
Abstract
Oxygen (O) is an inevitable impurity in bulk metallic glasses (BMGs) and its influence over the glass-forming ability (GFA) of BMGs is a longstanding controversy. The present ab initio molecular dynamics (AIMD) simulations indicate that the GFA decreases upon introducing 0.78 at. pct O in the amorphous Zr55Cu55Al9Be9 (at. pct), while examining the evolution of atomic configurations and kinetic properties in BMGs. This study includes a comprehensive analysis using pair correlation function (PCF), bond pair analysis (BPA), and Voronoi polyhedra construction. It is concluded that the incorporation of O leads to a decline in the closely packed icosahedral polyhedrons, where the atom O is coordinated with Be and Zr in the first nearest shell to form the O-centered clusters with enhanced ordering. Mean square displacement (MSD) analysis also shows that the trace O could induce remarkable acceleration of atomic mobility, therefore increasing crystallization tendency of the Zr55Cu55Al9Be9 alloy. The present results illuminate the role of O in the metallic glass-forming process and reveal the underlying role of O in the GFA of the Zr-Cu amorphous alloys.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The Zr-Cu based bulk metallic glasses (BMGs) represent an emerging class of structural as well as functional materials with unique physical and mechanical properties as a consequence of their amorphous nature.[1,2,3,4] It is known that oxygen (O) is an unavoidable impurity in BMGs, originating from both raw materials and surrounding atmospheric conditions. Even a small amount of O produces a dramatic effect on both crystalline phase fraction and mechanical properties of BMGs. The exact role of O on glass formation remains unknown despite considerable research on metallic glass synthesis, due to the complex interactions in multi-component alloys.[1,2,3,4] The objective of the present study is to identify the underlying mechanisms responsible for the effect of O content on the glass-forming ability (GFA) of Zr-Cu based BMGs. As outlined below, interpretation of the results from other recent studies remains controversial.
Generally, impurity O is regarded as a detrimental component to the GFA,[5] as it may promote the formation of crystalline phases. For instance, Qiu et al.[6] deduced that increasing O impurities (0.1 to 1.0 at. pct) in the Zr-Cu-Al-Ni alloys results in an increase of the primary crystallizing phases and hence deteriorates the GFA. Liu et al.[7] also demonstrated that a level of 0.3 at. pct O dramatically reduces the GFA and thus embrittles Zr-based BMGs. However, it was also reported that a tiny amount of O may enhance the GFA in Fe-based[8] and Zr-based[9] amorphous alloys by suppressing precipitation of the competitive primary phases and reinforcing liquid stability.[10,11] Wang et al.[12] found that the addition of O (from 0.015 to 0.075 wt pct) actually improves the GFA in the Zr-Cu alloys for some specific compositions (e.g., Zr53Cu47 and Zr54Cu46), resulting in a transition from the partial to fully amorphous condition.
It is apparent that the GFA depends strongly on the O content and the composition of BMG alloys.[13] The addition of O at fairly high levels (1.5 wt pct) was shown to preclude glass formation, but at low amounts (0.025 to 0.5 wt pct), amorphous structures were obtained.[14] Moreover, the introduction of new components (e.g., 0.1 at. pct B + 0.2 at. pct Si + 0.1 at. pct Pb in Zr-based BMGs) stabilizes the glass-phase matrix even though it contains 0.3 at. pct levels of O.[7]
Despite the numerous publications on GFA in BMG alloys, the influence of O on the GFA is still not well understood. According to the literature, the detrimental effect of O is normally attributed to the induced harmful oxides, acting as heterogeneous nucleation sites for crystalline phases. However, it is also known that the solubility of O in crystalline Zr is pretty high (e.g., ~ 30 at. pct at 200 °C).[15] A small amount of O is expected to dissolve uniformly into Zr alloys; accordingly Zr-containing oxides will not be formed.[12] Therefore, it is suspected that trace O influences the GFA of amorphous alloys through inducing or suppressing the formation of existing crystalline phases,[12] rather than heterogeneous nucleation of oxides.
Furthermore, the mechanism related to how O affects the GFA in the Zr-Cu-based alloys may be different from other BMG alloys. For example, the addition of O promotes the formation of the metastable Ti2Ni-type “Big Cube” phase in the Zr-Ni alloys,[14,16] which also depends on the local cooling conditions. However, the phases formed in the Zr-Cu BMG alloys are always ZrCu or Zr2Cu precipitates that can contain any level of O concentration.[12,14] Accordingly, the devitrification pathways may be quite different in the Zr-Cu amorphous system in comparison with the Zr-Ni system. It is evident that further work is required to understand the underlying science regarding the influence of O on the GFA of the Zr-Cu amorphous alloys, especially in the case of alloying with other dopants.
