
Abstract
Given the critical role that metastable retained austenite (RA) plays in advanced high-strength steel (AHSS), there is significant interest in obtaining a comprehensive understanding of its stability, to achieve excellent mechanical properties. Despite considerable attention and numerous studies, the significance of individual contributions of various microstructural factors (size, crystallographic orientation, surrounding phases, etc.) on the stability of RA remain unclear, partly due to the difficulty of isolating the direct effects of these factors. In this study, we examined the influence of microstructural factors while minimizing the effect of chemical composition on the mechanical stability of RA. We accomplished this by comparing the austenite (γ) stability in two distinct microstructures: a two-phase RA/martensite microstructure and a one-phase γ microstructure, both with nearly identical γ compositions. We employed in situ high-energy X-ray diffraction during uniaxial tensile testing conducted at both room temperature and 100 °C, facilitating the continuous monitoring of microstructural changes during the deformation process. By establishing a direct correlation between the macroscopic tensile load, phase load partitioning, and the γ/RA transformation, we aimed to understand the significance of the microstructural factors on the mechanical stability of the RA. The results indicate that very fine RA size and the surrounding hard martensitic matrix (aside from contributing to load partitioning) contribute less significantly to RA stability during deformation than expected. The findings of this study emphasize the critical and distinct influence of microstructure on γ/RA stability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The development of materials with superior mechanical properties, including high strength and ductility/toughness, has long been a demanding challenge, yet it remains an essential goal in the design of new materials or in the optimization of existing materials. A promising approach achieving such enhanced mechanical properties is through the development of multiphase microstructures with constituents of contrasting mechanical properties. Advanced high-strength steels (AHSS), such as dual-phase (DP) steels, transformation-induced plasticity (TRIP) steels, carbide-free bainitic steels, are a class of materials that incorporate such an approach and are renowned for their excellent mechanical properties.[1,2,3] In particular, the multiphase microstructure of these AHSS containing a metastable retained austenite (RA) is responsible for their excellent mechanical properties.[4,5] For instance, the transformation of the metastable RA to martensite during deformation contributes to a significant strain hardening and a delayed onset of necking, resulting in an excellent combination of high strength and high ductility. Nevertheless, controlling the stability of this RA is a critical factor that affects the performance of AHSS and has been extensively investigated in recent years,[6,7,8,9,10,11] with considerable research focused on evaluating the different factors governing the stability. Among the wide variety of factors, chemical composition, size, morphology, and crystallographic orientation of the RA, and micromechanical influence of surrounding microstructure have been identified as important factors influencing its stability. However, a systematic evaluation of the significance of these individual contributions to the stability of RA presents a significant challenge. This is due to the inherent difficulties present in multiphase microstructures in effectively deconvoluting them.
The effect of size on γ/RA stability (i.e., prior γ grain size for one-phase γ structure and RA region size for two-phase microstructure) has been extensively studied concerning the optimization of the mechanical properties. Most studies have indicated that small prior γ/RA sizes below 15 μm, particularly submicron RA, result in increased stability due to the inhibition effect it has on the γ to martensite transformation.[12,13,14,15,16] It has been suggested that small γ/RA have fewer preexisting martensite nuclei (in the case where defects within the γ were considered to be the preferred nucleation sites, rather than general grain boundaries), and thus there exists a lower probability of transformation.[17] It has also been argued from a mechanical aspect that with decreasing γ/RA size, the accommodation of plastic strain associated with γ to martensite transformation becomes more difficult, suppressing the martensitic transformation and thus making the γ/RA more stable.[7,18] Contrasting results, however, exist in the literature reporting that mechanical stability, in contrast to thermal stability, is lowered as its size decreases.[19] These observations regarding the contrasting role of size on the stability of RA have been correlated with the potential for formation of deformation twins in large RA regions. The activation of mechanical twinning promotes the development of in-grain deformation substructures, leading to an increased mechanical stability. Conversely, small RA regions present less favorable conditions for mechanical twinning, rendering them more susceptible to transforming into α′-martensite.[19] Interestingly, some other studies have also indicated that size (1 to 80 μm) does not influence the deformation-induced martensitic transformation behavior during tensile deformation, suggesting that the mechanical stability of γ/RA remains unaffected by size.[15] Despite numerous studies so far, the exact role of size and its underlying mechanisms responsible for the stability of γ/RA remains unclear.
Additional complexity arises from the constituent structures accompanying the RA due to load partitioning upon deformation (i.e., the local stress in the RA) and shielding effects that are inherent in multiphase microstructures with a hard matrix surrounding the RA. There are differing viewpoints in the literature regarding the role of the surrounding matrix. Some studies suggest that the volume expansion associated with the martensitic transformation of RA is resisted by the surrounding hard martensitic matrix, limiting the transformation.[7,20] While some other reports suggest that the stability of RA when surrounded by a softer ferrite phase is increased due to stress relaxation effects.[21] With these varying perspectives in literature, the role of the surrounding matrix on deformation-induced transformation of RA, the load partitioning behavior among different constituent structures, and their correlation with the mechanical stability of RA remain unclear.
In this study, we aim to overcome some of these challenges by examining the mechanical stabilities of γ/RA for the same γ composition in two medium manganese steels with distinct microstructures: a two-phase microstructure (constituting a martensitic matrix with thin film RA, \({\alpha }^{{{\prime}}}\)+\({\gamma }_{f}\)) and a one-phase equiaxed γ microstructure. This approach allows us to explore the different governing factors independently of composition while also reducing the complexity of the aforementioned micromechanical problem of multiphase microstructures. Prior to this work, we have also examined the thermal stabilities of these two steels.[22] The microstructures of these steels, characterized through laboratory XRD measurements and microscopy analysis are presented in Table I and Figure 1. The previous work revealed significant difference in the γ/RA thermal stabilities, with the RA in two-phase microstructure showing high thermal stability (Ms < − 196 °C), while its corresponding one-phase γ microstructure showed significantly lower thermal stability (Ms = − 8 °C). Building on this work, in the current study, we will further examine the γ/RA stabilities under an external load using in situ synchrotron X-ray diffraction during uniaxial tensile tests at room temperature (RT) and 100 °C. The use of synchrotron high-energy X-ray diffraction, as an advanced microstructural characterization tool, provides a method to directly monitor lattice plane spacing in multi-phase microstructures as a function of applied stress and/or strain during deformation and thus allows for real-time monitoring of microstructural changes. The correlation between the applied stress, load partitioning between phases and the γ to martensite transformation has been analyzed from the diffraction data of the two steels.

