
Abstract
U-Pu-Zr alloys are considered ideal metallic fuels for experimental breeder reactors because of their superior material properties and potential for increased burnup performance. However, significant constituent redistribution has been observed in these alloys when irradiated, or subject to a thermal gradient, resulting in inhomogeneity of both composition and phase, which, in turn, alters the fuel performance. The hybrid Potts-phase field method is reformulated for ternary alloys in a thermal gradient and utilized to simulate and predict constituent redistribution and phase transformations in the U-Pu-Zr nuclear fuel system. Simulated evolution profiles for the U-16Pu-23Zr (at. pct) alloy show concentric zones that are compared with published experimental results; discrepancies in zone size are attributed to thermal profile differences and assumptions related to the diffusivity values used. Twenty-one alloys, over the entire ternary compositional spectrum, are also simulated to investigate the effects of alloy composition on constituent redistribution and phase transformations. The U-40Pu-20Zr (at. pct) alloy shows the most potential for compositional uniformity and phase homogeneity, throughout a thermal gradient, while remaining in the compositional range of feasible alloys.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Metal fuels in nuclear reactors have many desirable properties: high thermal conductivity, high fissile and fertile atom density capability, and ease of fabrication.[1,2] However, metal fuels cannot survive the high temperatures that oxide fuels do and therefore must operate at lower temperatures. A decreased operating temperature lowers heat generation efficiency but increases the fuel safety because it limits the diffusion of fission gas bubbles, containing them within the fuel grains, and allows for a larger margin from the melting temperature.[3,4] The lower operating temperature also increases reactor safety as all structural and functional elements operate at a concomitantly lower temperature. In addition, metal fuels like U-Pu-Zr aid in the long-term management and disposition of plutonium[1,5] and other minor actinides,[6,7] thereby minimizing the amount of nuclear waste and access to weapons-usable material.
The U-Pu-Zr alloy has been the center of considerable study,[2] with renewed interest in recent years.[8,9] This alloy exhibits superior burnup performance in fast reactors and breeder reactors.[5,10,11,12] Plutonium concentration is determined from both the reactor design, such as breeding ratio, core size, and so on, and the fuel characteristics. The addition of plutonium into the fuel decreases the melting temperature of the U-Pu-Zr alloy and the eutectic temperature between the fuel and stainless steel cladding. As a result, plutonium is typically limited in concentration to 20 wt pct for the fast reactor design. Zirconium and other elements were initially tested in this alloy as a means to offset the low melting temperature of plutonium. However, zirconium was ultimately chosen because of its unique ability to (i) suppress the interdiffusion of components between the fuel and the stainless steel cladding, (ii) increase the melting temperature of the U-Pu-Zr alloy, (iii) remain essentially transparent to neutron transmission, and (iv) decrease swelling, which increases the safety margin of the fuel.[13] While zirconium successfully increases the liquidus of the alloy, it also increases the solidus. This higher solidus is problematic because of temperature constraints regarding the softening point of the injection casting molds of the fuel rods (usually SiO2-base mold is chosen). Consequently, it was determined that zirconium should be limited to about 10 wt pct for plutonium concentrations up to 20 wt pct.
In historical testing of the U-Pu-Zr alloy, three compositions were mainly investigated: U-10Zr, U-8Pu-10Zr, and U-19Pu-10Zr (wt pct).[13,14] Analysis of these alloys showed consistency in the quantity of fission gas released, burnup at which pores became interconnected, and anisotropic fuel swelling. Radial redistribution of constituents was expected and observed, with a seemingly related radial distribution of porosity as well. While limited characterization and performance data are available for the U-Pu-Zr system, it is clear that when commonly investigated compositions are irradiated, or subject to a thermal gradient, two or three concentric zones are formed, depending on the fuel center temperature.[13,14,15] Consistent with previous observations,[15] these radially concentric zones show a redistribution of the initially uniform alloy constituents, as well as the resulting inhomogeneity of phases and pore generation. Although metallic fuels have the potential for the highest fissile atom density, the resulting inhomogeneity in U-Pu-Zr alters the achieved fissile atom density and the thermal conductivity, and thus the fuel behavior and performance.[16]
Securing a better understanding of what drives the constituent redistribution in the U-Pu-Zr alloy is essential to analyzing and predicting its behavior as a nuclear fuel. Considerable efforts have been made to determine the necessary thermophysical properties for accurate modeling.[17,18,19] Further research has attempted to determine the kinetic values of the constituent diffusional coefficients and heat of transports via analytical models. Kim et al. investigated the kinetic and thermo-kinetic properties for irradiated U-Pu-Zr; they calculated the interdiffusion fluxes from experimental test results and used the fluxes to obtain the diffusion coefficients and heats of transport.[16]
Much of the modeling of the U-Pu-Zr system has focused on understanding experimental results by determining how material properties affect the distribution profiles via numerical methods. Ogawa et al. numerically solved a one-dimensional Fick’s law and hypothesized the effect of including Pu in a U-Zr alloy.[20,21] Ishida et al. extended the Marino model to the U-Pu-Zr system and, by assuming Pu was equally partitioned in U and Zr, defined the system as a quasi-binary system. The resulting model, however, predicted profiles that differed from experimental results, and the errors were attributed to temperature predictions that were too high.[21,22] Later work by Kim et al. used a pseudo-binary phase diagram, treating Pu as immobile, to calculate the redistribution of Zr.[23]
Using some of the next-generation modeling tools,[24,25] Galloway et al. modeled the 3D behavior of three U-Pu-Zr fuel rods subjected to different conditions with a single model.[26] As with the work of Kim et al.,[23] Galloway treated Pu as immobile, but they also included thermal diffusion (the Soret effect) and adapted their diffusion equations to ensure that compositions retained physical values (between 0 and 1) and that the compositions obeyed solubility limits of the appropriate phases. This formulation does lead to discontinuities in the composition and other variables that have to be corrected. However, after examining different scenarios and setting different variables as adjustable, they determined a single set of parameters that accurately predicts the constituent redistribution for a single fuel rod composition subjected to three different conditions. Their approach, focused so critically on validation with experimental data, demonstrates the necessity to obtain precise measurements of material properties in order to accurately simulate material behavior.