In the present work, the prime focus is on the topological properties and kinetics of Zr55Cu55Al9Be9 and Zr55Cu55Al9Be9O, in order to illustrate the effects of O on the cluster structures of Zr-Cu-Al-Be alloys when transforming from liquid to BMGs. Consequently, ab initio molecular dynamics (AIMD) simulations have been performed to examine the cooling process of these two BMG alloys. Furthermore, the present work also investigates how O influences the course of the BMG formation in Zr-Cu-Al-Be alloys. The study reveals the mechanism of the O-induced BMG crystallization from ancillary first-principles and phonon calculations based on density functional theory (DFT).
2 Calculational Methods
All DFT-based AIMD simulations in the present work were performed using the Vienna ab initio simulation package (VASP)[17,18] based on density functional theory (DFT). Note that ancillary first-principles and phonon calculations were also based on the VASP code and are presented in the Supplementary Material.
The generalized gradient approximation (GGA) by Perdew–Burke–Ernzerhof (PBE)[19] was employed as the exchange correlation functional. The AIMD simulations were conducted based on the NVT ensemble,[20,21] in which the number of atoms (N), volume (V), and temperature (T) kept constant. The Nosé–Hoover thermostat was utilized to control the temperature. The equation of ion motion was solved via the velocity Verlet algorithm with a time step of 2 fs. Only the Γ point in the Brillouin zone was applied. The plane-wave cutoff energies were adjusted by VASP, i.e., “PREC = low” for the general AIMD simulations and “PREC = Accurate” for the cases to estimate the zero external pressure. The convergence of electronic energy was smaller than 10−4 eV/atom.
The Zr-Cu-Al-Be BMG alloy was described by a 128-atom cubic supercell of Zr55Cu55Al9Be9. One O atom was introduced into this supercell, i.e., Zr55Cu55Al9Be9O1 with O content of 0.78 at. pct, to investigate the role of O in glass formation. In order to obtain the initial liquid configuration of Zr55Cu55Al9Be9 and Zr55Cu55Al9Be9O, the lattice parameters were set as 13.27 × 13.27 × 13.27 Å3 and 13.27 × 13.27 × 13.27 Å3, respectively, to coincide with experimentally measured density of the Zr55Cu30Ni5Al10 alloy[22] and also ensure the external pressure is zero. The initial structure was then equilibrated at 3000 K for at least 10,000 ionic steps to build a random liquid configuration. Then the cooling process was carried out sequentially from 1200 to 300 K with an interval of 100 K, each for 2000 steps (4 ps). The configuration at each temperature stage was all thermally annealed for at least 6000 steps (12 ps) to get equilibrium configurations; see detailed AIMD schemes in Figure 1. These annealed configurations also act as the input model for the next cooling process. Note that the equilibrium volumes of supercells at each temperature were adjusted to zero pressure using at least 3000 steps (6 ps); as summarized in Figure 2, the volumes decrease with decreasing temperature. Particularly, the simulated densities of Zr55Cu55Al9Be9 and Zr55Cu55Al9Be9-O at 300 K are 6.91 and 6.70 g/cm3, respectively, which agree with the experimental density around 6.799 g/cm3 of Zr55Cu30Ni5Al10.[22] It should be mentioned that we employed more supercomputer resources to perform these time-consuming AIMD simulations (see details in Acknowledgements). Roughly we can perform only 50 AIMD steps per hour by using 32 central processing units (CPUs).

Calculation steps including annealing and quenching for Zr55Cu55Al9Be9 and Zr55Cu55Al9Be9O, where the equilibrium volumes are adjusted at each temperature

Equilibrium volume per atom for Zr55Cu55Al9Be9 and Zr55Cu55Al9Be9O during the annealing processes
In order to characterize structural evolution and dynamic properties of Zr55Cu55Al9Be9 and Zr55Cu55Al9Be9O, various methodologies including the pair correlation function (PCF), bond pair analysis (BPA), coordination number (CN), Voronoi polyhedron, and mean square displacement (MSD) were systematically employed.
The pair correlation function (PCF) indicates the radial distribution for the atoms of interest, which provides the distance information between the nearest neighbors.[2324] The PCF is also normalized by the average density:
where g(r) stands for the PCF, n(r) the mean number of atoms in a shell with width of Δr at distance r, and ρ the average density of atoms in the system. All the PCF results involved in the present work were obtained by considering the average of the last 100 AIMD steps to ensure statistic accuracy. Moreover, the AIMD-simulated PCF, g(r), can be separated into the partial PCFs for all the possible atom pairs, with respect to a specific species. The uniqueness of PCF lies in its convenience in comparison with experimental results from such as X-ray and neutron diffractions. It also provides distance information (i.e., the cutoff value) between the nearest neighbors. However, the PCF is merely a statistical analysis of global structure, which cannot characterize the local structure motifs in both the liquid and amorphous states. To better quantify topological evolutions, other structural analysis methods were also used as detailed below.
