
Abstract
Phase equilibria in the SiO2-K2O-CaO system have been experimentally investigated in the SiO2-rich area. High-temperature equilibration, rapid quenching, and electron probe X-ray microanalysis (EPMA) techniques have been used in this study. K2O may vaporize during EPMA measurements causing significant uncertainties. In the present study, optimum EPMA operating conditions have been determined in order to accurately measure K2O concentrations in the quenched samples. The compositions of all phases present in the quenched sample were measured using EPMA with optimum operating parameters. The following primary phase fields were identified in the composition range investigated: SiO2, CaO·SiO2, 2CaO·SiO2, K2O·2CaO·2SiO2, and K2O·6CaO·4SiO2. The isotherms between 1273 K and 1473 K (1000 °C and 1200 °C) in these primary phase fields have been determined. The presence of the compounds K2O·2CaO·2SiO2 and K2O·6CaO·4SiO2 has been confirmed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The ternary SiO2-K2O-CaO system is important in various applications. Due to its low expansion rate and good chemical resistance, potassium–calcium-based glass has been produced for making beakers, bottles, and other containers for chemicals over hundreds of years.[1] SiO2, K2O, and CaO are predominant components in biomass ash,[2] which form slags during the production of bio-syngas. In iron blast furnace, the alkali calcium silicates often form on the linings.[3] SiO2, CaO, and K2O are also the main components in potassium silicate fertilizers[4] and glass coatings for biomedical applications with bioactivity.[5]
It is important to understand the phase equilibria of the SiO2-K2O-CaO system for the optimization of high-temperature processes mentioned above. However, it was found that there are significant discrepancies on the phase equilibria of this system. The first study in this system was undertaken by Morey et al.[6,7] in 1930s where they reported phase boundaries and isotherms in the SiO2-rich part. Seven potassium–calcium silicate compounds: 2K2O·CaO·3SiO2, K2O·CaO·SiO2, 2K2O·CaO·6SiO2, 4K2O·CaO·10SiO2, K2O·3CaO·6SiO2, K2O·2CaO·6SiO2, and K2O·2CaO·9SiO2 were reported in their studies.[6,7] The phase diagram was constructed by inspection of the crystals formed in the samples quenched from different temperatures using petrographic microscope. The pseudo-binaries K2SiO3-SiO2 and 2CaO·SiO2-K2O·CaO·SiO2 were later investigated using a similar method by Kracek et al.[8] and Taylor[9] in 1930s and 1940s, respectively. Arroyabe et al.[10] recently reported their doubts about the existence of K2CaSiO4 (K2O·CaO·SiO2). They found that K2CaSiO4 does not exist and may be a misinterpretation of K2Ca2Si2O7 (K2O·2CaO·2SiO2). The crystal K2Ca6Si4O15 (K2O·6CaO·4SiO2) was firstly mentioned by Ohsato et al.,[11] and later Arroyabe et al.[12] confirmed its existence and reported a complete structural and spectroscopic characterization. However, the phase relations of the K2CaSiO4 (K2O·CaO·SiO2) and K2Ca6Si4O15 (K2O·6CaO·4SiO2) have never been reported. Due to the limited thermodynamic data, little thermodynamic modeling work can be done in this system.[13,14]
Vaporization of K2O at high temperature results in the changes of the bulk compositions during the equilibration. This is the major experimental difficulty associated with the accurate determination of the phase equilibria in the SiO2-K2O-CaO system. The equilibration, quenching, and electron probe X-ray microanalysis (EPMA) techniques developed by the present authors allow the compositions of the phases present at high temperature to be accurately measured after the experiments. The changes in the bulk composition during high-temperature equilibration do not influence the phase compositions used for construction of the phase diagram. In the present study, the phase equilibria of the system SiO2-K2O-CaO in the SiO2-rich part have been investigated to characterize the potassium–calcium glass and biomass ashes and confirm the stabilities of potassium–calcium silicate compounds.
Experimental Procedure
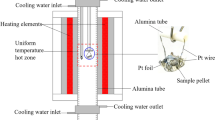
The experimental method used in the present study involves high-temperature equilibration in air, quenching, and EPMA.[15] The direct determination of the final compositions of the different phases present in the quenched samples by EPMA minimized the composition uncertainty caused by the vaporization of K2O.
Approximately 0.2 g pelletized mixture was used in each equilibration experiment. The mixtures were prepared by mixing the required amount of chemicals thoroughly in an agate mortar for 30 minutes. The chemicals used for preparation of the mixtures were powders of SiO2 (99.9 wt pct, Sigma-Aldrich), K2CO3 (≥99.0 wt pct, Sigma-Aldrich), and CaCO3 (≥99.0 wt pct, Sigma-Aldrich).
Equilibration experiments were carried out in a vertical tube furnace heated by lanthanum chromite (LaCrO3) elements. The reaction tube is an impervious recrystallized alumina tube with 30 mm inner diameter. A working thermocouple placed in a re-crystallized alumina sheath was kept inside the reaction tube to monitor the actual temperature of the sample. The working thermocouple was periodically calibrated against a standard thermocouple (supplied by the National Measurement Institute of Australia, NSW, Australia). The overall absolute temperature accuracy of the experiment was estimated to be ±3 K.
