
Abstract
This paper investigates the crystallization behavior of CaO-SiO2- and CaO-Al2O3-based mold fluxes for casting high-aluminum steels using single hot thermocouple technology, developed kinetic models, and scanning electron microscope. The results showed that the crystallization ability of the typical CaO-SiO2-based Flux A (CaO/SiO2 0.62, Al2O3 2 mass pct) is weaker than that of CaO-Al2O3-based Flux B (CaO/SiO2 4.11, Al2O3 31.9 mass pct) because of its higher initial crystallization temperature. The crystallization kinetics of Flux A was “surface nucleation and growth, interface reaction control” in the overall non-isothermal crystallization process, whereas that of Flux B was “constant nucleation rate, 1-dimensional growth, diffusion control, in the primary crystallization stage, and then transformed into constant nucleation rate, 3-dimensional growth, interface reaction control in the secondary crystallization stage.” The energy dispersive spectroscopy results for Flux B suggested that the variations in the crystallization kinetics for Flux B are due to different crystals precipitating in the primary (BaCa2Al8O15) and secondary (CaAl2O4) crystallization periods during the non-isothermal crystallization process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aluminum, as an effective additive, plays a key role in improving the tensile strength, yielding strength, and crash resistance of steel structures, and it tends to reduce the weight of the steel.[1,2,3] However, aluminum in molten steel may enter the mold flux as Al2O3 by reducing the components of the mold flux, such as SiO2 and MnO. In addition, the already existing Al2O3 inclusions formed in the primary and secondary steelmaking processes can be absorbed into the mold flux during casting. Such a pick-up of Al2O3 in the mold flux drastically deteriorates the thermal properties of the mold flux and leads to unstable heat transfer and poor mold lubrication, surface defects being introduced on the slab, or even the breakout of the molten steel.[4,5,6] To resolve these problems, two strategies have been proposed: (1) traditional mold flux with low CaO/SiO2, which contains 40 mass pct SiO2 to minimize the effects that the reaction between SiO2 (in the mold flux) and Al (from the molten steel) has on the mold flux properties,[7,8] and (2) a non-reactive mold flux, which is CaO-Al2O3-based to inhibit the slag metal reaction.[9]
A series of studies have been conducted to investigate the crystallization behavior of these two kinds of mold fluxes. Omoto et al.[7] designed a low CaO/SiO2-low viscosity mold flux for casting high-aluminum-containing electrical steel, and their results suggested that the crystallization ability of the mold flux became stronger with the addition of Li2O, whereas the slag rim became thicker. Seo et al.[10] indicated that a higher CaO/SiO2 ratio could enhance the crystallization capability of traditional mold fluxes but that B2O3 or Li2O would inhibit it. Ryu et al.[11] reported that the crystallization temperature increased, whereas the incubation time for crystallization decreased with the increase of the CaO/SiO2 mass ratio and alumina content. However, the change in the chemical composition of the reactive CaO-SiO2-based mold fluxes could not prevent the aluminum from the molten steel from migrating to the reactive mold flux. According to a report from ArcelorMittal,[12] the pick-up of Al2O3 in the spent mold flux could surpass 30 mass pct. during the continuous casting of high-aluminum transformation-induced plasticity (TRIP) steel. Therefore, to prevent a chemical reaction from occurring between the molten steel and the mold flux, a CaO-Al2O3-based non-reactive mold flux was designed. Zhou et al.[13] studied the effect of Al2O3 on the crystallization of a mold flux for casting high-aluminum steels and found that the crystallization ability was first enhanced and then inhibited with further increases in the Al2O3 content. Lu et al.[14] observed that Li2O tended to inhibit the crystallization ability of non-reactive mold fluxes and that Na2O played a similar role as Li2O. Xiao et al.[15] noted that the synergic effect of the combination of BaO and B2O3 tends to improve the mold flux crystallization in a non-reactive mold flux system. Shi et al.[16] reported that the addition of B2O3 suppressed the crystallization of lime-alumina-based mold fluxes by lowering the crystallization temperature, whereas Na2O had an opposite effect.