The bond pair analysis (BPA) offers specific bonding information between a central atom and its nearest neighbors.[25,26,27] According to the Honeycutt and Anderson’s formula,[28] the indexes (i, j, k, l) can be used to interpret atomic local circumstance in regard to a root pair that represents two atoms in the pair of interest for decomposing the first two peaks of the PCF. Here, i = 1 means there are bonds forming between the root pair; otherwise i = 2 indicates that atomic pairs are not the near neighbors. Parameter j denotes the number of atoms forming bonds with the root pair. Parameter k represents the number of bonds forming among j atoms. Parameter l is adopted to distinguish local structure when i, j, and k are the same. The typical bond pairs are related to specific ordering. For example, the (1551) bond pairs have fivefold symmetry, which gives a measure of perfect icosahedral ordering. The (1431) and (1541) indicate distorted icosahedral configuration. The (1421) and (1422) reflect the characteristics of bond pairs of face-centered cubic (fcc) and hexagonal-close-packed (hcp) structures. Moreover, the (1441) and (1661) pairs refer to body-centered cubic (bcc) structure. The other types of bond pairs are usually classified as random structures. The BPA can distinguish various inherent structures like icosahedral, fcc, hcp, and bcc clusters; nevertheless, it cannot concretely tell sufficient feature differences between the fcc and hcp structures.
The Voronoi tessellation method is a useful tool to analyze the local atomic environment,[29,30] which describes the possible clusters relative to a central atom spatially and visually. The Voronoi clusters are described by utilizing the 4-integer index 〈n3, n4, n5, n6〉, where ni represents the number of i-edge polygons.[31] Tiny surfaces and ultra-short edges are ignored for model simplification. The \( \sum n_{i} \) denotes the total coordination number (CN), which measures the number of the contacted neighboring atoms. The general atomic packing principle in BMGs/liquids is to maximize the fivefold packing,[32] and therefore the indexes with larger n5 values are more prone to stabilize the amorphous structure. For example, the Voronoi index of 〈0, 0, 12, 0〉 corresponds to a polyhedron of ideal icosahedral type. Moreover, the icosahedral clusters also tend to form the 〈0, 2, 8, 1〉 , 〈0, 1, 10, 2〉, 〈0, 2, 8, 2 〉 and 〈0, 3, 6, 3 〉 polyhedrons, which are usually regarded as the icosahedra-like structures. In terms of the ideal fcc/hcp structures, it has an index of 〈0, 12, 0, 0〉 and the 〈0, 6, 0, 8〉 is related to bcc clusters.[33,34] The uniqueness of the Voronoi polyhedron is that it is a three-dimensional (3D) approach and can be utilized to analyze the medium-range ordering based on the connection of coordination polyhedra. Nevertheless, this methodology cannot indicate the saturability of the bonding pairs, and there are some deviations in the description of local structures with high symmetries to some extent.
The diffusion behavior of BMG during liquid cooling is further investigated by exploring the mean squared displacement (MSD) values. Note that the MSD is a measure of the deviation of the position of a particle with respect to a reference position over time:[24]
where \( \overrightarrow {R}_{i} \) stands for the position of atom i, N the number of atoms to be averaged, and ns the number of time origins.
3 Results and discussion
3.1 Pair Correlation Function (PCF) and Understanding Metal-Oxygen Bonding
Figure 3 summarizes the partial PCFs of the O-centered bonds in Zr55Cu55Al9Be9O at different temperatures. The first major peak on the O-related PCF curves depicts the nearest neighbors of O atom. The peak value and the corresponding distance (bond distance) can be used to characterize atomic stacking configuration and reflect chemical affinity between atoms. At 1100 K in Figure 3(a), Zr has the largest probability to coordinate with O, with the nearest bond distance of 2.15 Å and the highest peak value. The static structure of the O-centered cluster is stabilized as temperature further cooling down from 1000 K to 500 K, with no significant change in the PCF distribution. At 1000 K, Be atoms aggregate around O efficiently and form the strong O-Be bonds, as proved by the rapid decrease of the O-Be distance (1.55 Å at the peak PCF position) and the incline of the related peak g(r) value. Meanwhile, the O-Zr bonds are weakened slightly (corresponding to the decreased peak value and enlarged bond distance), since the O-related bonding is partially assigned to Be. This bonding transition can be interpreted from the alternation or adjustment of atoms due to the requirement of atomic dense packing and chemical effect. It is obvious that O-Be forms the strongest bonds in the temperature range from 1000 K to 500 K, because the O-Be curve possesses the maximal intensity of the major peak among the O-related partial PCF curves; see Figures 3(b) through (d). In addition, this strong chemical affinity can also be illustrated by the average bond distance between O-Be at the peak RDF position, valuing 1.60 Å in the range of 1000 K to 500 K, which is almost equal to the sum of covalent radii of O (0.66 Å[35]) and Be (0.96 Å[35]). Besides, it also presents a strong bonding tendency between Zr and O due to the corresponding small bond distance (2.27 Å), though Zr possesses a relatively large covalent radius of 1.75 Å[35] as compared to Be.