SEM micrographs of heat-treated samples (a) Steel 1 and (b) Steel 2. The inset in (b) is the chemical etched surface of Steel 2 showing the ε-martensite and α′-martensite. γF thin film RA, γ:austenite, α′: bcc-martensite, ε: hcp-martensite
2 Experimental Methods
2.1 Material and Tensile Samples
The chemical composition of the two medium manganese steels used in this study is presented in Table II. Detailed information on the heat treatment processes applied and the characterization of their microstructures can be found in reference.[22] For Steel 1, an initial austenization was performed at 900 °C for 15 minutes, followed by rapid quenching in water to achieve a fully martensitic microstructure. Subsequently, an intercritical annealing (IA) at 650 °C for 3 hours was applied, resulting in the development of a two-phase microstructure consisting of a martensitic matrix (\({\alpha }^{{{\prime}}}\)-martensite) with thin film RA (\({\gamma }_{f}\)) present between the martensite laths, as shown in Figure 1. The average composition of the RA at the end of the IA was determined to be 0.69C-7.3Mn (See Table I) using a combination of transmission electron microscopy-energy dispersive X-ray spectroscopy (TEM-EDX) and X-ray diffraction (XRD) techniques.[23] TEM-EDX was used to analyze the manganese content, while the complementary XRD estimated the carbon content from the lattice parameter measurement considering the contribution from manganese as determined from TEM-EDX. Similarly, Steel 2 with nominal composition 0.68C-7.3Mn was subjected to an identical austenization at 900 °C for 15 minutes, followed by water quenching to room temperature. The resulting microstructure of Steel 2 was \(\upgamma \) with a small amount of ε-martensite and α'-martensite. This α′-martensite as described in our previous work[22] is the sample preparation induced α′-martensite from ε-martensite present in the microstructure. Flat dog-bone-shaped tensile specimens were carefully prepared by electrical discharge machining from the heat-treated steels. The cross-section of the tensile samples was 1 × 0.8 mm2 and the gauge length was 3 mm. Prior to the in situ tensile loading experiments, the samples were electropolished (20 V in voltage, at room temperature) using a solution of 10 volume pct HCIO4- ethanol to remove any mechanically induced surface alterations of the microstructure during the preparation.
2.2 In Situ High-Energy X-ray Diffraction
The in situ high-energy X-ray diffraction (HEXRD) measurements were performed at the FAST ID3A beamline of the Cornell High-Energy Synchrotron Source (Ithaca), using a monochromatic X-ray beam of energy 61.332 keV (wavelength λ = 0.0202153 nm) in transmission geometry, see the schematic illustration of the experimental setup in Figure 2. To investigate the mechanical stability of the RA, the tensile samples were mounted on a Rotation and Axial Motion system (RAMS2)[24] load frame with the load axis perpendicular to the incoming X-ray beam of 2.2 x 0.6 mm2 size. The RAMS2 load frame provided precise alignment of the sample in three directions with respect to the X-ray beam and a rotation around the loading axis of the sample. A continuous strain rate of 2 × 10−5 s−1 was applied while the specimens were continuously rotated around the loading axis until the specimen fractured. Two-dimensional diffraction patterns were recorded by two area detectors (Dexela 2923, 3888 × 3072 pixels, 74.8 × 74.8 µm2 per pixel and sample-to-detector distance of approximately 1000 mm) using an exposure time of 0.25 s, in rotation steps of Δω = 2.5 deg. The detector and instrument parameters for the X-ray diffraction setup were calibrated using a standard CeO2 sample (NIST SRM 674b). The macroscopic strain was determined by measuring the change in distance between points within the pattern observed on the surface on the gauge of the tensile specimen by digital image correlation (DIC) technique, using custom MATLAB code.

Schematic illustration of experimental setup used in the in situ high-energy X-ray diffraction experiments. The samples were mounted on a RAMS2 tensile load frame and subjected to a continuous strain rate while rotating the sample around the loading axis
2.3 Diffraction Data Analysis
The experimental data consisted of a series of two-dimensional Debye-Scherrer diffraction patterns as a function of sample elongation and rotation angle (ω). Calibration and integration of the diffraction data were performed using the GSAS-II software.[25] The 2D diffraction intensities in the two Dexela panels were integrated along the azimuthal angle in the range of 0 to 180 deg and merged to obtain the 1D diffraction patterns. These patterns were subsequently analyzed by the least square fitting of pseudo-Voigt function using LIPRAS.[26] Crystallographic reflections belonging to the face-centered cubic (fcc), body-centered cubic (bcc) and hexagonal closed pack (hcp) phases were identified. The volume fractions of fcc (γ/RA), bcc (α′-martensite) and hcp (ε-martensite) were calculated based on the fitted intensities from the one-dimensional diffraction patterns at the selected deformation steps, using the direct comparison method[27]
where \(n\) is the number of peaks examined for a particular phase, \({f}_{i}\), \({I}_{i}\) and \({R}_{i}\) are respectively the volume fraction, integrated intensity, and theoretical intensity of phase, i.
To investigate the lattice strain, sectors of 10 deg—centered around the loading direction—were analyzed from 2D diffraction images. This analysis focused on reflections: γ-111 and γ-200 of γ/RA, as well as α-110 and α-200 of the α'-martensitic matrix. The lattice plane strain for the hkl reflection was calculated by
where \({d}_{\text{h}kl}^{\phi }\text{ and }{d}_{\text{h}kl}^{ref,\phi }\) are the lattice plane distances at the moment of observation and prior to the onset of macroscopic tensile loading, respectively.