Further, some investigations on the kinetics and/or mechanism(s) behind mold flux crystallization have been conducted. Zhou et al.[17] studied the effect of CaO/SiO2 on the mold flux crystallization using the JMA isothermal crystallization theory and found that the crystallization kinetics of the mold flux changed from one-dimensional to three-dimensional growth for a constant number of nuclei when the CaO/SiO2 ratio of the mold flux varied from 0.8 to 1.2. Qi et al.[18] investigated the effect of TiO2 on the activation energy of the non-isothermal crystallization of a mold flux using the Kissinger method and revealed that the activation energy of mold fluxes diminished with the addition of TiO2. Lei et al.[19] studied the effect of MnO on the non-isothermal crystallization of mold fluxes using the Kissinger–Akahira–Sunose (KAS) method and found that as more MnO was added, higher activation energies were required to initiate crystallization. Yang et al.[20] explored the kinetics of non-isothermal crystallization of mold fluxes for the casting of high Al steels using the Avrami and Friedman methods and indicated that Na2O-free mold fluxes containing high Li2O were beneficial for improving crystallization. Recently, Shi et al.[21] investigated the non-isothermal crystallization kinetics of CaO-Al2O3-based mold fluxes using the Avrami–Ozawa and Friedman methods.
Although research on the crystallization of mold flux for casting high-aluminum steels has been carried out and kinetic models have been adopted to analyze the crystallization behavior of mold flux, very few of them have elucidated the difference between the crystallization kinetics of CaO-SiO2-based traditional mold flux and CaO-Al2O3-based non-reactive mold flux. Therefore, in this study, Continuous Cooling Transformation (CCT) diagrams of the typical CaO-SiO2-based and CaO-Al2O3-based mold fluxes are constructed using the Single Hot Thermocouple Technique (SHTT). Then, the crystallization behavior of these two mold fluxes is investigated via a kinetics study using the Johnson–Mehl–Avrami (JMA), Ozawa, and Mo models as well as Scanning Electron Microscopy (SEM) observations.
Experimental Apparatus and Method
Sample Preparation
The compositions of the mold fluxes for this study are shown in Table I, where Flux A is a conventional CaO-SiO2-based mold flux with a low CaO/SiO2 ratio of 0.62 and Flux B is a newly developed CaO-Al2O3-based mold flux with an Al2O3 content of 31.9 mass pct. The samples were first prepared via mixing chemical reagents, such as CaCO3, SiO2, Al2O3, MgCO3, B2O3, CaF2, Na2CO3, Li2CO3, and BaCO3. Then, the sample powders were heated in an induction furnace at 1773 K (1500 °C) for 10 minutes to homogenize their chemical compositions. Next, the molten slags were poured onto a water-cooled copper mold to achieve a fully glassy phase. Subsequently, the glassy slags were crushed and ground into sample powders for the Single Hot Thermocouple Technique (SHTT) tests.
Single Hot Thermocouple Tests
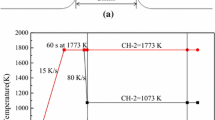
CCT (Continuous Cooling Transformation) tests of the mold fluxes were conducted using SHTT. The details of the SHTT apparatus are described in previous papers.[17,22] A schematic of the experimental apparatus and temperature control profile for the CCT test are shown in Figures 1((a) and (b)), respectively.

Schematic of the SHTT and temperature control profile: (a) the SHTT apparatus, (b) the temperature control profile for CCT test
During the CCT test, a B-type thermocouple with approximately 60 mg of sample powder was heated at a constant rate of 900 K/min from room temperature to 1773 K (1500 °C). The sample was held at this temperature for 5 minutes to melt the sample powders completely and eliminate any bubbles. Subsequently, the liquid sample was cooled at a pre-set cooling rate to room temperature. In this study, the pre-set cooling rate was set to 2, 4, 6, and 8 K/min for Flux A and 20, 30, 40, and 50 K/min for Flux B. The variations in the mold flux temperature vs time during the test were recorded by the thermocouple, and the evolution of the crystalline fraction was obtained from the CCD camera. Therefore, the non-isothermal kinetics analysis for mold flux crystallization was carried out based on those time, temperature, and relative crystalline fraction data obtained from the SHTT tests.[17]
Non-isothermal Kinetics Study of Crystallization Process
The non-isothermal kinetic models originated from the isothermal kinetic models. The Johnson–Mehl–Avrami (JMA) model is one of the classical models for describing an isothermal crystallization process.[23,24,25] According to the JMA model, the crystal fraction is given by
where t is the crystallization time; X(t) is the crystalline fraction at a given time t; n is the Avrami exponent associated with the nucleation and growth kinetics; and Z is the effective crystallization rate constant (including nucleation and growth).