It is noted that while the use of binary or pseudo-binary systems yields significant insight, a true ternary model of the system would allow a computational investigation of constituent redistribution over all possible alloy compositions. This would allow alloy designers to determine whether constituent redistribution can be minimized for different alloy compositions that might therefore exhibit superior fuel performance.
This research investigates constituent redistribution in U-Pu-Zr alloys, which is driven by thermal gradients in the nuclear fuel. The work details the extension of the hybrid Potts-phase field method[27] to ternary alloy systems. The Potts-phase field method, which is capable of simultaneously evolving both the microstructure and composition, utilizes a thermodynamic database of the U-Pu-Zr system[28] to drive the system evolution. The model is first applied to an alloy composition of U-16Pu-23Zr (at. pct) in order to compare the method results to previous work. The model is then used to investigate the composition and phase evolution of the U-Pu-Zr fuel over the entire compositional spectrum. It is noted that this work represents an important step forward through full ternary modeling of the constituent redistribution in the U-Pu-Zr fuel. The work is intended to provide general trends in the constituent redistribution rather than detailed analysis of specific alloy compositions.
2 Methods
2.1 Potts-Phase Field Method
Simulating constituent redistribution and phase transformations in multiphase materials like the U-Pu-Zr fuel present a particular challenge. While microstructure and composition are, in reality, interconnected and dependent upon each other, models used to predict microstructural and compositional evolution are often performed separately. Thus, the goal to simulate the U-Pu-Zr system requires the selection of an appropriate materials model.
The hybrid Potts-phase field model is a modeling technique that is capable of simultaneous microstructural–compositional evolution.[27] The modeling technique joins the Monte Carlo Potts model, which simulates the microstructure evolution, with the phase field method, which is used to simulate the compositional evolution. While both of these methods have proven particularly useful in modeling various microstructural phenomena, the combination provides a nice balance between resolution and efficiency. In short, the Monte Carlo Potts model uses discrete integer values to represent microstructural characteristics, such as grain orientation and phase, while the phase field method uses a continuum variable to simulate the composition.
The coupling between the two methods occurs through a free energy functional, which sums the volumetric free energy at a given location with the interfacial free energy terms from the two different methods:
In Eq. [1], Ev is the volumetric energy term as a function of both particle state, qi, and composition, Ci. The interfacial energy terms \(J\left( {q_{i} ,q_{j} } \right)\) and \(\kappa_{C} \left( {\nabla C_{i} } \right)^{2}\) are the traditional interfacial energy terms from Monte Carlo Potts and phase field, respectively. The Monte Carlo Potts interface energy defines grain boundary or phase boundary energy due to differing particle state values (i.e., when neighboring particle states are identical, they have no contribution, and when they are different, the interfacial energy is defined as equal to the grain or phase boundary energy). The phase field term originates from the Cahn–Hilliard compositional gradient energy.[29]
Recently, the Potts-phase field method was extended to account for thermal diffusion (the Soret Effect) and to incorporate volumetric energy defined by a thermodynamic database.[30] The latter has the particular advantage of simplifying simulation for a large range of alloys.
The present work extends the Potts-phase field framework to ternary alloy systems to model the U-Pu-Zr nuclear fuel. The generalization of the framework to ternary alloys has revealed that extending the modeling framework to higher-order alloy systems should be relatively straight forward.
2.2 Microstructure and Phase Evolution
The statistical mechanical Monte Carlo Potts model evolves a discrete set of particles on a lattice.[31] This set of particles represents the microstructure for each site with an integer spin number. This spin number can represent any given microstructural feature, such as grain orientation, phase, dislocation density, or any other feature that is critical for the model of interest. Boltzmann statistics are used to systematically attempt spin changes for each site to one of its neighboring sites. The probability, P, of a site changing its spin is given by
where ∆E is the change in energy associated with a given spin change, kB is Boltzmann’s constant, and T is temperature in Kelvin. In this way, the sites evolve to a lower overall energy by transforming the microstructure and grain boundary network.
2.3 Composition Evolution
The composition of the system is evolved using Fick’s 2nd law of diffusion,
where J, the diffusional flux, is defined as,
Here, M and µ are the mobility and the chemical potential of the constituent, respectively. The chemical potential is defined according to the Cahn–Hilliard approach[29] as
where µ* is the partial Gibbs energy,[32] also referred to as the homogenous free energy,[33] and the second term \(\varepsilon \nabla^{2}C\) characterizes the concentration gradient contribution to the interfacial free energy as defined by Cahn and Hilliard,[29,33] where ε is the gradient energy coefficient that controls the interface width. Assuming M is constant, the time rate change of composition (Eq. [2]) becomes,
which is the generic form of the Cahn–Hilliard equation commonly employed in phase field models.[34]
2.4 Ternary System
In extending the Potts-phase field method to simulate mass and heat transport in a ternary alloy, special attention was paid to standard methods.[35] Accordingly, the flux equations for mass and heat transport in a three-component, closed, network-constrained system are given as
where J1, J2, J3, and JQ are the fluxes of the three constituents and the heat flux, respectively. The direct and coupling coefficients between the various driving forces and fluxes are given by the different Mij (e.g., M11, MQ1, MQQ) coefficients. As noted above, μi is the chemical potential of the ith constituent, and T is the temperature. For a closed, network-constrained system, one can utilize the following relations,
as well as the Onsager Symmetry Principle[35] to reduce the number of mobility coefficients to four direct mobility coefficients (M11, M22, M33, and MQQ) and two coupling coefficients (M1Q and M2Q). The direct coefficient, MQQ, can be replaced by the thermal conductivity according to \( K = M_{\text{QQ}} /T ,\)[35] and the coupling coefficients M1Q and M2Q are frequently related to the heat of transport.[36]
In order to evolve the temperature in the system, we utilize the relationship between the heat flux and the definition of enthalpy, which gives
where h is the enthalpy, t is time, and cp is the specific heat.[35]
The final partial differential equations controlling constituent and temperature evolution in the system are given by
where µ is defined according to the Cahn–Hilliard approach in Eq. [5].