3 Results and Discussion
3.1 Macroscopic Stress–Strain Responses
The tensile stress-strain responses measured at RT and 100 °C during in situ HEXRD experiments of Steels 1 and 2 are shown in Figure 3. The relevant parameters that characterize the macroscopic mechanical behavior of both steels are detailed in Table III. At RT, Steel 1 displayed a yield strength of 460 ± 10 MPa, an ultimate tensile strength of 720 ± 10 MPa, and a total elongation of 15.6 pct. When the temperature increased to 100 °C, the yield strength of Steel 1 remained constant at 460 ± 10 MPa, while its ultimate tensile strength decreased to 650 ± 10 MPa and the total elongation increased to 21.2 pct. On the other hand, Steel 2 exhibited an unexpected brittle behavior at RT with a tensile strength of 300 ± 10 MPa, fracturing without exhibiting any plastic deformation. When the temperature increased to 100 °C, the yield strength was 280 ± 10 MPa, while the ultimate tensile strength and total elongation were 680 ± 10 MPa and 17.6 pct, respectively.

Macroscopic stress-strain behavior of Steels 1 and 2 during in situ high-energy X-ray diffraction tensile measurements at RT and 100 °C. The solid and dashed blue curves represent Steel 1, while the solid and dashed green curves represent Steel 2. Insert presents an enlarged version of Steel 2 at RT (Color figure online)
The significant differences in the mechanical behaviors of Steels 1 and 2 can be attributed to their distinct microstructures, i.e., the two-phase \(({\alpha }^{{{\prime}}}\)+\({\gamma }_{f})\) and one-phase (γ) microstructures. In Steel 1, the yield strength remained unchanged with an increase in temperature, suggesting that it is insensitive to temperature. This behavior, although unusual for conventional steels, has been previously reported in some TRIP-assisted steels and it is attributed to the γ stability.[28,29,30] Regarding variation in the plastic behavior at the two temperatures, considerable changes were observed for the two steels. In Steel 1, the total elongation increased from 15.6 to 21.2 pct, and in Steel 2, it increased from 0.2 to 17.6 pct. Moreover, the ultimate tensile strength decreased in Steel 1 from 720 to 650 MPa. While, in Steel 2, the ultimate tensile strength increased from 300 to 680 MPa. The influence of temperature on macroscopic plastic behavior (total elongation) of both steels appears to be in accordance with plastic behavior of conventional steel, which primarily deforms through dislocation slip. However, considering the presence of metastable γ in both steels, its transformation/deformation mechanisms, such as martensitic transformation and mechanical twinning, could be concurrently active with the dislocation slip. The stability of the γ/RA in the two microstructures as well as the changes in the deformation mechanisms during plastic deformation are thus the key factors in understanding the observed mechanical behavior of these steels.
3.2 In Situ High Energy X-Ray Diffraction Results
The 1-D diffraction patterns obtained during the tensile tests at different deformation steps, integrated over all ω values, for the two steels are shown in Figure 4. The diffraction patterns are shown for both RT and 100 °C, with a magnified view to emphasize low-intensity reflections. For Steel 1 consisting of a martensitic matrix and thin film RA (\({\alpha }^{{{\prime}}}\)+\({\gamma }_{f}\)) after IA, the initial patterns at both temperatures show expected reflections from RA, specifically γ-111 and γ-200, and reflections from α′-matrix, α-110 and α-200. The initial volume fraction of the RA and α′-matrix in Steel 1 were determined to be 0.29 and 0.71, respectively at both temperatures, consistent with the phase quantification obtained from laboratory XRD measurements performed at RT (Table I). Within the elastic deformation regime, the peak intensities in the patterns remained constant. However, in the plastic deformation regime, the patterns showed a gradual decrease in the intensity of the γ-111 and γ-200 reflections at both temperatures with increasing deformation. This gradual decrease in RA reflections with simultaneous increase in α′ reflections during the plastic deformation process indicates the occurrence of a plastic-strain-induced transformation of the RA to α′. During deformation at RT, RA reflections disappeared; however, at 100 °C, noticeable RA reflections were still present (corresponding to a volume fraction of RA of approximately 0.04) at the fracture (see Figure 4). This difference in transformation kinetics and thus RA stabilities is expected due to a decrease in driving force for the γ to α′-martensite transformation with an increase in temperature. Furthermore, for Steel 1 at RT, noticeable reflections associated with ε-martensite (hcp) formed during plastic deformation. The evolution of the amount of the different phases during deformation in Steel 1 at RT is shown in Figure 5. For Steel 1 deformed at 100 °C, no noticeable ε-martensite reflections were observed (Figure 4), possibly due to a lower driving force for ε-martensite formation at 100 °C compared to at RT.