Partial PCFs for O-containing bonds in Zr55Cu55Al9Be9O at different temperatures: (a) 1100 K, (b) 1000 K, (c) 900 K, (d) 800 K, (e) 700 K, and (f) 500K
As for O-Cu and O-Al, they both have larger bond distances at the peak PCF positions than the sum of the corresponding covalent radii (Cu: 1.32 Å, Al: 1.21 Å, O: 0.66 Å; see Table I), indicating there is no signal to form O-containing bonds with Cu and Al (see Figure 5 for the reason from force constants). The intensity of the first peak of the O-Al PCF is larger than that of the O-Cu PCF, indicating a stronger chemical affinity for the former case. Based on the above analyses, we conclude that O is surrounded by Be and Zr in an O-centered cluster at 500 K, while keeping Cu and Al from the inner shell.
To further reveal the O-related bonding tendency in the Zr-Cu-Al-Be-O system from the electronic structure, we calculated the projected electronic densities of states using one of the AIMD configurations at 500 K as depicted in Figure 4. It can be seen that both the Zr-total DOS (mainly from d-electrons) and the Be-p DOS have peaks aligned with those of oxygen, indicating their strong hybridization with O atoms.

Electronic DOS for Zr55Cu55Al9Be9O at 500 K (Zr- and Cu-states are mainly from d-electrons)
The existence of Be in Zr-Cu-based metallic glasses significantly reduces the probability of bonding between Zr and O due to the larger chemical affinity of Be with O. As for the Zr-Cu metallic glasses without Be, Wang et al.[13] reported the effects of oxygen impurities on glass-forming ability in the Zr2Cu alloy by ab initio molecular dynamics. They reported that the partial PCF of Zr-O shows the first sharp peak at both 1156 and 300 K in Zr-33Cu-1O (at.pct), indicating the formation of Zr-O bonds. It should be noted that the amplitudes of the Zr-O partial PCFs are around 7.5 and 11 at 1156 and 300 K in their work. In the present Zr-Cu-Al-Be, the amplitudes of the Zr-O partial PCFs are 4.8 and 5.2 at 1100 and 500 K, respectively. Chemical affinities between the oxygen-metal (O-M) atoms can be understood using (i) the global properties of oxides such as bulk modulus (B0), melting points (Tm), and the negative values of enthalpy of formation at 298 K (\( - \Delta H_{298} \)), and (ii) the local O-M bonding strength in terms of the stretching force constants (fSFC) from phonon calculations.[36] It is believed that the larger the values of those properties, the stronger the O-M bonding energy ε will be,[36]
where V0 is the equilibrium volume, R the gas constant, and dOM the bond length of O-M atomic pair. These properties are listed in Table I and the Supplementary Table SI based on the data in the literature and the present first-principles and phonon calculations; see Supplementary Material. It is worth mentioning that the O-M bonding strength decreases rapidly with increasing the dOM value. Figure 5 shows that the fSFC values of the O-M bonds decrease from around 5 to 7 eV/Å2 to zero with increasing the O-M bond length. When the bond length is greater than 2.3 Å at 0 K, there is almost no O-M bonding. Table II summarizes the trends of O-M bonding energy, i.e., the M-O affinity. It shows that the O-Zr bond possesses the largest affinity based on all the criteria in Eq. [3], indicating the O-Zr pairs are dominant around oxygen in Zr55Cu55Al9Be9O than the Cu-O pairs (see Figure 3), albeit the covalent radius of Zr (1.75 Å) is quite larger that of Cu (1.32 Å). For the minor alloying elements Al and Be in Zr55Cu55Al9Be9O, Table II shows different O-Be(Al) bonding trends based on different criteria. However, Be has the smallest covalent radius (0.96 Å), making it easy to diffuse and hence forming the Be-O bonds in Zr55Cu55Al9Be9O (approved by the present AIMD study, see below). According to the results in Figure 5 and Table I together with the simulated dMO (AIMD) values in Figure 3, we conclude that only the atoms Zr and especially Be are attracted by oxygen and forming the O-Be and O-Zr bonds in Zr55Cu55Al9Be9O, while the interactions of Al and Cu with oxygen are weaker and they will not form the O-Al and O-Cu bonds.

Predicted oxygen-metal (O-M) stretching force constants based on the present phonon calculations; see details in Supplementary Material
The snapshots of Zr55Cu55Al9Be9 and Zr55Cu55Al9Be9O at 500 K are illustrated in Figure 6. Both of them are amorphous due to the random distribution of atoms under the cooling rate of 2.5 × 1013 K/s. Figure 6(b) shows that O addition affects the local cluster structure significantly. According to the atomic configuration, Be (yellow) and Zr (green) atoms are gathered around O (red) as the nearest neighbors, while Cu (blue) and Al (purple) atoms are in the outer shell. Local distribution of O-centered cluster is in accordance with the results of O-related partial PCFs, see Figure 3, which are affected by the compositions in BMG, atomic sizes, and the O-M bond strengths. Since Zr is 6 times the concentration of Be in this BMG system, the O atom is most likely to bond with Zr atoms.