2.5 Incorporating a Thermodynamic Database
Simulation of composition evolution by the phase field method requires the definition of free energy as a function of composition for each phase of interest. This free energy is frequently defined by analytic functionals, but here we elect to utilize a thermodynamic database to achieve a more accurate response for any given alloy, an approach introduced by the authors in Reference 30. In this manner, the framework developed in this work can be applied to any number of alloys that have the necessary thermodynamic data, such as can be obtained from the CALculation of PHase Diagrams (CALPHAD) method.[37]
In the present work, the free energy at any location is given by the rule-of-mixtures, defined as
where qp is the phase fraction.
The partial Gibbs energy \( \mu^{*}\) for each phase is given (visually) as the value at which a tangent line (or plane or hyper-plane, depending upon the order of the alloy) of the free energy intersects the component axes. For a ternary system, this is defined mathematically as
where the subscripts refer to the three constituents.
Due to the additive nature of Eq. [11], the partial Gibbs energy for each component in a phase becomes a sum over the appropriate G and \( \partial G/\partial C\) terms in Eq. [12]. However, since both the partial Gibbs energy and the phase fraction can vary in space, the Laplacian of the chemical potential defined in Eq. [5] becomes
2.6 Implementation
In the present work, the simulation of constituent redistribution in U-Pu-Zr alloys examines only the microstructural and compositional aspects. There is no consideration of additional aspects known to affect fuel performance, such as porosity and swelling;[38,39,40] degradation or alteration of the material properties during simulation;[23,25] chemical interactions with the cladding;[41,42] power generation;[26] or a handful of other factors that can play a role in fuel performance.[23,25] While all of these factors are important, the present work is concentrated on evaluating constituent redistribution over a range of compositions, examining only composition and microstructure.
Implementing the Potts-phase field model to simulate constituent redistribution in U-Pu-Zr alloys requires numerous material properties and kinetic constants, whose values and origins are discussed below. In addition, a handful of implementation decisions had to be made to optimize the simulation of the alloy performance and are also described below.
The fuel rod performance is simulated in a 2D rectangular cross-section of a fuel rod, with a width of 4.32 mm, selected to match the diameter of the rods in References 15,16. The height of the simulation is 2.16 mm to give a 2:1 width:height ratio, though this ratio is selected for convenience only. This region is mapped onto a discretized system of 100 by 50 sites. The simulations are fully periodic. Each site has an area of a2, where a is the simulation diameter divided by the number of sites, 4.32 mm/100 sites or 0.0432 mm. For volumetric energy calculations, the simulation is quasi-3D and each site is given a depth of a, resulting in a volume of a3.
The Gibbs energy for this work utilizes the thermodynamic database developed by Kurata for the U-Pu-Zr system.[28] The database follows the standard CALPHAD approach for the 14 phases of this alloy found in the database. The database may not account for all recent advances in the understanding of phase behaviors in U-Pu-Zr,[2,17,43,44,45,46] but the database captures the general behaviors well,[28] and any advances to the database can easily be incorporated into the modeling framework.
While it is possible to directly connect the software to Thermo-Calc or use the analytic functions to calculate the energies during the course of the simulation, a tabulated form is the most efficient and provides the most flexibility for the future implementation of other alloy systems. For this work, a table of Gibbs free energy values is loaded as a function of composition in 0.01 at. pct steps and temperatures ranging from 768 K to 998 K (495 °C to 725 °C) in 5 K steps. The partial Gibbs energy, \( \mu^{*}\), for each constituent is also tabulated. During the simulation, values of G and \( \mu^{*}\) are interpolated from the tables using three-dimensional linear interpolation.
Occasionally, terminal phases do not exhibit a minimum in the free energy surface near the terminal composition (i.e., \( \partial^{2} G/\partial C^{2} \ne 0 \)), rather the energy has a minimum value at the terminal composition but still has a non-zero slope at this point (\( \partial G/\partial C \ne 0 \)). This is problematic for numerical simulations because the non-zero slope can drive the composition to values that are unphysical (e.g., negative or greater than unity). To correct this, a quadratic energy penalty is simply applied to all composition values that are unphysical, thereby driving the system back to realistic compositions for the terminal phase. For a few terminal compositions, the combination of the non-zero slope (\( \partial G/\partial C \ne 0 \)) and the quadratic correction results in a “miscibility gap” of sorts, forming a single-phase region with two compositions. Fortunately, these terminal systems are not of particular interest in the alloy development of the U-Pu-Zr system discussed here. Furthermore, future work will eliminate these artifacts by requiring continuity of the correction term with the thermodynamic data.
The phase fraction for all sites, qi, is defined using only zeros and ones. In other words, each site in the simulation has only one phase present at any given time.