The sample wrapped in Pt foil (99.998 pct, 0.025 mm thick, Johnson Matthey Ltd) was suspended by a platinum wire (0.5 mm diameter) and introduced from the bottom of the reaction tube. The sample was raised slowly (30 to 60 seconds) from the bottom to the hot-zone of the furnace. All samples were premelted at 1523 K (1250 °C) for 10 minutes and followed by equilibration at the target temperature for 4 to 72 hours depending on the composition and temperature.
After equilibration, the sample was dropped directly into the water by pulling up the platinum wire. The specimen was dried on a hot plate, mounted in epoxy resin, and polished for examinations. Water-based polishing abrasives are commonly used in the polishing process for metallography samples. However, K2O can react with water resulting in composition changes. In the present study, kerosene was used as the polishing medium to obtain proper samples for EPMA.
EPMA Measurements of K2O-Containing Samples
EPMA has been widely used for quantitative elemental analyses in phase equilibrium studies.[15,16] The common EPMA operating parameters include acceleration voltage of 15 kV, probe current of 15 nA, probe diameter (PD) of “0,” peak measuring time (PMT) of 30 s, background positions of ±5 μm, and background measuring time of 10 seconds. It should be noted that the PD “0” stands for the smallest operating PD from the focused electron beam. However, it was found that the K2O concentrations measured at normal EPMA parameters for the present system are significantly lower than the actual values. Lineweaver[17] mentioned that the static electric field caused by electron beam drives the oxygen outgassing and alkali metal cations moving inwards the sample. The other possibility is that K2O is decomposed and vaporized under continuous radiation of the electron beam. Under high vacuum condition inside the EPMA, the vaporization will be more serious. Accurate determination of the phase composition is the base of the present study. The optimum operating parameters for EPMA measurements have been carefully investigated.
Two glasses with compositions 63.9 wt pct SiO2-7.8 wt pct CaO-28.3 wt pct K2O and 52.5 wt pct SiO2-8.1 wt pct CaO-39.4 wt pct K2O were carefully prepared and used as the reference materials. The SiO2, CaCO3, and K2CO3 powders were accurately weighed and mixed thoroughly. The pellet of approximately 1 g was placed in a Pt crucible and heated at 1273 K (1000 °C) for 20 hours in air. The weight losses of the samples were accurately determined during the heat treatment so that the glass compositions can be ascertained. Less than 0.3 wt pct losses were identified during the preparation of the homogeneous glasses and the glass compositions were adjusted by assuming that the weight losses were only associated with K2O. As the viscosity of the liquid was high, it was easy to quench the liquid phase to homogeneous glass. The polished samples were coated with carbon using a JEOL (Japan Electron Optics Ltd) Carbon Coater for electron microscopic examination. A JXA 8200 Electron Probe Micro-analyser with wavelength-dispersive detectors was used for microstructure and composition analysis. The standards used for analysis were from Charles M. Taylor Co. (Stanford, California): KAlSi3O8 for K and CaSiO3 for Ca and Si. The ZAF correction procedure included in JEOL software was applied.
The parameters in EPMA measurements include probe current, acceleration voltage, beam diameter, and measuring time. Each condition was tested separately ,while other parameters were fixed. The effects of probe current and acceleration voltage were firstly evaluated. It was found that at acceleration voltage 15 kV, PD “0,” and PMT 30 seconds, 25 and 40 pct lower K2O concentrations were measured compared to the actual K2O concentration when the probe current was 7.5 and 15 nA, respectively. The concentrations of SiO2 and CaO were increased proportionally which confirms that the inaccuracies of the measurements were only associated with K2O. The composition measured at a lower probe current 7.5 nA is better but it is still not acceptable. Similarly, it was found that at fixed probe current 15 nA, PD “0,” and measurement time 30 seconds, the K2O concentrations measured were 70 and 40 pct lower than the actual value at the acceleration voltage of 7.5 and 15 kV, respectively. It seems that the adjustment of probe current and acceleration voltage cannot significantly increase the measurement accuracy. The acceleration voltage and probe current were selected to be 15 kV and 15 nA, respectively. In addition, it was found that variation of the background measurement time between 3 and 10 seconds did not have a significant effect on the measured K2O concentration. The background positions were kept at ±5 μm as there are no overlaps between the three elements. The experimental conditions were then focused on the PD and PMT to find optimum parameters for K2O measurement.
The lengths of PMT were chosen to be 10, 20, and 30 seconds and the PD were “0”, 5, 10, and 15 µm. Figure 1 shows the effects of PMT on K2O concentrations measured by EPMA at fixed PD. It can be seen from the figure that at a given PD, the K2O concentration measured decreases with increasing PMT. For the sample containing lower K2O (28.3 pct), the satisfying results (<1 wt pct compared to the actual concentration) can be achieved at 10 seconds measurement time and PD 10 and 15 µm. If the measurement time is 20 seconds, the PD must be 15 µm to obtain a satisfying result. For the sample containing higher K2O (39.4 pct), the satisfying result can only be achieved at 10 seconds measurement time and PD 15 µm.