Snapshots of AIMD simulations at 500 K for the (a) Zr55Cu55Al9Be9 and (b) Zr55Cu55Al9Be9O alloys
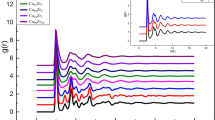
As Zr and Cu are the master components in the present alloys, we investigate the total (g(r)) and partial (gZr-Cu(r), gZr-Zr(r) and gCu-Cu(r)) PCFs in Zr55Cu55Al9Be9 and Zr55Cu55Al9Be9O at different temperatures, as displayed in Figure 7. The PCF curves exhibit the BMG character due to dispersive distributions of atoms.[2,27] In Figure 7(a), the first peak of g(r) becomes narrower and higher with decreasing temperature, which is a signal of local ordering.[2,27] Note that the total PCF curves shift to the right slightly. This originates from the expansion of supercell volume (per atom) induced by O addition (Figure 2). Similar phenomena have also been found in the partial PCFs. Moreover, the second peaks of gZr-Cu(r) (Figure 7(b)), gZr-Zr(r) (Figure 7(c)), and gCu-Cu(r) (Figure 7(d)) show a larger tendency to split in Zr55Cu55Al9Be9 (solid lines) compared to those in Zr55Cu55Al9Be9O (dashed lines), especially for the Cu-Cu interactions.

(a) Total and (b) through (d) partial PCFs in Zr55Cu55Al9Be9 (solid lines) and Zr55Cu55Al9Be9O (dash lines) at different temperatures: (b) Zr-Cu, (c) Zr-Zr, and (d) Cu-Cu for Zr55Cu55Al9Be9 (filled square) and Zr55Cu55Al9Be9O (open circle) at different temperatures
The splitting of the second peak is believed to be inherent and not related to the new local ordering during BMG transformation.[24,34] However, the differences in the second peaks of gZr-Cu(r), gZr-Zr(r), and gCu-Cu(r) are distinct between the alloys with and without O even at low temperatures, implying that there exists a large probability of short-range ordering in Zr55Cu55Al9Be9, which is a signal of BMG formation. In other words, the addition of O deteriorates the GFA to some extent. This deduction will be further analyzed using the BPA and Voronoi polyhedron below.
3.2 Bond Pair Analysis (BPA)
Figure 8 presents the percentages of bond pair types for Zr55Cu55Al9Be9 (filled symbols) and Zr55Cu55Al9Be9O (open symbols) as a function of temperature. The statistic results for BPA originate from the average of the last 100 AIMD steps. For both alloys, the icosahedral (ICO) bonds are the largest portion of total amounts. Moreover, the percentage of icosahedral bonds increases gradually in the cooling process, while that of the random types reversely decrease. In terms of crystal characteristics of bcc, fcc, or hcp, the proportion fluctuates with no obvious trend with changing temperature. Accordingly, the icosahedron-type bonds are predicted to transform from the random bonds with decreasing temperature. Note that the icosahedral ordering exhibits compact atomic ordering compared to the remaining clusters in the liquid, and therefore plays a critical role in the formation of amorphous structure. Hence, the increased fraction of icosahedral bonds implies a transition to BMG structure for both alloys at low temperatures.

Bond Pair Analyses (BPA) at different temperatures in Zr55Cu55Al9Be9O (filled symbols) and Zr55Cu55Al9Be9O (open symbols)
In addition, the percentage of icosahedral bonds is smaller in Zr55Cu55Al9Be9O than those in Zr55Cu55Al9Be9 over the entire temperature range. However, the random-type bonds occupy larger proportions in alloy with O than those without O. With regard to the crystal bcc, fcc, and hcp bonds, the amounts are close between the two alloys, and the variations are not significant during the solidification process. This observation indicates that though amorphous structures still exist when O is introduced, the GFA of Zr55Cu55Al9Be9O declines to some extent. These BPA results verify the above analyses by PCF.
3.3 Coordination Number (CN) and Voronoi Polyhedron
Figure 9(a) and (b) illustrates the distributions of CN for Zr55Cu55Al9Be9 (filled bars) and Zr55Cu55Al9Be9O (open bars) at selected temperatures. The CN proportions are counted based on the average of the last 100 AIMD steps. It shows that CN = 11 and 12 are dominant, followed by CN = 14, 15, and 13. The clusters with CN > 13 are mainly contributed by the Zr-centered polyhedrons, while clusters with CN < 13 mainly originate from the Be-, Cu-, and Al-centered configurations. The different distributions of CN derive from the size differences between the smaller atoms of Be, Cu, Al, and the large one of Zr. Moreover, the broad distribution of the CN values, ranging from 8 to 16, suggests that the local arrangements cannot be modeled by a uniquely prescribed stereochemical structure.