Performing a double logarithm of Eq. [1], it would be rearranged as Eq. [2].
The plot for \( \ln \ln \left( {\frac{1}{1 - X(t)}} \right) \) vs \( \ln (t) \) is nearly a straight line for an isothermal crystallization process, and the slope of the straight line is the Avrami exponent n.
Although the JMA model is applicable to an isothermal crystallization process, it is not suitable for non-isothermal crystallization processes. Ozawa extended the JMA model to non-isothermal crystallization based on the mathematical derivation from Evans.[26] The final Ozawa equation is written as
where C(T) is the crystalline fraction at a given temperature T; β is the heating/cooling rate, which can be expressed as \( \beta = \frac{{\left| {T - T_{0} } \right|}}{t} \) (where T 0 is the temperature when t = 0 second); K is a function related to the overall crystallization rate; and m is the Ozawa exponent and holds an identical physical meaning as the Avrami exponent n for isothermal crystallization.[21]
Performing a double logarithm on Eqs. [3] and [4] is obtained as
Therefore, if the Ozawa equation can describe non-isothermal crystallization behavior precisely, the Ozawa exponent m should be obtained from the slope of the straight line by plotting \( \ln \ln \left( {\frac{1}{1 - C\left( T \right)}} \right) \) vs \( { \ln }\beta \). However, many works[21,27,28,31] suggest that the Ozawa plots would deviate from linearity because the crystalline fraction C(T) chosen at a given crystallization temperature T may include values corresponding to both the initial and final stages of crystallization due to variations in the cooling rates. The later stage may be associated with variations of the chemistry in the remaining liquid matrix due to successive crystal formation, which causes the deviation from linearity in the Ozawa plots.
Using the failure of the JMA and Ozawa models to describe the nucleation and growth of a non-isothermal crystallization process, Mo[29,30,31] derived Eq. [5] by combining the JMA Eq. [2] and the Ozawa Eq. [4].
where the parameter \( F\left( T \right) = \left( {\frac{K}{Z}} \right)^{{{\raise0.7ex\hbox{$1$} \!\mathord{\left/ {\vphantom {1 m}}\right.\kern-0pt} \!\lower0.7ex\hbox{$m$}}}} \)refers to the cooling rate that must be selected within a unit of crystallization time when the measured system reaches a certain crystalline fraction and b is the Mo exponent, which is the ratio of the effective Avrami exponent n′ (here, the effective Avrami exponent n′ is used for the Avrami exponent obtained for the non-isothermal conditions differently from the isothermal crystallization process) over the Ozawa exponent m, i.e., b = n′/m. According to Eq. [5], the plot of ln β vs ln t yields a series of straight lines at given crystalline fractions. The Mo exponent b can be estimated from the slope of these lines.
Therefore, in this study, to obtain the Ozawa exponent m, the Mo exponent b is first determined from Eq. [5]. Then, the effective Avrami exponent n′ is estimated by applying the JMA Eq. [2] (also known as the pseudo-JMA, as it was used in the non-isothermal process) to the non-isothermal crystallization process. Finally, the Ozawa exponent m, which has an identical physical meaning to the Avrami exponent n for isothermal crystallization, is obtained from m = n′/b.[30,31] The crystalline fraction X(t) or C(T) at a certain time t or T was calculated using X(t) = A c/A T or C(T) = A c/A T,[17] where A c is the crystal area and A T is the total mold flux area. The values of A c and A T were obtained by image analysis, as shown in Figure 2.

Snapshots for estimating the crystalline fraction of mold flux: (a) X(t) = 0 pct, (b) X(t) = 8.51 pct
SEM and XRD Tests of Mold Fluxes Crystallization
After the SHTT test, the samples were observed by a Scanning Electron Microscope (JSM-6360LV, Japanese Electronics Company) with an acceleration voltage of 20 kW to identify the crystallization kinetics of the mold fluxes. In addition, the composition of the crystals precipitated in the mold fluxes was analyzed by Energy Dispersive Spectroscopy (EDS).