1-D diffraction patterns during tensile deformation of Steel 1 and Steel 2 at RT and 100 °C. (b, d, f, h) present enlarged views of corresponding regions in (a, c, e, g), highlighting low-intensity reflections of ε-martensite

Evolution of RA, ε-martensite, α′-martensite with the engineering stress for Steel 1 at RT
For Steel 2, the initial diffraction patterns at RT and 100 °C (Figure 4) showed the expected dominant γ reflections, γ-111 and γ-200, and noticeable ε-martensite reflections. The initial ε-martensite, as explained in previous work,[22] is thermally induced and formed during quenching from the austenization step, unlike in Steel 1 at RT where it is formed during tensile deformation. The initial RT patterns also showed a noticeable reflection (α-110) of the bcc α'-martensite. In our previous work,[22] we also observed the presence of initial α'-martensite in steel 2, which we attributed to the sample preparation-induced transformation. This was evidenced by the detection of α'-martensite in SEM and EBSD analyses of non-electropolished samples, while XRD analysis of electropolished samples consistently did not show any α'-martensite. This suggests that the α'-martensite is primarily confined to the sample surface and can be removed by electropolishing. In the present work, there is still some deformation-induced α'-martensite present, but the fraction is very low (< 0.005) and it is therefore considered to not affect the subsequent results and is excluded in the analysis. Thus, the volume fraction of γ and ε-martensite in Steel 2 were determined to be 0.98 and 0.02, respectively, consistent with the laboratory XRD measurements of this steel.[22] With increasing deformation until fracture at both temperatures, the diffraction patterns continued to predominantly show the γ-111 and γ-200 reflections and no new α′-martensite reflections were observed during the deformation, indicating higher γ phase stability against deformation-induced transformation compared to Steel 1. In addition, in the case of Steel 2 deformed at 100 °C, γ remains untransformed even at a high plastic strain of 17.6 pct, however, some of the initially observed ε-martensite reflection disappeared after the macroscopic yield stress while a new ε-martensite reflection concurrently emerged (Figure 4), indicating either strain-induced formation of ε-martensite or reorientation of the ε-martensite at 100 °C. All these observations demonstrate that Steel 2 with equiaxed γ microstructure exhibits significantly different stabilities and transformation behavior compared to the RA in Steel 1.
3.3 Lattice Plane Strains and Load Partitioning
Figure 6 shows the evolution of lattice strains for different hkl planes, determined using Eq. [2], as a function of the engineering stress for RA and α′-matrix of Steel 1, and for bulk γ of Steel 2, both at RT and 100 °C during tensile deformation. The lattice strain vs engineering stress for Steel 1 shows two distinct deformation stages, Stage 1 and 2, separated by a black dashed line in Figures 6(a) and (c), corresponding to the macroscopic yield stress of Steel. In Stage 1, the hkl lattice plane strains for the RA and α′-matrix exhibited mainly a linear relationship with engineering stress at both temperatures. The difference in slopes of the hkl lattice plane strains within each constituent is due to the anisotropic (i.e., orientation dependent) elastic stiffness and consistent with the expected behavior by the elastic anisotropy factor (Ahkl) for the cubic crystallographic structure where A111, γ < A200, γ and A200, α < A110, α.[31,32] In Stage 2, which begins with the plastic deformation of the lower strength constituent (RA), load redistribution between RA and α′-matrix becomes apparent, indicated by the change in slope of RA lattice strains. During this stage, the (elastic) lattice strain for the α-110 and α-200 planes of the α′-matrix, as well as the γ-200 plane, continued increasing with engineering stress, while γ-111 showed an anomalous behavior of decreasing plane strain with increasing engineering stress, compared to the expected increase. For Steel 2, a linearity between the γ lattice plane strains and engineering stress beyond the yield stress, at 100 °C, is observed. This effect is commonly reported for one-phase materials during tensile deformation.[33]

Evolution of the individual lattice strains in the γ/RA and α′-matrix with engineering stress in (a) Steel 1, RT, (b) Steel 2, RT, (c) Steel 1, 100 °C, (d) Steel 2, 100 °C with the plane normal along the loading direction
The decrease in γ-111 lattice plane strains of Steel 1, after exceeding the macroscopic yield stress is likely due to various factors. First, the partial or complete overlapping of diffraction peaks, specifically α-110 with γ-111, during plastic deformation could lead to errors in the peak position identification, and consequently, inaccuracies in lattice plane strain measurement. However, these peaks were sufficiently distinguishable early in the plastic deformation stage, reinforcing the reliability of the compressive trend. Second, the deformation-induced transformation of RA, leading to a decrease in RA fraction redistributes the load, and reduces the effective load on the remaining RA. In this case, the redistribution must influence all RA lattice planes uniformly, however, the significant decrease in γ-111 lattice plane strains, in contrast to γ-200, suggests that it is not due to load redistribution alone but likely combined with other phenomena, such as stresses as a result of the RA transformation to martensite. In general, the transformation of γ into martensite results in a net increase in volume. As martensite forms within the γ, its expansion is constrained by the surrounding untransformed γ. This constraint deforms the untransformed γ to accommodate the increased volume. The observations in this work indicate that this deformation is not uniform but rather the γ lattice experiences different types of stress on different planes. Specifically, the direction parallel to the normal of γ-111 lattices planes are compressed, while the direction normal to γ-200 planes are stretched. This heterogeneous yield of the remaining γ is a result of the internal stresses generated by the martensitic transformation, as also suggested in.[34]
The stress experienced by the RA (phase-specific stress) during the tensile deformation can be evaluated from the lattice strains using Hooke’s law. While lattice strains averaged over several reflections is more appropriate for determining phase-specific stress,[34,35,36] such an approach was not feasible in the present study due to the limited probed reflections. As an alternative, lattice strain information of single γ-200 reflection was used to evaluate the stress within the γ/RA during transformation. This method, while constrained by the number of hkl reflections, provides a meaningful degree of insight into the RA stress condition during the deformation.
where the Young’s modulus (\({\text{E}}_{200}\)) was taken from the literature as 147 GPa for γ, as experimentally determined from the γ-200 reflection at RT.[37] It is also critical to acknowledge that this approach involves certain assumptions and introduces a degree of approximation. Thus, the results are to be interpreted with understanding of this limitation.
The stress heterogeneity in steel 1 after yielding was attributed to the martensitic transformation, although it is not the sole significant contributor and this phenomenon even exists above Md, the maximum temperature at which deformation-induced martensite can form. The presence of RA, which differs in terms of mechanical properties from the surrounding martensite matrix, as well as the anisotropic elastic and plastic properties of the individual phases plays crucial roles. This difference in strength leads to uneven stress distribution and localized stress concentrations. Additionally, the different work-hardening behavior of martensite and RA also contributes to this behavior.
