interface. The Ca content near the interface of A1 and B1 was close to 1 pct by mass. In the Fe-Ca binary phase diagram,[13] the Ca solubility in γ-Fe was below 0.02 pct by mass at 1723 K (1450 °C). The measured high Ca content by EPMA analysis may be caused by the significantly small inclusions formed during heat treatment. The Ca content in the metal was not considered because CaO in the oxide cannot be thermodynamically reduced by either element in the metal. The changes in Fe, Mg, and Al were not remarkable as shown in Figures 3(d) through (f). Although the concentrations of Fe in steel were scattered in Figure 3(d), the averaged concentrations of A1, B1, and C1 were, respectively, 96.4, 97.3, and 97.8 mass pct with less than 2.0 pct of measured errors. By the sum of the averaged contents of Fe, and the contents Mn and Si in Table I, the total compositions are 100 mass pct. On the other hand, the FeO concentrations at a distance of 5 μm from the interface were below 1 pct by mass and then decreased drastically near the interface as shown in Figure 4(d). Furthermore, the concentrations of Mg and Al in solid steel were scattered and below 0.05 mass pct. In the present work, the MnO in the slag can be thermodynamically reduced by the Si in the steel. This indicates that the diffusion of CaO, MgO, and Al2O3 from slag to steel were unremarkable as shown in Figures 4(c), (f), and (e). Each composition in the slag was slightly changed as the MnO decreased and as the SiO2 increased. Based on the diffusion behaviors of Mn and Si, the diffusion length was determined by 300 μm, and the diffusion behavior of Ca, Fe, Mg, and Al were not considered in this study.
In contrast, despite the similar initial concentration of Mn, the Mn content in the metal increased in the following order: A1, B1, and C1. As shown in Figure 3(b), the concentrations of Si in the metal decreased after heat treatment compared to each initial composition. In particular, the changes in Mn and Si in the metal of C1 showed the concentration gradients near the interface of metal. When the Si content in metal was high, such as in A1 and B1, the concentration gradients of Mn and Si near the interface were not observed, but their concentrations were changed compared to the initial values.
Figure 4 shows the composition changes in MnO (a), SiO2 (b), CaO (c), FeO (d), MgO (e), and Al2O3 (f) in the oxides of runs A1, B1, and C1 as a function of the distance from the interface. The concentration gradients of all the elements, except for FeO in the oxide, were barely detectable. This finding indicated that the mass transfer rates of all the elements in the liquid oxide were very rapid. The FeO concentration near the interface was not considered because its value was greatly influenced by the Fe signal from the metal phase because of the limited beam diameter of the EPMA. As mentioned above, the composition changes of CaO, MgO, and Al2O3 were not remarkable compared with the initial concentrations of these oxides. However, the MnO content after heat treatment decreased compared with the initial content of oxide 1. The decrease of the MnO concentration after heat treatment was substantial and exhibited a dependence on the increase of the Si content in the metal of runs A1, B1, and C1. This decrease in MnO in the oxide caused the increase in Mn in the metal as mentioned in Figure 3(a). Furthermore, the content of SiO2 in the oxide increased slightly in the order of A1, B1, and C1, because the Si in the metal was diffused as shown in Figure 3(b).
Figure 5 shows the composition changes in Mn (a) and Si (b) in the metal of runs C1 through C3 as a function of the distance from the interface. Since the Fe contents of C2 and C3 were almost constant, and the changes in Ca, Mg, and Al of C2 and C3 were below 0.1 pct by mass as mentioned above, the diffusion behaviors of Fe, Ca, Mg, and Al were not considered in this study. However, the concentration gradients of Mn and Si were observed to depend on the concentration of MnO in the oxides. The concentration gradient and concentrations of Mn near the interface of the metal increased with increases in the MnO content in the contacting oxide. In contrast, the concentrations of Si near the interface of the metal decreased compared with the initial Si content. Therefore, it was determined that the Mn diffused into the metal from the oxide, while the diffusion of Si occurred in the opposite direction. These diffusion tendencies of Mn and Si were observed also in the composition changes in the oxides.