Coordination numbers (CN) at different temperatures in (a) Zr55Cu55Al9Be9 (filled symbols) and (b) Zr55Cu55Al9Be9O (open symbols)
Furthermore, the major Voronoi polyhedrons are examined for Zr55Cu55Al9Be9 (filled bars) and Zr55Cu55Al9Be9O (open bars) at selected temperatures, as shown in Figure 10(a) and b, respectively. In accordance with the CN analysis, the fractions of polyhedrons with CN = 12 and 11 are the largest. Of all polyhedrons, the icosahedra-like CN:11 < 0, 2, 8, 1>, CN:12 <0, 2, 8, 2>, CN:12 <0, 3, 6, 3> as well as the ideal icosahedron CN:12 <0, 0, 12, 0> are the predominant structures in liquid. The icosahedral clusters are still dominant in the cooling process, implying a tendency to stabilize the liquid structure. Besides, one can see the five-edged faces are also overwhelming among Voronoi polyhedrons with CN = 10, 13, 14 and 15, e.g., represented by <0, 1, 10, 2>, <0, 2, 8, 0>, and <0, 1, 10, 3>. The existence of Voronoi icosahedrons with high fractions indicates the formation of BMG for both alloys, which is in consistent with the above PCF and BPA results.

Voronoi polyhedra at different temperatures in (a) Zr55Cu55Al9Be9 (filled bars) and (b) Zr55Cu55Al9Be9O (open bars)
By comparison, the dominant icosahedral clusters, e.g., CN:11 <0, 2, 8, 1>, CN:12 <0, 2, 8, 2>, CN:12 <0, 0, 12, 0> have higher fractions in Zr55Cu55Al9Be9 (filled red column) than those in Zr55Cu55Al9Be9O (open red column) at 1100 K. With decreasing temperature, though the fractions of different Voronoi polyhedrons fluctuate, the alloys without O (filled blue column) exhibit larger probability to form icosahedrons in general, such as <0, 0, 12, 0> and <0, 2, 8, 2> at 500 K. This implies that the addition of 0.78 at. pct O may diminish the GFA in Zr55Cu55Al9Be9, by decreasing the fraction of the icosahedral clusters. Furthermore, the fractions of the FCC type polyhedrons with Voronoi indexes of <0, 3, 6, 4> and <0, 3, 6, 5> increase with the addition of oxygen, which could reduce the GFA by lowering the barrier for crystallization. As such, the studies of atomic configurations provide a basis for the analysis of diffusion kinetics from the liquid state to the glass state.
3.4 Diffusion Kinetics
To explore the diffusion behavior of BMG during cooling, the mean squared displacement (MSD) of Zr, Al, Cu, Be and O in Zr55Cu55Al9Be9 and Zr55Cu55Al9Be9O are studied and summarized as a function of time in Figure 11. The entire cooling processes are considered, where the configurations at different temperatures are marked by various colors. According to the Einstein relation, the self-diffusion coefficient is proportional to the first derivative of the corresponding MSD values relative to time. Some previous researches illustrate that the MSD needs to be over 4 Å2 in order to obtain any statistically significant information on the self-diffusivity coefficient.[37] To eliminate misunderstanding, we use the trends of the MSD values to compare diffusivity rather than self-diffusion coefficient in the present work.

Mean squared displacements (MSD) at different temperatures in Zr55Cu55Al9Be9 and Zr55Cu55Al9Be9O
Noticeably, the MSD values of Zr, Al, Cu, Be and O increase over time in both Zr55Cu55Al9Be9 and Zr55Cu55Al9Be9O due to their diffusions. There exists a transition point around 5 ps when applicable in all cases, before which the MSD values climb rapidly and nearly linearly, while afterward the increasing rates slow down. This phenomenon roughly corresponds to the transformation from liquid to glass phases, as the self-diffusion rate in a liquid is much higher than in BMGs. With regard to Zr55Cu55Al9Be9, the transition temperature of MSD appears at around 1100 K for Zr, Al and Cu, and at about 900 K for Be. In terms of Zr55Cu55Al9Be9O, the turning points shift to 800 K for the Zr- and Cu-related MSD curves, while at around 900 K for Al and Be. Therefore, we conclude that the introduction of 0.78 at. pct O decreases the glass formation temperature, which is not beneficial to freeze the liquid structure. This is because the liquid structure lasts for a longer period of time once doping one O atom, which provides more chances for the cluster ordering and hence increases crystallization potency. In addition, the MSD values of Zr, Al, Cu and Be are larger in Zr55Cu55Al9Be9O than those in Zr55Cu55Al9Be9 in the whole cooling process. It is known that the glass state is suppressed, given that the crystallization process is facilitated in undercooled liquid. It is found that 0.78 at. pct O acts as stimulus to diffusion for all components in Zr55Cu55Al9Be9, which plays a positive role for the nucleation and growth of crystalline phases. Therefore, it is detrimental to the GFA of the Zr-Cu amorphous glasses by the incorporation of trace O.
The diffusion rate in a liquid is sequenced as Be > Cu > Al > Zr for both Zr55Cu55Al9Be9O and Zr55Cu55Al9Be9, which corresponds to the increasing order of B0V0, i.e., O-Be < O-Cu < O-Al < O-Zr; see Tables I and II. Nevertheless, no significant differences in solid diffusion rates are shown between elements, since the rates during solid diffusion are pretty low. The variation of O-related MSD does not have a strong regularity, as there is only one O atom acting as a statistical object. However, it is found the MSD values of O are very low, indicating the inactive diffusion of O. This may be due to the strong bonding of O with its neighboring atoms.