3.4 Analysis of Critical Stresses and Driving Forces for Martensitic Transformation
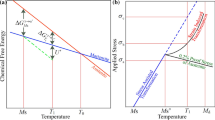
From a thermodynamic point of view, γ transforms into martensite when the available driving force exceeds a certain barrier. It happens spontaneously upon cooling at Ms when the difference in Gibbs energy between the γ and α′-martensite, i.e., the chemical driving force \(( (\Delta {\text{G}}_{\text{chem}} = {\text{G}}_{{\upgamma}} - G_{\alpha } \)) exceeds the transformation barrier (\(\Delta {\text{G}}^{*})\). Since martensitic transformation is a diffusionless shear process involving coordinated movements of atoms (i.e., mechanical in nature), it can also be initiated by applying an external stress at temperatures above Ms but below Md, the maximum temperature below which martensite transformation can be induced by deformation. The \(\Delta {\text{G}}_{\text{chem}}\) is then complemented by a mechanical driving force (\(\Delta {\text{G}}_{\text{mech}}\)) resulting from the applied load to overcome \(\Delta {\text{G}}^{*}\) , as illustrated by T1 and T2 in Figure 7. At temperatures above Ms but below \({\text{M}}_{\text{s}}^{\upsigma }\) (the transition temperature from stress induced to strain induced martensitic transformation), see Figure 7, the stress required for the plastic deformation of γ exceeds the stress required for the martensitic transformation (\({\upsigma }_{\text{transformation}}<{\upsigma }_{\text{austenite}}\)). As the temperature increases, the chemical driving force decreases, and higher applied stress is needed to form martensite. Above \({\text{M}}_{\text{s}}^{\upsigma }\) the required stress for the martensitic transformation is higher than the required stress for plastic deformation of γ (\({\upsigma }_{\text{transformation}}>{\upsigma }_{\text{austenite}}\)).

Schematic illustrations of (a) the influence of stress and temperature on the martensitic transformation and (b) driving force of the martensitic transformation and temperature
In this study, we evaluated stabilities of γ/RA in the two steels using a thermodynamic-based expression,
\(\Delta {\text{G}}_{\text{chem}}\) was determined from the difference in Gibbs energy between the γ and the α′ phase (\({G}_{\gamma }- {G}_{\alpha^{\prime} }\)) using Thermo-Calc and the TCFE6 database.[38] \(\Delta {\text{G}}_{\text{mech}}\) was determined with the empirical relation given in[39] using the stress experienced by the constituent γ/RA during the transformation/fracture.
where σ is the equivalent stress (MPa), ∆v/v is the fractional volume change upon transformation, and \({\upsigma }_{\text{h}}\) is the hydrostatic stress (MPa) and is equivalent to 1/3σ for uniaxial tension.
Based on previous work[22] which included cooling to liquid nitrogen (LN) temperature, for Steel 1, \(\Delta {\text{G}}^{*}\) was determined based on the available chemical driving force at the LN temperature since the RA, when no deformation was applied, was observed to be stable upon cooling in LN, and for Steel 2, it was determined based on the available chemical driving force at the measured Ms temperature[22] (Table IV).
The stress required for the transformation of RA to martensite in Steel 1 was evaluated for the two different temperatures used during the in situ HEXRD measurements using Eq.3 and the associated γ-200 lattice plane strain at which a decrease in RA amount was observed (see Figure 8). This critical strain also corresponds to the onset of the decrease in the γ-111 lattice plane strain. It should be noted that lattice plane strains beyond this critical point must be interpreted with caution due to inherent complexity in separating the influence of the deformation-induced martensite on the lattice strains of the martensitic matrix and the untransformed γ.

(a) Evolution of lattice plane strains in the RA in Steel 1 with engineering stress; (b) Evolution of RA with engineering stress, during the tensile deformation at RT and 100 °C
The uncertainty margin associated with determining the critical stresses is considered as it accounts for variations in estimates that could affect the precise determination of the critical stress needed to initiate the transformation. The uncertainty in the initiation of the RA transformation depends on the distances between the experimental data points shown in Figure 8a. To determine this objectively, we identified the initial point of RA volume reduction and considered the inter-point distance as an uncertainty in determining the critical stress. Table IV presents the critical γ-200 lattice strain in Steels 1 and 2 at the two temperatures, along with the corresponding critical stresses and mechanical driving forces evaluated from Eq.5. The critical (transformation) stresses of the RA in Steel 1 were determined to be 617 ± 161 MPa at RT and 676 ± 118 MPa at 100 °C. In contrast for Steel 2, where no martensitic transformation was observed, the critical stress was directly taken as the fracture stress, which represents the maximum stress experienced by γ in Steel 2. The critical (fracture) stress of γ in Steel 2 was determined to be 147 ± 29 MPa at RT and 985 ± 29 MPa at 100 °C.