Results and Discussion
The Continuous Cooling Transformation of Mold Fluxes
The non-isothermal CCT diagrams for Flux A and Flux B are shown in Figures 3((a) and (b)), and the initial crystallization temperatures of the two mold fluxes at different cooling rates are shown in Figure 3(c). Figure 3 shows that the initial crystallization temperature of mold flux decreased as the cooling rate increased. The reason for that is that it takes time for the mold flux to initiate crystallization, which is called the incubation time. The temperature will drop lower during the incubation time when the cooling rate is lager, resulting in a lower initial crystallization temperature.

CCT diagrams and initial crystallization temperature of mold fluxes. (a) CCT diagram for CaO-SiO2-based Flux A, (b) CCT diagram for CaO-Al2O3-based Flux B, (c) the initial crystallization temperature for two samples
Additionally, Figure 3(c) shows that the initial crystallization temperature of Flux A at small cooling rates (2 to 8 K/min) is much lower than for Flux B at relatively larger cooling rates (20 to 50 K/min). This may be because Flux A with a low CaO/SiO2 ratio (CaO/SiO2 = 0.62) has a weaker crystallization ability, whereas Flux B, which has a high alumina content and high CaO/SiO2 ratio, exhibits a much stronger crystallization ability. The results obtained here are consistent with previous studies.[13]
Analysis from the Mo, Pseudo-JMA, and Ozawa Models
The plots of \( \ln \beta \;vs\;\ln t \)at various crystalline fractions for Flux A and Flux B, according to Mo’s equation [5], are shown in Figures 4((a) and (b)), respectively. Figure 4 shows that all plots exhibit a good linear relationship between\( {\text{ln }}\beta \) and \( {\text{ln }}t \), suggesting that Mo’s equation can be used to describe the non-isothermal crystallization of these mold fluxes. The Mo exponent b and the parameter F(T) obtained from the slope and intercept of the linear fitted straight lines in Figure 4 are listed in Table II. Table II shows that F(T) increases with the crystalline fraction, which suggests that a larger cooling rate \( \beta \) is needed if more precipitate crystals are required in the molten mold flux when the crystallization time is fixed. In fact, it is reasonable that a lower temperature can enhance the driving force for crystallization, resulting in a larger crystalline fraction.

Plots of ln β vs ln t for non-isothermal crystallization of mold fluxes at various crystalline fractions X(t). (a) Flux A, (b) Flux B
Figure 5 shows the plots of \( \ln [ - \ln (1 - X(t))] \) vs \( {\text{ln }}t \) at various cooling rates when the pseudo-JMA model is applied to the non-isothermal crystallization process. The plots of CaO-SiO2-based Flux A are roughly single lines with some deviations, as shown in Figure 5(a), whereas the plots of the CaO-Al2O3-based Flux B constitute two linear sections along with \( {\text{ln }}t \)-axis, as shown in Figure 5(b). The changes in the slopes for Flux B are caused by the different crystalline phases that precipitate during the primary and secondary stages of the crystallization process, which is discussed in detail in Section III–C.

Plots of ln[−ln(1 − X(t))] vs ln t for non-isothermal crystallization of mold fluxes at different cooling rates. (a) Flux A, (b) Flux B
The values of the effective Avrami exponent n′ can be estimated from the slopes of the fitted lines in Figure 5, and they are summarized in Table III, where the n′ for Flux A varies in a narrow range from 1.589 to 1.743 for cooling rates from 2 to 8 K/min and the n′ for Flux B varies within two narrow ranges from 1.667 to 1.900 for the primary crystallization stage and 4.575 to 4.865 for the secondary crystallization stage when the cooling rates are in the range from 20 to 50 K/min. The average value of n′ is 1.656 for Flux A, but it is 1.775 and 4.720 for the primary and secondary crystallizations of Flux B, respectively. The variation of n′ for Flux B suggests that the crystallization kinetics in the primary and secondary crystallization stages are different.