Effects of peak measuring time on K2O concentrations measured by EPMA at fixed probe diameters (PD), acceleration voltage 15 kV, and probe current 15 nA
Figure 2 shows the EPMA measurements of K2O concentrations as a function of PD. It can be seen that K2O concentrations measured by EPMA increase with increasing PD at a given measurement time. The optimum parameters obtained from Figure 1 are confirmed in Figure 2.

Effects of peak diameter on K2O concentrations measured by EPMA at fixed measuring time, acceleration voltage 15 kV, and probe current 15 nA
Orthogonal experiments with varying both PD and PMT were performed to find the optimum condition. All the conditions shown in Figures 1 and 2 are summarized in Table I. The average deviations of the measured K2O concentration from the two compositions are summarized in Figure 3. It can be seen clearly that condition 10 (PD = 15 µm, PMT = 10 seconds), which has the largest PD and shortest PMT, shows a lowest deviation among all the conditions.

Deviation of the measured K2O concentrations from actual K2O value at different conditions
The final parameters used in the EPMA measurements of this study were selected to be acceleration voltage 15 kV, probe current 1.5 × 10−8 A, PD 15 µm, PMT 10 seconds, background positions ±5 μm, and background measuring time 10 seconds.
Results and Discussion
The liquidus temperatures in the SiO2-K2O-CaO system have been determined in the temperature range between 1273 K and 1473 K (1000 °C and 1200 °C). The primary phase fields present in the composition range investigated include SiO2, CaO·SiO2, 2CaO·SiO2, K2O·6CaO·4SiO2, and K2O·2CaO·2SiO2. The typical microstructures of the quenched samples are shown in Figure 4. Figure 4(a) shows the equilibrium of liquid with SiO2 and Figure 4(b) shows the equilibrium of liquid with CaO·SiO2. In Figure 4(c), the liquid phase was in equilibrium with 2CaO·SiO2 and Figure 4(d) shows the equilibrium of liquid with K2O·6CaO·4SiO2. Figure 4(e) shows the equilibrium of liquid with K2O·2CaO·2SiO2. Figure 4(f) represents the typical microstructure showing the equilibrium of liquid with two solid phases K2O·6CaO·4SiO2 and K2O·2CaO·2SiO2.

Typical microstructures of quenched samples showing equilibrium of liquid with (a) SiO2, (b) CaO·SiO2, (c) 2CaO·SiO2, (d) K2O·6CaO·4SiO2, (e) K2O·2CaO·2SiO2, and (f) K2O·6CaO·4SiO2 + K2O·2CaO·2SiO2
The phase compositions measured by EPMA are summarized in Table II. The phase diagram of the system SiO2-K2O-CaO is constructed based on the experimental data given in Table II and is shown in Figure 5. The thick solid lines in the figure indicate experimentally determined boundaries between the primary phase fields, while the thick dashed lines indicate approximate boundaries. The thin solid and dashed lines indicate the experimentally determined and approximate isotherms, respectively. The data in SiO2-K2O system were derived from a previously reported phase diagram.[8]

Experimentally determined phase diagram in the SiO2-K2O-CaO system; the binary data were adopted from Ref. [8]
The phase diagram (solid lines) presented here is compared with the previous one reported by Morey et al.[6,7] (dashed lines) in Figure 6. It can be seen clearly that the phase diagram determined in the present study is significantly different from those determined in the previous investigations.[6,7] The K2O·CaO·SiO2 primary phase field was reported by Morey et al.,[6,7] but it does not appear in the present diagram. Two new primary phase fields K2O·2CaO·2SiO2 and K2O·6CaO·4SiO2 are determined in the present study. It is the first time to report these two new primary phase fields and their liquidus temperatures. It can be seen that liquidus temperatures in the SiO2 primary phase field increase with increasing SiO2 concentration. In contrast, liquidus temperatures in the primary phase fields of K2O·2CaO·2SiO2 and K2O·6CaO·4SiO2 decrease with increasing SiO2 concentration. The liquidus temperatures in the CaO·SiO2 primary phase field increase with increasing CaO/K2O ratio.