Material properties for the U-Pu-Zr system are determined as follows. The thermal conductivity and specific heat, given for each constituent in Table I, are averaged during evolution calculations by taking a compositionally weighted average. The Potts interface (grain boundary) energy is set to 0.2 J/m2,[47] and the Cahn–Hilliard energy term, ϵ, is set to unity, similar to other works.[33] The higher-order Cahn–Hilliard energy terms are calculated according to methods described in Reference 48. It is noted that, in this work, the Cahn–Hilliard contribution to the evolution of the phases is typically small, though it plays an important role in the evolution of the composition through diffusion. The Gibbs free energy, Potts interface energy, and Cahn–Hilliard energy values are calculated as extensive quantities by multiplying the appropriate volumetric, molar, or specific values by the appropriate volume/area/mass associated with each site. It is noted that the inclusion of these extensive quantities requires an additional multiplication factor for the Potts interface energy due to numerical precision issues. The Potts interface energy is scaled by a factor of 104 to ensure a meaningful contribution in the energy calculations for the volumes simulated in this work. To ensure that this scaling factor does not overcompensate, a smaller discretization scheme not requiring the scaling factor was utilized to confirm similar behaviors. Due to computational expense, this smaller discretization scheme is not used in the majority of simulations in this work.
Kinetic quantities required to simulate the U-Pu-Zr system are defined as follows. Kim et al. derived diffusivities for each constituent in each of the three concentric zones, though not for individual phases.[16, 23] However, since this model framework assumes constant values of M across the simulation cell to conserve mass, all phases must use the same diffusivity values. Thus, this work uses an average value of the diffusivities of each constituent from Reference 16 regardless of the phase in which diffusion is calculated. These values are given in Table I. The direct mobility terms are calculated from the averaged diffusivities according to
where T, in this case, is the average temperature across the simulation (again required because M must be constant for each step).
As a means to simulate the correct grain growth rate, a grain boundary mobility term is used to convert the probability of grain boundary (GB) motion by the Monte Carlo Potts model (Eq. [2]) to a rate. The GB mobility for this work is set to 0.001, which results in GB velocities of approximately 10−6 m/s, which is in the range of experimental results.[49] As seen in previous work, the kBT value in the Potts model (Eq. [2]) is critical to preventing grain growth stagnation[27] and is set accordingly for this work.
To simulate the thermal gradients that drive so much of the phase and constituent evolution, a temperature profile similar, but not identical, to that described in Reference 16 is utilized. This profile is obtained and maintained at steady state by constantly adding heat to all the sites in the model. This addition of heat, which simulates the heat generated by fission events, in combination with the boundary condition that the outermost sites are maintained at 823 K (550 °C), results in a quadratically shaped (negative curvature) temperature profile with a peak temperature of 943 K (670 °C). Maintaining the outermost sites at 823 K (550 °C) effectively acts as the sink for the heat transferred out of the fuel. This matches certain temperatures of an actual fuel rod, though the temperature profile is not an exact match.
Each simulation is tracked over 24,000 Monte Carlo steps (MCS), with 100 sweeps of the concentration field for each MCS. All hybrid Potts-phase field model simulations are performed using the Stochastic Parallel PARticle Kinetic Simulator (SPPARKS). SPPARKS is an open-source, parallel Monte Carlo code for on/off lattice models maintained by Sandia National Laboratories.[50] A hybrid Potts-phase field application style was created in SPPARKS.[27]
3 Simulation Results and Discussion
3.1 Simulation of U-16Pu-23Zr (At. Pct)
As a means to benchmark the ternary phase and composition modeling of U-Pu-Zr alloys, a system composition of U-16Pu-23Zr (at. pct), or U-19Pu-10Zr (wt pct), is simulated. This initial composition is chosen in order to compare to a detailed analysis of the U-19Pu-10Zr (wt pct) alloy published by Kim et al.[16] of experimental results.[51,52] The initial conditions for phase fraction and phase composition are determined from the isothermal phase diagram at 823 K (550 °C), the low temperature boundary condition for the simulations. At this temperature, the equilibrium phases are δ and ζ, which have phase fractions of 27 pct and 73 pct, respectively, and compositions of U-10Pu-70Zr and U-18Pu-6Zr (at. pct), respectively. The simulation cell is randomly and uniformly seeded using these phase fractions and compositions.
In the presence of a thermal gradient, some regions of the material will want to change phase. To simulate phase transformations, nucleation of all possible phases is attempted with a frequency of 0.00001 for every attempted Potts spin change. In other words, for every 100,000 attempts to change a spin using the Potts model, one of the sites will be randomly assigned to any of the possible 14 phases. This leads to the attempted nucleation of many phases, but the energetic cost of nucleating non-equilibrium phases causes the majority of these nucleation events to disappear in a subsequent spin change of that site. Only the energetically favorable phases persist following nucleation.
Figure 1 shows the evolution of grains, phases, average phase fractions, composition, average constituent composition, and temperature profile for this system, with the left and right edges representing the fuel rod center and fuel rod surface, respectively. The entire diameter of the fuel rod was simulated (r/R from − 1 to 1), but the images are cut in half, and only half of the simulation result is presented (r/R from 0 to 1) due to space limitations in the figure. The maps are designed to mimic the radial cross-section of a U-Pu-Zr fuel rod.

Evolution of U-16Pu-23Zr alloy at various steps throughout the simulation. The columns from left to right identify the microstructure map (with individual grain coloring), the phase map (colored according to the key below), the Y-averaged phase fraction, the composition map (colored according to the key below), the Y-averaged composition, and the temperature profile (in Kelvin) enforced as a boundary condition. The maps are designed to mimic the radial cross-section of a U-Pu-Zr fuel rod, with the left edge corresponding to the centerline of the fuel and the right edge corresponding to the free surface (Color figure online)
In the last step of the simulation, Figure 1 shows a strong dependence of both phase and composition on the radial distance from the fuel rod center, as is expected for the U-Pu-Zr fuel. The BCC-γ phase nucleates and becomes the dominant phase in the high-temperature center zone. The δ and ζ phases are present at the lower-temperature outer zones. The composition profiles show a slightly Zr-enriched and slightly U-depleted center zone. At the lower-temperature outer zones, there is significant U enrichment and Zr depletion. Minimal plutonium redistribution is also seen, with a slight decrease in composition from fuel center to surface, as observed in experiments.[53] Interestingly, early in the simulation at 3200 MCS, prior to the formation of large single-phase regions, very different concentration profile with significant U enrichment and Zr depletion near the fuel surface are seen. This transient is accompanied by phase transformations, but this early observation may be attributed to initial conditions and boundary conditions more than being an indication of what happens in real fuels at this point.