Figure 6 shows the composition changes in MnO (a) and SiO2 (b) in the oxide of runs C1 through C3 as a function of the distance from the interface. Since the mass transfer rate in the liquid oxide is larger than that in the solid alloy, the concentration gradients in the oxide were not observed except for FeO. In addition, the composition changes of FeO, CaO, MgO, and Al2O3 showed similar tendency to those represented in Figure 4, and the changes in CaO, MgO, and Al2O3 were not remarkable compared to the initial concentrations. Although similar tendencies for MnO and SiO2 in oxide were also observed, the decrease in the MnO concentration in the oxide depended on the initial concentration of the MnO before heat treatment. Furthermore, the content of SiO2 in the oxide increased under the conditions in runs C2 and C3, as observed in Figure 6(b). For run C1, although the increase of SiO2 was not detectable, a small amount of Si diffused from the metal, as observed in Figure 5(b). Therefore, it was presumed that the MnO in the oxide was exchanged with Si in the metal.
Discussion
Based on the results, the diffusion of Mn and Si between the oxide and metal was more remarkable than that of the other elements. Figure 7 represents the schematic image of Mn and Si diffusion between the solid alloy and the liquid oxide.

Schematic image of diffusion between solid alloy and liquid oxide
The Mn diffused from the oxide into the metal as much as the decrement of MnO in oxide. In contrast, the diffusion direction of Si was the opposite from that of Mn, and the SiO2 content in oxide increased with decreasing Si in the metal. This finding indicated that the MnO in the oxide was reduced by the Si in the metal at the interface. The relationship between the diffusion of Mn and Si could be derived using the following equations:
In Eq. [2], the standard free energy change at 1473 K was calculated by extrapolating the thermodynamic data.[14,15] Based on this reaction, the local equilibrium retaliation at the interface can be derived by the following equation:
where a i , γ i , and xi are the activity, activity coefficient, and mole fraction of component i in Eq. [1], respectively. The measured values by EPMA presented in Figures 4 and 6 were used as the mole fractions of each oxide component in the oxide phase at the interface. The γMnO and \( \gamma_{{{\text{SiO}}_{2} }} \) were derived using the regular solution model.[16] The equilibrium constant, K(1), can be determined using Eq. [2]. In this study, the equilibrium parameter of KMnSi was used to express the activity ratio of Mn and Si at the interface.
Figure 8 presents the equilibrium ratio (KMnSi) obtained from the equilibrium relationship for runs A1, B1, C1 through C3 as a function of the mass ratio of (pct MnO)/(pct SiO2) in the oxide. In addition, the ratios of [pct Mn]2/[pct Si] in the matrix of the metal before heat treatment and the measured value at a distance of 5 μm from the metal after heat treatment are shown in Figure 8. As the mass ratio of (pct MnO)/(pct SiO2) increased, KMnSi increased exponentially; this tendency was similar to that of the [pct Mn]2/[pct Si] ratio using the analyzed values at a distance of 5 μm from the metal side. Furthermore, the difference in [pct Mn]2/[pct Si] at the interface and in the matrix of the metal increased upon increasing the (pct MnO)/(pct SiO2) ratio in the oxide. The Mn content at the interface was considered sufficiently high to move into the metal after heat treatment for 10 hours. Therefore, this difference in concentrations would be related to the driving force for the diffusion of Mn and Si between the oxide and metal. In the cases of runs A1 and B1, when the mass ratio of (pct MnO)/(pct SiO2) was below 0.1, the difference in [pct Mn]2/[pct Si] before and after the heat treatment was small, and the driving force of diffusion was also small. Although the concentration gradients of Mn and Si in the vicinity of the interface on the metal side were not observed for runs A1 and B1 (see Figures 3(a) and (b)), the concentration changes of Mn and Si in both phases occurred after heating for 10 hours.