EPMA measurements of the quenched samples enable the compositions of the solid phase to be accurately determined together with the liquid in equilibrium. The microstructures and formulated compositions confirm that K2O·2CaO·2SiO2 and K2O·6CaO·4SiO2 are present in the SiO2-K2O-CaO system. It can be seen from Table III that two ternary compounds have narrow composition ranges close to their theoretical compositions.
Conclusions
The phase diagram of the SiO2-K2O-CaO system in the SiO2-rich area has been constructed using equilibration/quenching/EPMA technique. The EPMA operating parameters have been thoroughly investigated and the optimum condition has been determined to accurately measure K2O concentrations. The liquidus temperatures between 1273 K and 1473 K (1000 °C and 1200 °C) have been determined in the primary phase fields of SiO2, CaO·SiO2, 2CaO·SiO2, K2O·6CaO·4SiO2, and K2O·6CaO·4SiO2. The phase diagram determined in the present study is significantly different from those reported previously. The K2O·CaO·SiO2 primary phase field reported previously was not observed in the present study. Two new compounds K2O·2CaO·2SiO2 and K2O·6CaO·4SiO2 are confirmed and their primary phase fields have been determined for the first time.
References
W. Stern and Y. Gerber: Archaeometry, 2004, vol. 46, pp. 137-56.
S. V. Vassilev, D. Baxter, L. K. Andersen, and C. G. Vassileva: Fuel, 2010, vol. 89, pp. 913-33.
H. Hughes: Trans. Br. Ceram. Soc., 1966, vol. 65, pp. 661-79.
T. Akiyama, Y. Yao, and S. Matsuno: Jpn. J. Soil Sci. Plant Nutr., 2001, vol. 72, pp. 484-8.
C. Vitale-Brovarone and E. Verné: J. Mater. Sci. Mater. Med., 2005, vol. 16, pp. 863-71.
G. Morey, F. Kracek, and N. Bowen: J. Soc. Glass Technol., 1930, vol. 14, pp. 149-87.
G. Morey, F. Kracek, and N. Bowen: J. Soc. Glass Technol., 1931, vol. 14, pp. 57-8.
F. Kracek, N. Brown, and G. Morey: J. Phys. Chem., 1937, vol. 41, pp. 1183-93.
W. C. Taylor: J. Res. Natl. Bur Stand., 1941, vol. 27, pp. 311-23.
E. Arroyabe, R. Tessadri, D. M. Többens, and V. Kahlenberg: J. Am. Ceram. Soc., 2011, vol. 94, pp. 2652-5.
H. Ohsato, T. Sugimura, S. Hayashi, and T. Ogihara: Bull. Nagoya Inst. Technol., 1980, vol. 32, pp. 123-30.
E. Arroyabe, R. Kaindl, D. M. Többens, and V. Kahlenberg: J. Solid State Chem., 2009, vol. 182, pp. 3254-61.
L. Zhang, C. Schmetterer, and P. J. Masset: High Temp. Mater. Processes, 2013, vol. 32, pp. 223-8.
C. Bale, E. Bélisle, P. Chartrand, S. Degterov: Calphad, 2009, vol. 33, pp. 295-311.
M. Chen and B. Zhao: J. Am. Ceram. Soc., 2013, vol. 96, pp. 3631-6.
B. Zhao, P. C. Hayes, and E. Jak: Metall. Mater. Trans. B, 2010, vol. 41, pp. 374-85.
J. L. Lineweaver: J. Appl. Phys., 1963, vol. 34, pp. 1786-91.
Acknowledgments
The authors would like to thank Ms. Jie Yu for the lab assistance. The authors acknowledge the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, The University of Queensland, for providing facilities and scientific and technical assistance .
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted January 6, 2016.
Rights and permissions
About this article

Cite this article
Chen, M., Hou, X., Chen, J. et al. Phase Equilibria Studies in the SiO2-K2O-CaO System. Metall Mater Trans B 47, 1690–1696 (2016). https://doi.org/10.1007/s11663-016-0623-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-016-0623-z