It is noted, however, that the radially dependent average composition and phase fractions do not replicate those in experimental work[14] and subsequent analysis.[16,52] The previously published works suggest that there should be three well-defined zones: an inner (center) zone composed of the BCC-γ phase, an intermediate zone composed of the ζ and γ phases, and an outer zone composed of the δ and ζ phases.
While all phases are present in the simulation at appropriate locations, it is not clear whether there are two or three zones. Furthermore, different zone boundary locations could be determined depending on whether one examines the averaged phase fraction or the averaged composition. If one examines the averaged phase fraction, one might suggest that a center zone with the γ phase exists from r/R =0.0 to 0.7; that an intermediate zone exists from r/R = 0.7 to 0.85, where both the ζ and γ phases are present; and that an outer zone exists from r/R = 0.85 to 1.0, where both the δ and ζ phases are present. However, if one examines the averaged composition, one might suggest that the center zone exists from r/R = 0.0 to 0.7, where the U and Zr are relatively constant; and that an intermediate zone starts at r/R = 0.7 but may extend to either 0.95 or all the way to the outside (r/R = 1.0). The extent of the intermediate zone, and the possible existence of an outer zone, is dependent on whether one believes that there is a slight leveling of the U and Zr concentrations just prior to the outermost surface. It is not clear how to appropriately weigh the evidence of zones based on the phase fractions and compositions since they are not entirely consistent with the experiments.
There are several possible sources for the discrepancy between the present simulations and the experimental results. These include the temperature profile used in the simulations, the diffusivity values and approach used, the fact that the model does not consider some aspects of the experiments, and the thermodynamic database used.
The temperature profile used in this work is only an approximation of, and is not identical to, the profile used by Kim et al.[23] or Galloway et al.[26] More importantly, the profile is not a duplicate of that actually experienced during irradiation, where pore formation and the resulting inhomogeneity of thermal conductivity can have a significant effect on the temperature profile and corresponding phases that emerge. The shift of the uranium-rich region toward the fuel surface is similar to results presented by Kim et al.[23] when different temperature profiles are used. Galloway et al. also demonstrated that their model is extremely sensitive to the temperature profile used.[26] Some correction may be obtained by simply altering the temperature profile used, which may be one of the most significant factors causing the discrepancy.
Another factor causing the discrepancy may be the definitions used for constituent mobilities. As described in Section II–F, the mobility terms used in this model are based on diffusivities derived by Kim et al.,[23] who calculated constituent diffusivities uniquely for each experimentally measured concentric zone, but not for each phase. Unfortunately, constituent diffusivity values are not known for the majority of phases in the U-Pu-Zr system. We recognize that accurate simulation results require mobility terms for each constituent in each phase, but because of both the way conservation of mass was implemented in the model and the lack of diffusivity data for all phases, this work assumes the diffusivities to be the same in all phases. This assumption may give higher or lower diffusivities; thus changing the rate of constituent redistribution and accelerating or decelerating evolution. In fact, if the diffusivities assumed are far from the actual values, the direction of the component flux could be reversed and alter the location of the zone boundaries in the simulations. Accurate prediction of constituent redistribution will require diffusivities of each component in every phase.
As noted in the discussion of the temperature profile, the model does not consider pore formation or inhomogeneous thermal conductivity, nor does it consider the mechanical influence of these factors. In addition, the evolution varies significantly depending on the burnup, duration of irradiation, and position along the length of the fuel pin. Galloway et al. demonstrated just how much some of these factors influence a simulation,[26] and these factors are not considered in this work.
The thermodynamic database used for this work is impressive in its completeness,[28] but as with most calculated phase diagrams, there will be disagreement. Even Kim et al. had to adjust their pseudo-binary phase diagram to more accurately match the experimentally observed zone boundaries.[23] Upon detailed examination of Figure 1, it could be suggested that the intermediate zone is transforming into a single phase ζ zone, rather than a ζ–δ or ζ–γ two-phase zone.[16] Modeled in this way, the simulations results, while not directly consistent with experimental results, are consistent with predictions from the database used. Thus, the simulation technique reliably returns results consistent with the thermodynamic input.
In comparison to the models by Kim et al.[23] and Galloway et al.,[26] a full ternary system is modeled using a thermodynamic database. As a result, no assumptions about Pu diffusion, or the lack thereof, have to be made. Furthermore, the database overcomes problems encountered in the work of Galloway et al. related to solubility limits and the corresponding numerical instabilities. In contrast, both Kim et al. and Galloway et al. used more accurate diffusivity values that are not the same for all phases (zones) in the model. Furthermore, both Kim et al. and Galloway et al. went to greater lengths adjusting model parameters to validate their modeling techniques. Each of the different approaches has unique strengths and weaknesses that provide different insight into the U-Pu-Zr system.
In summary, the present modeling technique shows the same general trends in constituent redistribution and phase transformations as other published works, and even though it is not exact, the simulation results match the database inputs. In other words, this technique has great potential as a predictive tool. The limitations noted above can be corrected in future iterations, and as better material property data become available, its prediction ability will improve.