As already mentioned, experimental observation demonstrates that if no stress is applied, the RA in Steel 1 remained stable upon quenching in LN.[22] This suggests that the transformation barrier, ∆G*, must be greater than the available chemical driving force at LN temperature, \(\Delta {\text{G}}_{\text{chem},\text{ LN}},\) 3440 J/mol. The additional mechanical driving force required to initiate the RA transformation to martensite at RT should thus be greater than 1142 J/mol (\(\text{i}.\text{e}., \Delta {\text{G}}_{\text{additional},{\text{ RT}}} > \Delta {\text{G}}_{\text{chem},\text{ LN}}- \Delta {\text{G}}_{\text{chem},{\text{ RT}}}\) with the values given in Table IV). However, the mechanical driving force for the RA to martensite transformation, calculated using the critical stress and Eq. 5, gives only 343 ± 116 J/mol and is much lower than 1142 J/mol. Similarly, for Steel 1 at 100 °C, the extra driving force should exceed 1677 J/mol (i.e., \(\Delta {\text{G}}_{\text{additional}, 100\,\,{}^\circ {\text{C}}} > \Delta {\text{G}}_{\text{chem},\text{ LN}}\)- \(\Delta {\text{G}}_{{\text{chem}}, 100\,\,{}^\circ {\text{C}}}\)) to initiate the RA transformation but the calculated mechanical driving force, \( \Delta {\text{G}}_{{{\text{mech}},100\,\,{}*{\circ } {\text{C}}}} , \) corresponding to the critical (transformation) stress of the RA, is only 386 ± 87 J/mol. However, the experimental observations indicate that even if these calculated driving forces are below the transformation barrier the transformation takes place in Steel 1 at both temperatures. Interestingly, the total available driving forces (i.e., \(\Delta {\text{G}}_{\text{chem}}+ \Delta {\text{G}}_{\text{mech}}\)), 2641 ± 116 J/mol at RT and 2149 ± 87 J/mol at 100 °C, are close to the transformation barrier, \({\Delta{ \text{G}}}_{\text{Steel }2,{\text{ RT}}}^{*}\), 2531 J/mol, determined for γ in Steel 2 with the same chemical composition at the experimental Ms temperature (− 8 °C). This suggests that the RA transformation barrier during an applied stress in Steel 1 is primarily composition-dependent, and the microstructural effects, such as small size, which are expected to significantly increase the barrier, may be negligible. This size independence on RA stability during deformation is consistent with a previous study by Matsuoka et al.,[15] which did not show any significant difference in RA mechanical stability across different prior γ grain sizes. However, for Steel 1 at 100 °C, the total driving force is still below the transformation barrier of 2531 J/mol. This raises questions why the RA in Steel 1 transformed at this temperature despite the seemingly insufficient driving force. One hypothesis could be related to additional factors, not considered in this work, providing the deficit energy to surpass the transformation barrier. Strain-induced defects, including nucleation sites, stacking faults, and deformation twin effects, have been suggested in literature[40,41,42,43] as potential contributors to the transformation. While the exact mechanisms by which these factors contribute to overcome the barrier fall beyond the scope of this work, they could play a crucial role in facilitating the RA transformation at 100 °C.
For Steel 2 at RT, the available chemical driving force \(, \Delta {\text{G}}_{\text{chem},{\text{ RT}}},\) 2298 J/mol, is the same as for Steel 1 due to identical composition, while the mechanical driving force, \(\Delta {\text{G}}_{\text{mech},{\text{ RT}}} ,\) during deformation (corresponding to the fracture stress) is only 52 ± 13 J/mol, which is lower than the required driving force of 233 J/mol. The total driving force during deformation, 2350 ± 13 J/mol, is thus lower than the transformation barrier of 2531 J/mol. Similarly, for Steel 2 at 100 °C, the available chemical driving force, \( \Delta {\text{G}}_{{{\text{chem}},100\, \, {}^{\circ } {\text{C}}}} ,\) decreases to 1763 J/mol, indicating an additional driving force of 768 J/mol is needed for the transformation. The maximum mechanical driving force, \( \Delta {\text{G}}_{{{\text{mech}},100\, \, {}^{\circ } {\text{C}}}} , \) during the deformation is 621 ± 23 J/mol. The total driving force at 100 °C, 2384 ± 23 J/mol, is also thus lower than the transformation barrier of 2531 J/mol. These results indicate that during deformation, the γ in Steel 2 is unlikely to transform to martensite due to the absence of a sufficient driving force, and is consistent with experimental observations, which is in contrast to the Steel 1 at 100 °C. The difference in the microstructure between Steel 1 and 2 is the key to explaining the observations. The hypothesis regarding additional factors providing the deficit energy to surpass the transformation barrier in Steel 1 may not apply in the same way in Steel 2. The absence of transformation in Steel 2 suggests that these additional factors (i.e., strain induced defects) may either not play a significant role in this steel or may have retarded the initiation of the martensitic transformation process. A similar observation has been reported by Wang et al.[19] where large regions of RA showed higher mechanical stability compared to smaller regions of RA despite having the same chemical composition. As mentioned in the introduction, these observations are correlated with the formation of deformation twins in large regions of RA which can enhance the mechanical stability of the material. When mechanical twinning is activated, it leads to the creation of specific internal structures within the grains, improving the mechanical strength of the steel. On the other hand, smaller RA regions do not support mechanical twinning, making them more prone to transform into α′-martensite upon deformation.[19]
3.5 Influence of Microstructure on RA Stability: Thermal vs Mechanical Transformations
A significant result of this study is the observation that the thin film RA in Steel 1 exhibits relatively lower stability during deformation compared to the coarse equiaxed γ in Steel 2, despite having similar magnitude of total driving force. This suggests that microstructural factors, particularly size and the surrounding matrix, play a crucial role in governing the γ/RA stability. In addition, the thermal stability of RA in Steel 1 (Ms < − 196 °C), as previously mentioned, was very high compared to the thermal stability of γ in Steel 2 (Ms = − 8 °C),[22] indicating the contrasting roles of microstructure on thermal and mechanical stabilities. This observation potentially challenges a previously presumed direct correlation between microstructure and stability, suggesting that the correlation is more complex and multifaceted than initially perceived.
There are three possible reasons why the microstructural factors such as the size and surrounding matrix only influence the thermal stability but not the mechanical stability of RA: (1) different transformation barriers for thermally and mechanically induced martensite, (2) variation from multivariant to single-variant transformation for thermal and mechanical conditions, (3) lattice defects (dislocation formation).
First, when examining the transformation barrier from γ to martensite, it is important to consider the distinct influencing factors and mechanisms involved under thermal and mechanical conditions. Thermal condition here refers to transformation upon cooling without any external applied stress, while mechanical condition refers to transformation at a given temperature by an applied stress. For one-phase γ materials, the strength of the material increases with decreasing size due to the Hall-Petch effect. Consequently, during γ to martensite transformation, the adjacent untransformed γ grains, surrounding the transforming region, have higher strength with decreasing size and offer greater resistance in accommodating the transformation strain, thus increasing the transformation barrier. A similar effect also manifests in two-phase microstructures with a martensitic matrix where it differs significantly in strength from γ. With decreasing RA size, it becomes more challenging to deform the matrix. The size and thus the (relative) strength of the surrounding structure are influential factors in determining the transformation barrier under thermal conditions. However, under a mechanical load the strength of the matrix, and thus the transformation barrier, are affected differently for the same material than when only subjected to a temperature change. This is because the matrix surrounding the γ is subjected to a tensile stress due to the applied load, making it easier to accommodate the transformation strain even for small sizes, making it easier to overcome the barrier compared to thermal induced transformations.