Effect of MnO/SiO2 ratio on the change in the equilibrium ratio (KMnSi), [pct Mn]2/[pct Si] at the interface between the metal and oxide
Figure 9 shows the diffusion lengths of Mn and Si in the metal for runs C1 through C3 as a function of the differences between KMnSi and the [pct Mn]2/[pct Si] in the matrix before heat treatment. As illustrated in Figure 9, the diffusion lengths of both elements were logarithmically proportional to the differences of KMnSi and the [pct Mn]2/[pct Si] in the matrix. In general,[17] the diffusion length (LM) can be expressed by diffusion coefficient (DM) of Mn or Si, and the heating time (t) using the following equation:
In this equation, when the heating time is constant, the changes in diffusion length depend on the diffusion coefficient. This means that the diffusion coefficients for Mn and Si in the present work were dependent on the driving force as well as the difference in [pct Mn]2/[pct Si] between the interface and matrix. Furthermore, the diffusion length of Si was larger than that of Mn despite the same heating time. This result was caused by the larger diffusion coefficient of Si in solid Fe compared with that of Mn. Therefore, the diffusion of Mn in the metal was assumed to be the rate-limiting step in this system because the mass transfer in the liquid oxide was more rapid than that in the solid metal.

Comparison of diffusion lengths of Mn and Si for C1, C2, and C3 as a function of the difference in [pct Mn]2/[pct Si] between the interface and matrix
In order to clarify the relationship between the driving force by the thermodynamic parameter and the diffusion of Mn and Si, the diffusion coefficients of Mn and Si are necessary to be considered by the concentration difference between the interface and matrix. In Figure 7, the diffusive fluxes of Mn and Si are represented by classifying the liquid oxide and the solid metal based on different diffusion rates. The chemical diffusion coefficients of Mn and Si in the metal were obtained in the present study. As shown in Figure 7, the diffusive fluxes of Mn and Si, JMn and JSi, should be in balance between the oxide and the alloy, and the flux of element M in the solid alloy is represented by Fick’s second law:
where M is Mn and Si, and C, J, t, and y are the concentration, the diffusive flux, the time, and the diffusive distance, respectively. DMnSi is the inter-diffusion coefficient between the diffusive fluxes of Si and Mn. On the basis of Darken’s equation,[18] this coefficient can be represented by thermodynamic parameter as
where k, T, and BM are the Boltzmann’s constant, temperature, and the atom mobility of M element, respectively. From Eq. [6], the relationship between the chemical diffusion coefficient and the ideal diffusion coefficient can be obtained as
When the self-diffusivity of a given element was investigated by the movement of trace elements or isotopes, the diffusion coefficient was mainly dependent on the temperature because the activity coefficients of the elements are constants in Eq. [7]. Since the heating temperature and the heating time were constant in the present study, the diffusion behaviors of Mn and Si could not be explained by the self-diffusion coefficients. Therefore, it is necessary to obtain the diffusion coefficients of Mn and Si that vary with the driving force at the interface between the liquid oxide and solid metal. This diffusion coefficient was also called the “chemical diffusion coefficient.”[18,19]
From Eq. [4], the chemical diffusion coefficient of Mn and Si (\( D_{\text{Mn}}^{\text{C}} \) and \( D_{\text{Si}}^{\text{C}} \)) could be obtained at 36,000 seconds. Furthermore, the Mn and Si in solid γ-Fe were very low, and both mole fractions in Eq. [8] were assumed to be (1 − xFe). Therefore, from Eq. [8], the DMnSi was obtained as
Figure 10 shows the comparison of the chemical diffusion coefficients of Mn and Si with a self-diffusion coefficient as a function of the difference in [pct Mn]2/[pct Si] between the interface and matrix. In this figure, the self-diffusion coefficients of DMn in liquid oxide and DSi in γ-Fe were not represented as they were larger than ~ 10−5 mm2/s.[20,21] The values of \( D_{\text{Mn}}^{\text{C}} ,\;D_{\text{Si}}^{\text{C}} , \) and DMnSi were increased with an increase in the difference in concentration, when the \( D_{\text{Si}}^{\text{C}} \) was larger than the \( D_{\text{Mn}}^{\text{C}} . \) The DMnSi covered a range similar to the self-diffusion coefficient of Mn in γ-Fe (\( D_{\text{Mn}}^{\text{Self}} \)). It was determined that the rate-limiting step between the liquid oxide and the solid alloy was governed by the diffusion of Mn in the metal.