3.2 Survey of Composition Spectrum
Having established the ability of the model to capture the critical trends of constituent redistribution and phase transformation, the entire compositional spectrum of U-Pu-Zr is investigated. This is done by varying the composition of U, Pu, and Zr in 20 at. pct steps, according to the compositions of the 21 alloys detailed in Table II. These 21 compositions are also plotted on top of a ternary phase diagram in Figure 2. The underlying phase diagram shows the equilibrium phases at 823 K (550 °C), though only the single-phase regions are labeled to simplify the diagram. In addition to the 21 alloy compositions considered here, the compositions investigated by Pahl et al.[14] and Burkes et al.[54] are also plotted. The U-16Pu-23Zr (at. pct) composition detailed in the previous section is also indicated. This figure demonstrates a large range of compositions that have not been considered. The gray shading indicates regions that are less desirable due to the high concentrations of zirconium or plutonium, where the melting temperatures will be too high or low, respectively. Thus, the unshaded region is preferred for melting temperature reasons.

Phase diagram of U-Pu-Zr at 823 K (550 °C) overlaid with compositions investigated by previous and the present work. Only the single-phase regions are labeled to simplify the complex diagram. Gray shading shows low and high melting point compositions, while the white region shows melting points in a preferred range
The initial conditions for the 21 simulations covering the entire composition spectrum differ from those in the U-16Pu-23Zr (at. pct) benchmark, in that each site is assigned a randomly selected phase and given a composition equal to the overall composition (i.e., the initial composition is uniform). This ensures that all phases are initially present and that results are not dependent on phase nucleation, offering equal opportunity for each phase to exist.
Figure 3 shows the evolved phase maps, after 24,000 MCS, overlaid with the average constituent composition for each of the 21 simulations. The sequential evolution images for each of the simulations, similar to Figure 1, can be found in the supplemental materials (Supplemental Figures S1–S22). Figure 3 also repeats the isothermal phase diagram from Figure 2 beneath the microstructure and composition maps. This gives an indication of the possible phases and overall compositions that will be expected.

Twenty-one alloy simulations of varying initial compositions arranged according to their composition on a ternary phase diagram. The phase maps show the final configuration after 24,000 MCS, which started from a randomly seeded simulation and were subject to the same temperature profile as in Fig. 1. The simulations are designed to mimic the radial cross-section of a U-Pu-Zr fuel rod, with the left edge corresponding to the centerline of the fuel and the right edge corresponding to the free surface. Overlaid on each phase map is the Y-averaged composition revealing the constituent redistribution and phase regions formed. The coloring of the phases corresponds to the key in the upper-right corner. Images showing the evolution of each of these simulations are available in the supplemental material (Color figure online)
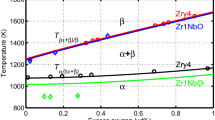
In an effort to validate the results of this survey, we compare our results to a recent study by Burkes et al.[54] In this work, Burkes et al. determined the phase transition temperatures of several U-Pu-Zr alloys, namely, U-24Pu-15Zr, U-36Pu-20Zr, U-39Pu-20Zr, U-34Pu-30Zr, U-35Pu-30Zr, and U-29Pu-40Zr, all in wt pct.Footnote 1 These alloys can be compared, most closely, to simulation 13 (U-20Pu-40Zr at. pct) or simulation 17 (U-20Pu-20Zr at. pct) for the first alloy, simulation 13 for the second and third alloys, and simulation 8 (U-20Pu-60Zr at. pct) for the last three alloys. By comparison, the results are consistent with Burke’s findings for two of the three transition temperatures. The ζ + γ → δ + ζ + γ and δ + ζ + γ → δ + ζ transitions, which occur in the first three alloys, are within the temperature ranges given by Burkes. However, the γ → ζ + γ transition in the simulation occurred at a noticeably lower temperature, 40 °C to 50 °C lower than that found by Burkes et al.[54] This lower simulated γ → ζ + γ transition temperature is attributed to the same discrepancies detailed in Section III–A, where the γ zone extends closer to the surface than experimental results but is consistent with the thermodynamic input.
Upon further examination of Figure 3, it is clear that significant constituent redistribution occurs in U-Pu-Zr alloys over a wide range of overall compositions. Nevertheless, careful selection of alloy composition can significantly reduce the amount of redistribution that occurs. Obviously, little-to-no constituent redistribution will occur in pure, or nearly pure, substances, such as simulations 1, 6, and 21. However, simulations 2, 5, 7, 10, 11, and 14 also showed little redistribution.
There are a number of factors to consider when picking an ideal alloy composition in the U-Pu-Zr system. First, there are specific composition ranges for the constituents. Alloys rich in plutonium have unacceptably low liquidus temperatures, while zirconium-rich alloys have unacceptably high solidus temperatures. As a result, most alloys balance these requirements.[54] Second, certain microstructural features can accelerate formation of pores that can lead to failure. Previous work has documented that highly distorted microstructure and large cavities, due to “tear-like” porosity, are seen between grain boundaries, as well as phase boundaries within grains, primarily in the outermost zone where the δ phase is present.[38] Finally, fuel performance and high achievable burnup percentages can be improved by maintaining uniformity of phase and compositions across the fuel rod.
When all of these constraints and considerations are combined, it appears that the U-40Pu-20Zr (at. pct) alloy (simulation 14) may warrant consideration as it exhibits a uniform composition profile and consists almost entirely of the γ phase. By sustaining an even distribution of constituents, the U-40Pu-20Zr (at. pct) alloy would preserve a consistent dispersal of fissile atoms and uniform thermal conductivity, thus limiting localized hot spots. Furthermore, a corresponding decrease in the δ phase may be possible for this alloy, which is important because the δ phase forms high surface area lamellae that has a large impact on swelling behavior.[55] Finally, reducing phase boundary area by maintaining a homogenous phase would likely decrease pore generation and microstructural tearing, further improving fuel performance. It is worth noting that since the simulation does not compare perfectly with experiments, identifying the exact fuel composition for compositional and phase stability may require iteration with experiments. Furthermore, this alloy exceeds the 20 wt pct Pu recommendation for fast breeder reactors. Nonetheless, this modeling technique provides a good estimate of the starting fuel composition and an opportunity to examine a range of compositions and determine optimal alloys that deserve further consideration. If new alloy compositions are identified by this method, more detailed simulations that focus heavily on validation[23,24,26] will be required.