Therefore, to explore the different non-isothermal crystallization kinetics between Flux A and Flux B, the Ozawa exponent m is estimated from the obtained Mo exponent b, and the effective Avrami exponent n′ is determined via the relationship b = n′/m. The calculated Ozawa exponent m corresponding to different crystalline fractions for the mold fluxes is listed in Table IV. In addition, because the Ozawa exponent m obtained in the non-isothermal crystallization process has an identical physical meaning to the Avrami exponent n for isothermal crystallization,[21] the corresponding relationship between n and the kinetics for the nucleation and growth of crystals are summarized in Table V according to Christian[32] and others.[33,34]
The Ozawa exponent m for Flux A for the overall non-isothermal crystallization process is determined to be nearly 1, as shown in Table IV. Therefore, by comparing the values for n in Table V, we conclude that there are three possible cases for the non-isothermal crystallization kinetics in Flux A:
Case (I): Constant number of nuclei, 2-dimensional growth, and diffusion control; Case (II): Constant number of nuclei, 1-dimensional growth, and interface reaction control; Case (III): Surface nucleation and interface reaction control.
Meanwhile, the Ozawa exponent m for Flux B for non-isothermal crystallization is determined to be approximately 1.5 during the primary crystallization stage and approaches 4.0 in the later stage, as shown in Table IV. Therefore, the possible cases for the non-isothermal crystallization kinetics for Flux B are as follows:
Case (i): Constant number of nuclei, 3-dimensional growth, and diffusion control in the primary stage, which transforms into a constant nucleation rate, 3-dimensional growth, and interface reaction control in the secondary crystallization stage. Case (ii): Constant nucleation rate, 1-dimensional growth, and diffusion control, which transforms into a constant nucleation rate, 3-dimensional growth, and interface reaction control in the secondary crystallization stage.
Observations from the SHTT Images and SEM
To further analyze the actual non-isothermal crystallization kinetics for Flux A and Flux B, the snapshots of the crystal precipitation at different crystalline fractions during the SHTT tests and SEM images after SHTT tests are shown in Figures 6 through 9. Among these, Figures 6 and 8 show the SHTT images for Fluxes A and B; Figures 7(a) and 9(a) show the general views of the samples after the SHTT tests; Figures 7(b), (c), and 9(b), (c) are magnifications of the boxed areas in Figures 7(a) and 9(a); and Figures 7(d), (e), and 9(d), (e) are the EDS results from the precipitated crystalline phases in Flux A and Flux B.

Crystallization evolution of Flux A when volume fraction is at (a) X 1 = 0.00 pct, (b) X 2 = 9.05 pct, (c) X 3 = 20.39 pct, (d) X 4 = 29.03 pct, (e) X 5 = 39.34 pct, (f) X6 = 49.05 pct, (g) X 7 = 60.42 pct, (h) X 8 = 70.40 pct, (i) X 9 = 79.91 pct

SEM and EDS analysis of Flux A after SHTT. (a) General view, (b) crystals in box areas of A-1, (c) crystals in box areas of A-2, (d) EDS results of A-I, (e) EDS results of A-II
Figure 6 shows that the crystals in Flux A first precipitate on the surface of the thermocouple wire as shown in Figures 6((a) and (b)). The crystals then grow toward the melt bath in the middle, as shown in Figures 6((c) through (i)). In reality, it is difficult to determine the exact crystallization kinetics using only images of the SHTT tests. This includes if it nucleates as constant number of nuclei and then grows upward (cases I and II) or if it just continuously nucleates first from the surface of thermocouple wire and then from the surface of the previous nuclei (case III).
However, further observations from the SEM in Figure 7 show that for Flux A after SHTT testing, many small bar-like crystals are oriented in the direction of the heat transfer, which suggests that it is impossible for crystals to grow up only from a constant number of nuclei. Therefore, combining the kinetic analysis from the models (Section III–B) and observations of the SHTT images in Figure 6 and SEM images in Figure 7, it can be concluded that the non-isothermal crystallization kinetics of Flux A is case (III): Surface nucleation and growth, interface reaction control. In addition, the EDS results listed in the table in Figure 7 show that there is only one kind of crystal phase with elements Si, Ca, and O, close to CaSiO3 that precipitated in Flux A.
Figure 8 shows the crystallization evolution of Flux B during the non-isothermal cooling SHTT test. In Figure 8, two stages occur during the crystallization process. The first stage covers Figures 8((a) through (d)), where a large columnar crystal forms at the bottom right side and grows into the center of the sample. The second stage includes Figures 8((e) to (i)), where new equiaxed crystals precipitate around the large columnar crystal and then grow upward. These two stages correspond to the primary and the secondary crystallization for Flux B, which is consistent with the results from the kinetics analysis (Section III–B). In order to determine the crystallization kinetics of Flux B, the crystallized sample is subjected to SEM analysis.