Combined with diffusion kinetics and analyses of atomic configurations, we conclude that the reacts are extremely sensitive with the incorporation of O in the melt. The O-centered cluster tends to be surrounded by Zr and Be in the inner shell, while keeping Al and Cu in the outer shell. The feature of Be around O, on the one hand, relies on the strong Be affinity with O and, on the other hand, is also contributed by the rapid diffusion rate of Be in liquid than those of other metals (Al, Cu, and Zr). Hence, though the Be component has a small percentage, the atomic ordering induced by Be diffusion is very significant, leading to the formation of Be-O bond pairs. Meanwhile, the segregation of Zr around O is mainly due to the strong affinity of Zr with O (see Reference 36 as well as Table I) and also the large proportion of Zr in BMG alloys. Moreover, the diffusion ability of Zr is also enhanced by O addition, as evidenced by the increased MSD in Figure 11. Therefore, the weakened glass formation ability with O contamination is attributed to the local segregation induced by the active diffusion behavior of solutes and the strong affinity of O-solutes, hence resulting in the formation of O-related clusters with enhanced ordering. In the present work, the strong chemical affinity of O with Zr and Be leads to the formation of O-centered coordination polyhedrons. We can reasonably assume that with increasing O content more O-centered coordination polyhedrons will appear in liquid, leading to a decline of the stable ICO polyhedrons and a low GFA due to higher crystallization tendency.
4 Conclusions
The topological and kinetic properties of Zr55Cu55Al9Be9 and Zr55Cu55Al9Be9O during solidification have been systematically investigated by AIMD simulations. The specific aim is to reveal the effects of O on the glass-forming ability (GFA) of the Zr55Cu55Al9Be9 alloy. It is evident that the amorphous structure exists in both Zr55Cu55Al9Be9 and Zr55Cu55Al9Be9O under the present cooling rate of 2.5 × 1013 K/s, implying the formation of BMG in both alloys. The introduction of trace O results in a decrease in the dense icosahedral polyhedrons and an increase in the crystallization potency of the current alloy. This is based on results from the pair correlation function (PCF), bond pair analysis (BPA), and Voronoi polyhedra construction. Considering the evolution of kinetic properties, the trace O addition increases the diffusivities of Be, Zr, Cu, and Al, implying that the O-containing liquid is less likely to be vitrified. The origin of the enhanced ordering in Zr55Cu55Al9Be9O stems from the formation of O-centered clusters with Be and Zr gathering around O as the nearest neighbors due to the strongest O-Zr bonding energy (compared to those of O-Al, O-Be, and O-Cu) and the smallest covalent radius of Be (compared to Al, Cu, and Zr), while Cu and Al locate in the outer shell. The results presented reveal the underlying mechanism of O-induced BMG crystallization, and provide a reference for the design of new BMG alloys with tailored GFA.
References
D. Granata, E. Fischer, and J.F. Lo: Scr. Mater., 2015, vol. 103, pp. 53–56.
X. Hui, H.Z. Fang, G.L. Chen, S.L. Shang, Y. Wang, J.Y. Qin, and Z.K. Liu: Acta Mater.,2009, vol.57, pp. 376–91.
W.H. Wang, C. Dong, and C.H. Shek: Mater. Sci. Eng. R, 2004, vol. 44, pp. 45–89.
L.S. Luo, B.B. Wang, F.Y. Dong, Y.Q. Su, E.Y. Guo, Y.J. Xu, M.Y. Wang, L. Wang,J.X. Yu, R.O. Ritchie, J.J. Guo, and H.Z. Fu: Acta Mater., 2019, vol.171, pp. 216–30.
Q. Cheng, X. Han, I. Kaban, I. Soldatov, W. Hua, Y. Hao, and J. Orava: Scr. Mater., 2020, vol.183, pp. 61–65.
F. Qiu, Y. Liu, R. Guo, Z. Bai, and Q. Jiang: Mater. Sci. Eng. A, 2013, vol.580, pp.13-20.
C.T. Liu, M.F. Chisholm, and M.K. Miller: Intermetallics, 2002, vol.10, pp.1105–12.
H.X. Li, J.E. Gao, Z.B. Jiao, Y. Wu, and Z.P. Lu: Appl. Phys. Lett., 2009, vol.95, pp.16-18.
D.J. Sordelet, X.Y. Yang, E.A. Rozhkova, M.F. Besser, and M.J. Kramer: Appl. Phys. Lett., 2003, vol.83, pp.69-71.
B.S. Murty, D.H. Ping, K. Hono, and A. Inoue: Appl. Phys. Lett., 2000, vol.76, pp.55-57.
B. Cantor, K.B. Kim, and P.J. Warren: Mater. Sci. Forum, 2002, vol.386, pp.27-32.
Y.X. Wang, H. Yang, G. Lim, and Y. Li: Scr. Mater., 2010, vol.62, pp.682-85.