4 Conclusions
The hybrid Potts-phase field method has been utilized to simulate constituent redistribution and phase transformations in the U-Pu-Zr nuclear fuel. The paper details the model development as well as the use of a thermodynamic database to inform the energetics and composition evolution.
Validation of this modeling approach was performed by comparison with experimentally examined fuel rods of U-16Pu-23Zr (at. pct). While the qualitative phase evolution and constituent redistribution was captured in the simulation, the quantitative differences are attributed to inexact matches with the thermal profiles and diffusivity values, leading to differences in the location of the observed zones. However, simulation results were consistent with the thermodynamic database, confirming the reliability of the modeling technique given the available data.
The effects of alloy composition were also investigated by modeling fuel evolution over the entire range of U-Pu-Zr compositions. Results showed some phase transition temperatures that were generally in agreement with previous work.
Constituent redistribution was found to be dependent on overall composition, and the U-40Pu-20Zr (at. pct) alloy showed the most promise to remain homogenous, in both phase and compositions, throughout a thermal gradient. Future work would seek to obtain accurate diffusivity values for each component in all critical phases and investigate the effects of thermal profiles.
The greatest challenges to accurately modeling the U-Pu-Zr system are related to balancing the simplicity of the model based on limited material properties, thermodynamic data, and kinetic quantities with the need to capture complex phenomena. This complexity is likely the reason that previous works focused on pseudo-binary systems.[23,26] The present model consistently matched the inputs, indicating the need to determine which material data must be obtained to improve the fidelity of the model.
The ternary modeling technique presented in this work is an important step forward in simulating complex alloy systems like U-Pu-Zr. The work can be readily expanded to quaternary or higher-order alloy systems. The modeling technique has the potential to be an effective predictive tool to explore wide ranges of alloy composition and complex conditions, such as the temperature profiles seen in this work. The accuracy of the model is limited mostly by the boundary conditions and material properties used as input.
Notes
The Americium and Neptunium in these alloys are not listed in these compositions; Americium is substitutional for Plutonium, and Neptunium is substitutional for both Uranium and Plutonium, and these are included accordingly.[54]
References
T.A. Lennox, D.N. Millington, and R.E. Sunderland: Prog Nucl Energ, 2007, vol. 49, pp. 589–96.
V. Ivanchenko and T. Pryadko: in Non-Ferrous Metal Systems. Part 4, G. Effenberg and S. Ilyenko, eds., Springer, Berlin 2007, pp. 454–72.
B.R. Seidel, L.C. Walters, and Y.I. Chang: JOM-J Met, 1987, vol. 39, pp. 10–13.
R.D. Leggett and L.C. Walters: J Nucl Mater, 1993, vol. 204, pp. 23–32.
D.C. Crawford, D.L. Porter, and S.L. Hayes: J Nucl Mater, 2007, vol. 371, pp. 202–31.
M.K. Meyer, S.L. Hayes, W.J. Carmack, and H. Tsai: J Nucl Mater, 2009, vol. 392, pp. 176–83.
L. Capriotti, S. Brémier, K. Inagaki, P. Pöml, D. Papaioannou, H. Ohta, T. Ogata, and V.V. Rondinella: Prog Nucl Energ, 2017, vol. 94, pp. 194–201.
W.J. Carmack, D.L. Porter, Y.I. Chang, S.L. Hayes, M.K. Meyer, D.E. Burkes, C.B. Lee, T. Mizuno, F. Delage, and J. Somers: J Nucl Mater, 2009, vol. 392, pp. 139–50.
J. Vujić, R.M. Bergmann, R. Škoda, and M. Miletić: Energy, 2012, vol. 45, pp. 288–95.
R.G. Pahl, D.L. Porter, D. C. Crawford, and L.C. Walters: J Nucl Mater, 1992, vol. 188, pp. 3–9.
L.C. Walters: J Nucl Mater, 1999, vol. 270, pp. 39–48.
T. Ogata: in Comprehensive Nuclear Materials, Rudy J M Konings, ed., Elsevier, Amsterdam, 2011, pp. 1–40.
G.L. Hofman, L.C. Walters, and T.H. Bauer: Prog Nucl Energ, 1997, vol. 31, pp. 83–110.
R.G. Pahl, C.E. Lahm, R. Villareal, W.N. Beck, and G.L. Hofman: in International Conference on Reliable Fuels for Liquid Metal Reactors, Tuscon, AZ, 1986, pp. 36–51.
W.F. Murphy, W.N. Beck, F.L. Brown, B.J. Koprowski, and L.A. Neimark: Postirradiation Examination of U-Pu-Zr Fuel Elements Irradiated in EBR-II to 4.5 Atomic Percent Burnup, Argonne National Laboratory, Report ANL-7602, 1969.
Y.S. Kim, G.L. Hofman, S.L. Hayes, and Y.H. Sohn: J Nucl Mater, 2004, vol. 327, pp. 27–36.
Y.S. Kim and G.L. Hofman: AAA Fuels Handbook, Argonne National Laboratory, Report ANL-AAA-068, 2003.
S. Kaity, J. Banerjee, M.R. Nair, K. Ravi, S. Dash, T.R.G. Kutty, A. Kumar, and R.P. Singh: J Nucl Mater, 2012, vol. 427, pp. 1–11.
Y.S. Kim, T.W. Cho, and D.-S. Sohn: J Nucl Mater, 2014, vol. 445, pp. 272–80.
T. Ogawa, T. Iwai, and M. Kurata: J Less-Common Met, 1991, vol. 175, pp. 59–69.
G.P. Marino: Nucl Sci Eng, 1972, vol. 49, pp. 93–98.