Crystallization evolution of Flux B when volume fraction is at (a) X 1 = 0.00 pct, (b) X 2 = 10.38 pct, (c) X 3 = 19.84 pct, (d) X 4 = 29.07 pct, (e) X 5 = 39.47 pct, (f) X 6 = 48.37 pct, (g) X 7 = 59.47 pct, (h) X 8 = 72.78 pct, (i) X 9 = 82.78 pct
From the kinetics results and SHTT images, the key point in determining whether case (i) or case (ii) describes the crystallization kinetics for Flux B is that the cloud-like crystalline aggregate that occurs during the primary crystallization stage is composed of some large crystals [case (i)] or large amounts of small crystals [case (ii)]. The magnified SEM image in Figure 9(b) shows that it is the latter. Therefore, we conclude that the non-isothermal crystallization kinetics for Flux B is case (ii): Constant nucleation rate, 1-dimensional growth, and diffusion control in the primary crystallization stage, which transforms into a constant nucleation rate, 3-dimensional growth, and interface reaction control.

SEM analyses of Flux B after SHTT. (a) General view, (b) crystals in box areas of B-1, (c) crystals in box areas of B-2, (d) EDS results of B-I, (e) EDS results of B-II
The EDS results in the table of Figure 9 show that the primary precipitated crystals contain the elements Ca, Si, Al, and Ba which is close to BaCa2Al8O15, whereas the secondary precipitated crystals contain the elements Ca, Si, and Al, which is close to CaAl2O4. Therefore, the different crystals precipitating in the primary and secondary crystallization stages cause the variations in the crystallization kinetics for Flux B during the non-isothermal crystallization process.
Conclusions
The non-isothermal crystallization kinetics of CaO-SiO2-based Flux A and CaO-Al2O3-based Flux B for casting high-aluminum steels was investigated using SHTT, kinetics models, and SEM. The main conclusions are summarized as follows:
-
(1)
The initial crystallization temperature of the mold flux decreased as the cooling rate increased, and the initial crystallization temperature of Flux A at a low cooling rate (2 to 8 K/min) is much lower than for Flux B at a large cooling rate (20 to 50 K/min), suggesting that the crystallization ability of Flux B is higher than that of Flux A.
-
(2)
Kinetic models were developed to describe the non-isothermal crystallization process of the two mold fluxes system. The results indicated that the models could be combined to determine the approximate range of the crystallization kinetics.
-
(3)
Combing the kinetic analysis with the models and observations from the SEM and SHTT, we find that the crystallization kinetics for Flux A is “surface nucleation and growth, interface reaction control” over the non-isothermal crystallization process, whereas the crystallization kinetics for Flux B are “constant nucleation rate, 1-dimensional growth, and diffusion control during the primary crystallization stage, which transforms into a constant nucleation rate, 3-dimensional growth, and interface reaction control during the secondary crystallization stage.”
-
(4)
The EDS results show that the precipitated crystals in Flux A are CaSiO3, whereas the primary and secondary precipitated phases in Flux B are BaCa2Al8O15 and CaAl2O4, respectively. These different precipitated crystal phases tend to cause variations in the crystallization kinetics of Flux B during non-isothermal crystallization processes.
References
O. Grässel, L. Krüger, G. Frommeyer, and L.W. Meyer: Int. J. Plasticity, 2000, vol. 16, pp. 1391-1409.
J.E. Jin, and Y.K. Lee: Acta mater., 2012, vol. 60, pp. 1680–88.
H. Peng, X. Song, A. Gao, and X. Ma: Mater. Lett., 2005, vol. 59, pp. 3330–32.
L. Zhou, W. Wang, and K. Zhou: Metals, 2016, vol. 6, pp. 139–53.
J.W. Cho, K. Blazek, M. Frazee, H. Yin, J.H. Park, and S.W. Moon: ISIJ Int., 2013, vol. 53, pp. 62–70.