Comparison of chemical diffusion coefficients of Mn and Si with the self-diffusion coefficient as a function of the difference in [pct Mn]2/[pct Si] between the interface and matrix
In order to compare the validity of DMnSi with the self-diffusion coefficient of Mn in γ-Fe (\( D_{\text{Mn}}^{\text{Self}} \)), the concentration changes in Mn and Si in γ-Fe were calculated by following equations:
where L is diffusion length, and t is set at 36,000 seconds. The Δ[pct Mn] is determined by Eq. [10]. Although the [pct M]* is the concentration of M element, it was regarded as the concentration at a distance of 5 μm from the metal. Table II represents the values of [pct M]Matrix and Δ[pct M] for the calculation of concentration changes in Mn and Si in γ-Fe. In this table, the DMnSi for each condition and the \( D_{\text{Mn}}^{\text{Self}} \) at 1473 K were also represented.
Figures 11 and 12 show the concentration changes of Mn and Si in γ-Fe calculated by the chemical diffusion coefficients (a) and self-diffusion coefficient (b), respectively. As shown in Figure 11(a), the calculated composition changes of Mn in γ-Fe showed good agreement with the experimental results compared to the calculated results of Figure 11(b). In particular, the calculation for the conditions in run C3 using the self-diffusion coefficient showed lower values compared to the experimental results of run C3, and the calculated composition gradients of C3 were close to that of C2 beyond 20 μm. In Figure 12(a), the calculated composition changes in Si in γ-Fe by DMnSi were in relatively good agreement with the experimental results. However, the calculated composition changes in Si in γ-Fe using the self-diffusion coefficient of Mn showed similar values for the conditions in runs C2 and C3.

Composition changes of Mn by calculation with chemical diffusion coefficients (a) and self-diffusion coefficient (b)

Composition changes of Si by calculation with chemical diffusion coefficients (a) and self-diffusion coefficient (b)
Figure 13 shows the composition changes of Mn (a) and Si (b) by calculation using DMnSi of A1, B1, and C1 in Table II. The values of DMnSi for runs A1 and B1 were obtained by the relationship between the DMnSi and KMnSi in Figure 10. In Table II, the [pct M]Matrix for the conditions in runs A1 and B1 were determined from the experimental results, because the Mn content in the metal was remarkably increased by diffusion of MnO from the liquid oxide reduced by the high Si content in the metal. As shown in Figure 13, the calculated results of Mn and Si content by DMnSi agreed with the experimental results. Therefore, the chemical diffusion coefficient with thermodynamic consideration at the interface was necessary to calculate the diffusions of Mn and Si between the oxide and the solid iron-based alloy.

Composition changes of Mn (a) and Si (b) by calculation using the DMnSi of runs A1, B1, and C1 in Table II
Conclusions
Based on heat treatment at 1473 K of a solid iron-based alloy and multi-component liquid oxides, the effect of the alloy and oxide compositions on the diffusion of the elements was investigated. The following conclusions were drawn:
-
(A)
After heat treatment, the diffusion of Mn and Si in both phases was more remarkable compared with that of the other elements (Al, Mg, Ca, and Fe). Mn diffused from the oxide into the metal, and Si diffused in the opposite direction. Although the concentration gradients of MnO and SiO2 in the oxide were not detected, those of Mn and Si on the metal side with low Si content were detected. In the case of the high Si-metal, the concentration changes of Mn and Si after heat treatment were varied in the entire matrix without concentration gradients near the interface.
-
(B)
With increases in the initial content of MnO in the oxide and Si in the metal, the MnO content in the oxide decreased and the Mn concentration of the metal near the interface increased after heat treatment. In contrast, the Si content of the metal decreased, and the SiO2 content in the oxide increased slightly.
-
(C)
The thermodynamic assessment presuming local equilibrium at the interface revealed that the [Mn]2/[Si] ratio at the interface increased upon increasing the MnO/SiO2 ratio in the oxide. Therefore, the driving force for the diffusion of Mn and Si was dependent on the activities of MnO and SiO2 in the oxide. The diffusion lengths of Mn and Si on the metal side were proportional to the difference of [Mn]2/[Si] between the interface and matrix. Furthermore, the chemical diffusion coefficients of Mn and Si in this study were obtained by the diffusion lengths of Mn and Si on the metal side. Using DMnSi based on the obtained coefficients, the calculated changes of Mn and Si content in the metal showed good agreement with the experimental results.