M. Ishida, T. Ogata, and M. Kinoshita: Nucl Technol, 1993, vol. 104, pp. 37–51.
Y.S. Kim, S.L. Hayes, G.L. Hofman, and A.M. Yacout: J Nucl Mater, 2006, vol. 359, pp. 17–28.
T. Ogata, Y.S. Kim, and A.M. Yacout: in Comprehensive Nuclear Materials, R.J.M. Konings, ed., Elsevier, Amsterdam, 2012, pp. 713–53.
R.L. Williamson, J.D. Hales, S.R. Novascone, M.R. Tonks, D.R. Gaston, C.J. Permann, D. Andrs, and R.C. Martineau: J Nucl Mater, 2012, vol. 423, pp. 149–63.
J. Galloway, C. Unal, N. Carlson, D. Porter, and S. Hayes: Nucl Eng Des, 2015, vol. 286, pp. 1–17.
E.R. Homer, V. Tikare, and E.A. Holm: Comp Mat Sci, 2013, vol. 69, pp. 414–23.
M. Kurata: IOP Conference Series: Materials Science and Engineering, 2010, vol. 9, p. 012022.
J.W. Cahn and J.E. Hilliard: J Chem Phys, 1958, vol. 28, pp. 258–67.
J.J. Cox, E.R. Homer, and V. Tikare: Mater Res Soc Symp Proc, 2013, vol. 1524, p. 165.
K.G.F. Janssens, D. Raabe, E. Kozeschnik, M.A. Miodownik, and B. Nestler: Computational Materials Engineering: An Introduction to Microstructure Evolution, Academic Press, Boston, 2007.
M. Hillert: Phase Equilibria, Phase Diagrams and Phase Transformations, Cambridge University Press, Cambridge, 2007.
L. Zhang, M.R. Tonks, P.C. Millett, Y. Zhang, K. Chockalingam, and B. Biner: Comp Mat Sci, 2012, vol. 56, pp. 161–65.
H. Emmerich: The Diffuse Interface Approach in Materials Science, Springer-Verlag, Berlin, 2003.
R.W. Balluffi, S.M. Allen, and W.C. Carter: Kinetics of Materials, John Wiley & Sons, Hoboken, 2005.
P.G. Shewmon: Diffusion in Solids, McGraw-Hill, New York, 1963.
H.L. Lukas, S.G. Fries, and B. Sundman: Computational Thermodynamics, Cambridge University Press, Cambridge, 2007.
D. Yun, J. Rest, G.L. Hofman, and A.M. Yacout: J Nucl Mater, 2013, vol. 435, pp. 153–63.
A. Karahan and N.C. Andrews: Nucl Eng Des, 2013, vol. 258, pp. 26–34.
[40] F.N. Kryukov, O.N. Nikitin, S.V. Kuzmin, A.V. Belyaeva, I.F. Gilmutdinov, P.I. Grin, and I. Yu Zhemkov: IOP Conference Series: Materials Science and Engineering, 2017, vol. 168, p. 012029.
A. Aitkaliyeva, J.W. Madden, B.D. Miller, C.A. Papesch, and J.I. Cole: Metall Mater Trans E, 2015, vol. 2, pp. 220–28.
C. Matthews, C. Unal, J. Galloway, D.D. Keiser Jr., and S.L. Hayes: Nucl Technol, 2017, vol. 198, pp. 231–59.
D.E. Janney, J.R. Kennedy, J.W. Madden, and T.P. O’Holleran: J Nucl Mater, 2014, vol. 448, pp. 109–12.
A. Aitkaliyeva, J.W. Madden, C.A. Papesch, and J.I. Cole: J Nucl Mater, 2016, vol. 473, pp. 75–82.
D.E. Janney, J.R. Kennedy, J.W. Madden, and T.P. O’Holleran: J Nucl Mater, 2015, vol. 456, pp. 46–53.
D.E. Janney and B.H. Sencer: J Nucl Mater, 2017, vol. 486, pp. 66–69.
Y. Shibuta, T. Sato, T. Suzuki, H. Ohta, and M. Kurata: J Nucl Mater, 2013, vol. 436, pp. 61–67.
D.A. Cogswell and W.C. Carter: Phys Rev E, 2011, vol. 83, p. 061602.
G. Gottstein and L.S. Shvindlerman: Grain Boundary Migration in Metals, CRC Press, Boca Raton, 2010.
S.J. Plimpton, C.C. Battaile, M. Chandross, E.A. Holm, A. Thompson, V. Tikare, G. Wagner, E. Webb, and X. Zhou: Sandia National Laboratories, Report SAND2009–6226, 2009.
R.G. Pahl, D.L. Porter, C.E. Lahm, and G.L. Hofman: Metall Trans A, 1990, vol. 21, pp. 1863–70.
D.L. Porter, C.E. Lahm, and R.G. Pahl: Metall Trans A, 1990, vol. 21, pp. 1871–76.
Y.H. Sohn, M.A. Dayananda, G.L. Hofman, R.V. Strain, and S.L. Hayes: J Nucl Mater, 2000, vol. 279, pp. 317–29.
D.E. Burkes, J.R. Kennedy, T. Hartmann, C.A. Papesch, and D.D. Keiser Jr: J Nucl Mater, 2010, vol. 396, pp. 49–56.
G.L. Hofman, R.G. Pahl, C.E. Lahm, and D.L. Porter: Metall Trans A, 1990, vol. 21, pp. 517–28.
Acknowledgments
Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy’s National Security Administration under contract DE-NA0003525.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted December 31, 2017.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cox, J.J., Homer, E.R., Tikare, V. et al. Simulated Microstructural and Compositional Evolution of U-Pu-Zr Alloys Using the Potts-Phase Field Modeling Technique. Metall Mater Trans A 49, 6457–6468 (2018). https://doi.org/10.1007/s11661-018-4922-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-018-4922-7