K. Blazek, H. Yin, G. Skoczylas, M. Mcclymonds, and M. Frazee: World Iron & Steel, 2013.
J. Gao, G. Wen, Q. Sun, P. Tang, and Q. Liu: Metall. Mater. Trans. B, 2015, vol. 46, pp. 1850–59.
J. Liao, Y. Zhang, S. Sridhar, X. Wang, and Z. Zhang: ISIJ Int., 2012, vol. 52, pp. 753–58.
S. Jung and I. Sohn: Metall. Mater. Trans. B, 2012, vol. 43, pp. 1530–39.
M.D. Seo, C. Shi, J.W. Cho, and S.H. Kim: Metall. Mater. Trans. B, 2014, vol. 45, pp. 1874–86.
H.G. Ryu, Z. Zhang, J.W. Cho, G.H. Wen, and S. Sridhar: ISIJ Int., 2010, vol. 50, pp. 1142–50.
K. Blazek, Y. Hong-bin, G. Skoczylas, M. McClymonds, and M. Frazee: Iron Steel Technol. 2011, vol. 8, pp. 232–40.
L. Zhou, W. Wang, and K. Zhou: Metall. Mater. Trans. E, 2015, vol. 2, pp. 99–108.
B. Lu, and W. Wang: Metall. Mater. Trans. B, 2014, vol. 45, pp. 1496–1509.
D. Xiao, W. Wang, and B. Lu: Metall. Mater. Trans. B, 2015, vol. 46, pp. 1–9.
C. Shi, M.D. Seo, J.W. Cho, and S.H. Kim: Metall. Mater. Trans. B, 2014, vol. 45, pp. 1081–97.
L. Zhou, W. Wang, F. Ma, J. Li, J. Wei, H. Matsuura, and F. Tsukihashi: Metall. Mater. Trans. B, 2012, vol. 43, pp. 354–62.
X. Qi, G. Wen, and P. Tang: J. Non-Cryst. Solids, 2008, vol. 354, pp. 5444–52.
Y. Lei, B. Xie, F. Qi, and J. Diao: Journal of Iron & Steel Research International, 2008, pp. 732–36.
C. Yang, G. Wen, and P. Tang: Steel Res. Int., 2015, vol. 87, pp. 880-89.
C. Shi, M. Seo, H. Wang, J.W. Cho, and S.H. Kim: Metall. Mater. Trans. B, 2015, vol. 46, pp. 345-56.
W. Wang, X. Yan, L. Zhou, S. Xie, and D. Huang: Metall. Mater. Trans. B, 2016, vol. 47, pp. 963-73.
M. Avrami: J. Chem. Phys., 1939, vol. 7, pp. 1103-12.
M. Avrami: J. Chem. Phys., 1940, vol. 8, pp. 212-24.
M. Avrami: J. Chem. Phys., 1941, vol. 9, pp. 177-84.
T. Ozawa: Polymer, 1971, vol. 12, pp. 150-58.
M. Abareshi, S.M. Zebarjad, and E.K. Goharshadi: B. Mater. Sci., 2014, vol. 37, pp. 1113-21.
D.W. Henderson: J. Therm. Anal. Calori., 1979, vol. 15, pp. 325-31.
R.M.R. Wellen, and E.L. Canedo: J. Mater. Res., 2016, vol. 31, pp. 1-11.
T. Liu, Z. Mo, and H. Zhang: J. Appl. Polym. Sci., 1998, vol. 67, pp. 815–21.
T. Liu, Z. Mo, S. Wang, and H. Zhang: J. Appl. Polym. Sci., 2000, vol. 37, pp. 568-75.
J.W. Christian: The Theory of Transformations in Metals and Alloys, 3rd ed., Pergamon Press Ltd, London, UK, 2002.
K. Prapakorn: Ph.D. Dissertation, Carnegie Mellon University, Pittsburgh, PA, 2003
D.R. MacFarlane and M. Fragoulis: Phys. Chem. Glasses, 1986, vol. 37, pp. 228-34
Acknowledgments
The financial support from the National Science Foundation of China (51504294, 51528402) and the Opening Foundation of Ministry of Education Key Laboratory of Metallurgical Emission Reduction & Resources Recycling (Anhui University of Technology) is greatly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted April 24, 2017.
Rights and permissions
About this article

Cite this article
Zhou, L., Li, H., Wang, W. et al. Non-isothermal Crystallization Kinetics of Mold Fluxes for Casting High-Aluminum Steels. Metall Mater Trans B 48, 2949–2960 (2017). https://doi.org/10.1007/s11663-017-1117-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-017-1117-3