A second explanation is the variations in the number of martensitic variants, as explained by Matsuoka et al.[15] For thermally induced martensitic transformation, large grains favor multivariant transformation, where 24 variants are equally formed to minimize anisotropic transformation strain, while small grains undergo only single-variant transformation due to spatial limitations. The strain from martensitic transformation cannot be effectively minimized through a single-variant transformation, leading to the suppression of martensitic transformation in small grains. In the case of deformation-induced martensite, specific variants are selected to release the uniaxial tensile strain. In this case, the suppression of martensitic transformation by grain refinement becomes invalid because multi-variant transformation is no longer necessary.[15]
A third explanation involves lattice defects. These defects play a crucial role, as their presence can either facilitate or retard the transformation process depending on their density within the microstructure.[44] When subjected to a mechanical load, the deformation introduces a higher density of dislocations and may promote martensitic transformation by assisting martensite nucleation and growth. Conversely, under thermal conditions, the limited number of dislocations due to lack of deformation results in a low probability of martensitic transformation. Furthermore defects, such as stacking faults and twins, also influence the stability of γ/RA. In undeformed state, γ/RA phase contains few stacking faults. However, during plastic deformation, the accumulation of partial dislocations and regular arrays of stacking faults occurs in the γ/RA. Larger regions of γ/RA tend to develop stacking faults and exhibit mechanical twining, enhancing mechanical stability. In contrast, smaller regions of γ/RA are less prone to mechanical twining and may transform into α′ martensite even with minimal deformation.[19] Several studies have consistently shown a decrease in mechanical twinning with grain refinement. For instance, Ueji et al. observed in a deformed TWIP steel that the percentage of grains containing deformation twins decreased from 50 to 17 pct when the grain size was reduced from 49.6 to 1.8 μm.[45] Gutierrez-Urrutia et al. reported a significant decrease in the twin area fraction due to grain refinement, from 0.2 for a grain size of 50 μm to 0.1 for a grain size of 3 μm.[46] This behavior is generally attributed to increased critical twinning stress with decreasing grain size, often described by a Hall-Petch type relationship.
4 Conclusion
The study investigated the stability of γ/RA in two medium manganese steels with different microstructures using in situ high energy X-ray diffraction. Steel 1, with a two-phase microstructure, showed lower RA stability during deformation, in contrast to the thermal stability, compared to the equiaxed γ in Steel 2, despite similar γ composition, smaller size, and a thin film morphology. The lower stability in Steel 1 can be largely attributed to load partitioning between the martensitic matrix and RA, directing a significant portion of the load to RA, increasing the driving force for martensitic transformation. It highlights the need for micromechanical models to simulate the average response of the phases and load partitioning, considering martensite formation. The variation in stability between the thin film RA in Steel 1 and the coarse equiaxed γ in Steel 2, despite similar total driving forces, emphasizes the critical influence of microstructural factors on γ/RA stability. The limited role of these additional factors in Steel 2 indicates the complexity of the underlying mechanisms. Additionally, the distinct effects of microstructure on the thermal and mechanical stabilities of RA further highlight the importance of comprehensively understanding the microstructural effects on γ/RA stability. This requires knowledge of the transformation barrier, chemical driving force, mechanical driving force, and consideration of load distribution and phase interaction in multiphase microstructures and defects formation.
References
C.C. Tasan, M. Diehl, D. Yan, M. Bechtold, F. Roters, L. Schemmann, C. Zheng, N. Peranio, D. Ponge, M. Koyama, K. Tsuzaki, and D. Raabe: Annu. Rev. Mater. Res., 2015, vol. 45, pp. 391–431.
S. Zhang and K.O. Findley: Acta Mater., 2013, vol. 61, pp. 1895–1903.
S.M. Hasan, M. Ghosh, D. Chakrabarti, and S.B. Singh: Mater. Sci. Eng. A, 2020, vol. 771, pp. 1–12.
H. Luo, J. Shi, C. Wang, W. Cao, X. Sun, and H. Dong: Acta Mater., 2011, vol. 59, pp. 4002–14.
L. Liu, B. He, and M. Huang: Adv. Eng. Mater., 2018, vol. 20, pp. 1–17.
D. De Knijf, C. Föjer, L.A.I. Kestens, and R. Petrov: Mater. Sci. Eng. A, 2015, vol. 638, pp. 219–27.
X.C. Xiong, B. Chen, M.X. Huang, J.F. Wang, and L. Wang: Scr. Mater., 2013, vol. 68, pp. 321–24.
I. Tamura: Met. Sci., 1982, vol. 16, pp. 245–53.
B. He: Materials, 2020, vol. 13, pp. 1–31.
A. Arlazarov, M. Gouné, O. Bouaziz, and A. Hazotte: Philos. Mag. Lett., 2017, vol. 97, pp. 125–31.
A. Saha Podder and H.K.D.H. Bhadeshia: Mater. Sci. Eng. A, 2010, vol. 527, pp. 2121–28.
E. Jimenez-Melero, N.H. van Dijk, L. Zhao, J. Sietsma, S.E. Offerman, J.P. Wright, and S. van der Zwaag: Acta Mater., 2007, vol. 55, pp. 6713–23.
Z.H. Cai, H. Ding, R.D.K. Misra, and Z.Y. Ying: Acta Mater., 2015, vol. 84, pp. 229–36.