Z. Wang, L. Huang, G.Q. Yue, B. Shen, F. Dong, R.J. Zhang, Y.X. Zheng, S.Y. Wang, C.Z. Wang, M.J. Kramer, K.M. Ho, and L.Y. Chen: J. Phys. Chem. B, 2016, 120, 9223-9229.
X.Y. Yang, Y.Y. Ye, M.J. Kramer, and D.J. Sordelet: J. Alloys Compd., 2009, vol.484, pp. 914–19.
M. Miyake, M. Uno, and S. Yamanaka: J. Nucl. Mater., 1999, vol.270, pp.233–41.
A. Gebert, J. Eckert, and L. Schultz: Acta Mater., 1998, vol.46, pp. 5475–82.
G. Kresse, and J. Furthmuller: Comput. Mater. Sci., 1996, vol.6, pp.15–50.
G. Kresse, and J. Furthmuller: Phys. Rev. B., 1996, vol. 54, pp. 11169–86.
M. Ernzerhof, and G.E. Scuseria: J. Chem. Phys., 1999, vol.110, pp.5029-36.
S. Nosé: J. Chem. Phys., 1984, vol.81, pp. 511-19.
W.G. Hoover: Phys. Rev. A., 1985, vol.31, pp.1695-97.
O. Haruyama, Y. Nakayama, R. Wada, H. Tokunaga, J. Okada, and T. Ishikawa: Acta Mater., 2010, vol.58, pp.1829–36.
X. Hui, R. Gao, G.L. Chen, S.L. Shang, Y. Wang, and Z.K. Liu: Phys. Lett. A, 2008, vol.372, pp.3078-84.
Y.Q. Cheng, and E. Ma: Prog. Mater. Sci., 2011, vol.56, pp.379–473.
F.A. Celik: Phys. Lett. A, 2014, vol.378, pp.2151-56.
W.Y. Wang, J.J. Han, H.Z. Fang, J. Wang, Y.F. Liang, S.L. Shang, Y. Wang, X.J. Liu, L.J. Kecskes, S.N. Mathaudhu, X. Hui, and Z.K. Liu: Acta Mater., 2015, vol.97, pp.75-85.
J. Ding, and E. Ma: NPJ Comput. Mater., 2017, 3, 91-912.
J.D. Honeycutt, and H.C. Andemen: J. Phys. Chem., 1987, vol.91, pp.4950–63.
W. Brostow, J.P. Dussault, and B.L. Fox: J. Comput. Phys., 1978, vol.29, pp.81–92.
P. Taylor, Local atomic arrangements in polytetrahedral materials II . Coordination polyhedra with 14 and 15 atoms, 2009, pp.37–41.
Y.D. Wei, P. Peng, Z.Z. Yan, L.T. Kong, Z.A. Tian, K.J. Dong, and R.S. Liu: Comput. Mater. Sci., 2016, vol.123, pp.214-23.
Y.C. Hu, F.X. Li, M.Z. Li, H.Y. Bai, and W.H. Wang (2015) Nat. Commun., 6, 83101-83108.
W.Y. Wang, S.L. Shang, H.Z. Fang, H. Zhang, Y. Wang, S.N. Mathaudhu, X. Hui, and Z.K. Liu (2012) Metall. Trans. A, 43A, 3471-80.
X.J. Liu, S.D. Wang, H.Y. Fan, Y.F. Ye, H. Wang, Y. Wu, and Z.P. Lu: Intermetallics, 2018, vol.101, pp.133-43.
Wolfram Mathematica: Modern Technical Computing, (n.d.). https://www.wolfram.com/mathematica. Accessed March 16, 2020).
S.L. Shang, Y. Wang, B. Gleeson, and Z.K. Liu: Scr. Mater., 2018, vol.150, pp.139–42.
J. Han, W.Y. Wang, C. Wang, X. Hui, X. Liu, and Z.-K. Liu: Intermetallics, 2014, vol.46, pp.29-39.
Verlag Berlin Heidelberg: Thermodynamic Properties of Inorganic Materials, Landolt-Boernstein New Ser. Gr. IV, Springer, Scientific Group Thermodata Europe (SGTE), 1999.
Acknowledgments
This work was funded by the U.S. Department of Energy with award number of DE-NE0008757. BB and ZKL were supposed by a NASA Space Technology Research Fellowship (grant 80NSSC18K1168). First-principles calculations were performed partially on the Roar supercomputer at the Pennsylvania State University’s Institute for Computational and Data Sciences (ICDS), partially on the resources of the National Energy Research Scientific Computing Center (NERSC) supported by the U.S. Department of Energy Office of Science User Facility operated under Contract No. DE-AC02-05CH11231, and partially on the resources of the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the National Science Foundation with Grant No. ACI-1548562.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted October 14, 2020; accepted March 10, 2021.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, C., Shang, SL., You, J. et al. Understanding the Effect of Oxygen on the Glass-Forming Ability of Zr55Cu55Al9Be9 Bulk Metallic Glass by ab initio Molecular Dynamics Simulations. Metall Mater Trans A 52, 2501–2511 (2021). https://doi.org/10.1007/s11661-021-06242-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-021-06242-4