These conclusions suggest the feasibility to simulate the composition change in the small oxide inclusions by using chemical diffusion coefficients between liquid oxide and solid metal. In order to simulate the mass transfer in the liquid oxide, we are currently studying the time dependency of the diffusion of Mn and Si between the liquid oxide and a Si-deoxidized alloy in greater detail.
References
T. Mimura: 182/183rd Nishiyama Kinnen Kouza ISIJ, 2004, pp. 127–50.
S. Okushima: 126/127th Nishiyama Kinnen Kouza ISIJ, 1988, pp. 147–66.
A. Yoshimochi, K. Yokoe, T. Shibata, and Y. Oki: Wire J. Int., 1983, vol. 16, pp. 224–26, 228, 231.
4. E. Stmpa, and M. Cipparrone: Wire Journal Int., 1987, vol. 20, pp. 44-55.
5. H. Iwai, B. Tsujino, S. Isa, and T. Ao: Tetsu-to-Hagane, 1968, vol. 54, pp.1037-1046.
6. H. Iwai, B. Tsujino, S. Isa, and T. Ao; Tetsu-to-Hagane, 1969, vol. 55, pp.887-900.
7. K. Takano, R. Nakao, S. Fukumoto, T. Tsuchiyama, and S. Takaki: Tetsu-to-Hagane, 2003, vol. 89, pp. 616-622.
8. W. Choi, H. Matsuura, and F. Tsukihashi: ISIJ Int., 2011, vol. 51, pp. 1951-1956.
9. H. Shibata, K. Kimura, T. Tanaka, and S. Kitamura: ISIJ Int., 2011, vol. 51, pp.1944-1950.
10. Y. Ren, L. Zhang and P.C. Pistorius: Metall. Mater. Trans. B, 2017, vol. 48, pp. 2281-2292
11. K.-H. Kim, S.-J.Kim, H. Shibata, and S. Kitamura: ISIJ Int., 2014, vol. 54, pp. 2144-2153.
12. K.-H. Kim, H. Shibata, and S. Kitamura: ISIJ Int., 2014, vol. 54, pp. 2678-2686.
13. H. Okamoto: J. Phase Equilibria and Diffusion, 2010, vol. 31 pp. 88-90.
14. O. Knacke, O. Kubaschewski, and K. Hesselmann: Thermochemical Properties of Inorganic Substances, 2nd ed., Springer-Verlag, Verlag Stahleisen, 1991.
15. E.T. Turkdogan: Physical Chemistry of High Temperature Technology, Academic Press, NewYork, USA, 1980 pp. 81.
16. S. Banya: ISIJ Int., 1993, vol. 33 pp. 2-11.
17. P.G. Shewmon: Diffusion in Solids, McGraw-Hill, NewYork, USA, 1963, pp 8.
18. L.S. Darken: Trans. AIME, 1948, vol. 175 pp. 184.
19. L. Bolzmann:, Ann. Phys.,1984, vol. 53 pp. 959-964.
Tekko-Binran, Handbook of Iron and Steel, 3rd ed., ISIJ, Maruzen, Tokyo, 1981, pp. 350.
21. K. Nagata, N. Sata, and K. Goto: Tetsu-to-Hagane, 1982, vol. 68, pp. 1694-1705.
Acknowledgments
The ISIJ Research Promotion Grant of Iron and Steel Institute of Japan is acknowledged for partial financial support for this study. And this study was supported by research fund from Chosun University, 2016. We would like to appreciate Professor Katsunari Oikawa in Tohoku University for valuable comments about the diffusion coefficient and the thermodynamics.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted July 11, 2017.
Rights and permissions
About this article

Cite this article
Kim, SJ., Tago, H., Kim, KH. et al. Diffusion Behavior of Mn and Si Between Liquid Oxide Inclusions and Solid Iron-Based Alloy at 1473 K. Metall Mater Trans B 49, 977–987 (2018). https://doi.org/10.1007/s11663-018-1233-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-018-1233-8