S. Lee, S.J. Lee, and B.C. De Cooman: Scr. Mater., 2012, vol. 66, pp. 832–33.
Y. Matsuoka, T. Iwasaki, N. Nakada, T. Tsuchiyama, and S. Takaki: ISIJ Int., 2013, vol. 53, pp. 1224–30.
H. Koohdar, M. Nili-Ahmadabadi, M. Habibi-Parsa, H.R. Jafarian, T. Bhattacharjee, and N. Tsuji: Metall. Mater. Trans. A Phys. Metall. Mater. Sci., 2017, vol. 48A, pp. 5244–57.
Y. Inokuti and B. Cantor: Acta Metall., 1982, vol. 30, pp. 343–56.
S.M.C. van Bohemen and L. Morsdorf: Acta Mater., 2017, vol. 125, pp. 401–15.
M.M. Wang, C.C. Tasan, D. Ponge, A. Kostka, and D. Raabe: Acta Mater., 2014, vol. 79, pp. 268–81.
J. Hidalgo, K.O. Findley, and M.J. Santofimia: Mater. Sci. Eng. A, 2017, vol. 690, pp. 337–47.
Y. Tomota, P. Lukáš, D. Neov, S. Harjo, and Y.R. Abe: Acta Mater., 2003, vol. 51, pp. 805–17.
J. Kumpati, S.M. Hasan, M.B. Rolland, and A. Borgenstam: Metall. Mater. Trans. A Phys. Metall. Mater. Sci., 2024, vol. 55A, pp. 466–76.
F. Huyan: Computational Materials Design of Medium Mn Steels, 2018.
P.A. Shade, B. Blank, J.C. Schuren, T.J. Turner, P. Kenesei, K. Goetze, R.M. Suter, J.V. Bernier, S.F. Li, J. Lind, U. Lienert, and J. Almer: Rev. Sci. Instrum., 2015, vol. 86, pp. 1–9.
B.H. Toby and R.B. Von Dreele: J. Appl. Crystallogr., 2013, vol. 46, pp. 544–49.
Line-Profile Analysis Software (LIPRAS). https://mse.ncsu.edu/jones/tools/. Accessed 20 Aug 2024.
A.K. De, D.C. Murdock, M.C. Mataya, J.G. Speer, and D.K. Matlock: Scr. Mater., 2004, vol. 50, pp. 1445–49.
X.D. Wang, B.X. Huang, Y.H. Rong, and L. Wang: Mater. Sci. Eng. A, 2006, vol. 438–440, pp. 300–05.
M.R. Berrahmoune, S. Berveiller, K. Inal, A. Moulin, and E. Patoor: Mater. Sci. Eng. A, 2004, vol. 378, pp. 304–07.
H.C. Shin, K. Ha, and Y.W. Chang: Scr. Mater., 2001, vol. 45, pp. 823–29.
R.K. Dutta, R.M. Huizenga, M. Amirthalingam, A. King, H. Gao, M.J.M. Hermans, and I.M. Richardson: Scr. Mater., 2013, vol. 69, pp. 187–90.
K. Yan, K.D. Liss, I.B. Timokhina, and E.V. Pereloma: Mater. Sci. Eng. A, 2016, vol. 662, pp. 185–97.
E.C. Oliver, M.R. Daymond, and P.J. Withers: Acta Mater., 2004, vol. 52, pp. 1937–51.
M. Villa, F. Niessen, and M.A.J. Somers: Metall. Mater. Trans. A Phys. Metall. Mater. Sci., 2018, vol. 49A, pp. 28–40.
A.N. Ezeilo, G.A. Webster, P.J. Webster, and X. Wang: Physica B Condens Matter, 1992, vol. 180–181, pp. 1044–46.
B. Clausen, T. Lorentzen, and T. Leffers: Acta Mater., 1998, vol. 46, pp. 3087–98.
M.R. Daymond, C.N. Tomé, and M.A.M. Bourke: Acta Mater., 2000, vol. 48, pp. 553–64.
J.O. Andersson, T. Helander, L. Höglund, P. Shi, and B. Sundman: Calphad, 2002, vol. 26, pp. 273–312.
Z.D. Feinberg: Design and Optimization of an Austenitic TRIP Steel for Blast and Fragment Protection, 2012.
G.B. Olson and M. Cohen: J. Less-Common Met., 1972, vol. 28, pp. 107–18.
H. Fujita and T. Katayama: JIM, 1992, vol. 33, pp. 243–52.
L. Bracke, L. Kestens, and J. Penning: Scr. Mater., 2007, vol. 57, pp. 385–88.
X.S. Yang, S. Sun, and T.Y. Zhang: Acta Mater., 2015, vol. 95, pp. 264–73.
S. Chatterjee, H.S. Wang, J.R. Yang, and H.K.D.H. Bhadeshia: Mater. Sci. Technol., 2006, vol. 22, pp. 641–44.
R. Ueji, N. Tsuchida, D. Terada, N. Tsuji, Y. Tanaka, A. Takemura, and K. Kunishige: Scr. Mater., 2008, vol. 59, pp. 963–66.
I. Gutierrez-Urrutia, S. Zaefferer, and D. Raabe: Mater. Sci. Eng. A, 2010, vol. 527, pp. 3552–60.
Acknowledgment
This work was performed within the Competence Centre Hero-m 2i financed by VINNOVA (the Swedish Governmental Agency for Innovation Systems), Swedish industry, and KTH Royal Institute of Technology. This work is based on research conducted at the Center for High-Energy X-ray Sciences (CHEXS), which is supported by the National Science Foundation (BIO, ENG and MPS Directorates) under award DMR-1829070.
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Funding
Open access funding provided by Royal Institute of Technology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kumpati, J., Rolland, M.B., Hasan, S.M. et al. Deconstructing the Retained Austenite Stability: In Situ Observations on the Austenite Stability in One- and Two-Phase Bulk Microstructures During Uniaxial Tensile Tests. Metall Mater Trans A (2024). https://doi.org/10.1007/s11661-024-07569-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11661-024-07569-4