
Abstract
Petiveria alliacea L. is a medicinal plant originating from the Amazon region. This study describes an efficient cryopreservation protocol for somatic embryos (SEs) produced from roots of P. alliacea based on the comparison of vitrification, encapsulation-dehydration, and D cryo-plate techniques. With the vitrification technique, SEs treated with PVS2 solution (0.4 M sucrose, 3.3 M glycerol, 2.4 M ethylene glycol, and 1.9 M DMSO) for 30 min displayed high viability (85%) and intermediate proliferation recovery (about 12 adventitious SEs produced from original SEs [SEs/SE] after 90 d of culture). With the encapsulation-dehydration technique, lower viability (70%) and very low proliferation recovery (about two SEs/SE) were achieved with cryopreserved SEs dehydrated for 10 min in a laminar air flow cabinet. The D cryo-plate technique led to high viability (85%) and proliferation recovery (19 SEs/SE) of cryopreserved SEs after 90 min dehydration. In the experimental conditions tested, the D cryo-plate method was the most efficient technique for cryopreservation of P. alliacea SEs.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Petiveria alliacea L. (Phytolaccacea) is a medicinal plant known as guinea, among other names (Kubec and Musah 2001). It is native to the Amazon region but can be found from Central America, including the Caribbean, to South America, as well as in some countries of the African continent with tropical climates (Luz et al. 2016). This species is used in traditional medicine (Germano et al. 1993), and various pharmacological uses have been scientifically proven (Sertié et al. 1995; Lopes-Martins et al. 2002), such as its potential antitumor properties (Mata-Greenwood et al. 2001). Several chemical compounds have been characterized in this species including polysulfides (De Sousa et al. 1990). Among them, Dibenzyltrisulfide (DTS) has neurotoxic and cytostatic effects, which prevent proliferation of a range of cancer cell lines (Williams et al. 1997; Rosner et al. 2001).
Biotechnological studies have shown that large DTS quantities can be produced from P. alliacea somatic embryos (SEs) (Webster et al. 2008). Therefore, somatic embryogenesis protocols have been established for this species from leaf (Cantelmo et al. 2013) and root segments (Soares 2016). Due to the great pharmacological interest of this species, the conservation of lineages with high biosynthetic capacity is essential, and cryopreservation has been the safest method for the long-term conservation of these materials. The metabolic blockade induced by the ultra-low temperatures of the liquid nitrogen allows the storage of material without genetic alterations. In addition, many samples can be kept in a small physical space (Engelmann 2011).
The vitrification process occurs when there is the formation of an amorphous (vitreous) solid in dehydrated tissues and subjected to ultra-low temperatures (Fahy et al. 1984). It has served as the basis for the development of many techniques, with specific adaptations for different plant tissues (Sakai and Engelmann 2007; Reed 2008). In all the developed techniques, the previous dehydration of the tissues is fundamental and can be conducted by osmotic or evaporative treatments (Engelmann 2011).
The original vitrification technique involved the treatment of the samples with cryoprotective solutions in increasing concentrations (Matsumoto et al. 1994). The PVS2 solution was one of the first to be developed for plants (Sakai et al. 1990), and is still the most used in conjunction with ultra-cooling and rapid rewarming. This procedure was standardized for apices, cell suspensions, and SE of several different species (Sakai and Engelmann 2007; Sakai et al. 2008).
In the encapsulation-dehydration technique, explants are imbedded in calcium alginate beads, then dehydrated to approximately 20% of the initial water content before supercooling. This technique has been applied to apices of numerous species from temperate and tropical origins and to cell suspensions and SEs of several species (González-Arnao and Engelmann 2006; Engelmann et al. 2008).
Recently, the cryo-plate dehydration technique (D cryo-plate) (Niino et al. 2013) was developed, based on the technique of droplet-vitrification (Panis et al. 2005), with the adaptation of adhering the explants to aluminum plates that are subjected to all treatments of dehydration, cryoprotection, and rewarming after the withdrawal of liquid nitrogen. This technique allows the manipulation of many explants at the same time and minimizes the variations that can occur when the explants are treated individually (Engelmann 2014). The D cryo-plate protocol has been tested with basal stem buds and lateral buds of Lomandra longifolia Mat rush (Niino et al. 2013, 2014), Phoenix dactylifera (date palm) polyembryonic masses (Salma et al. 2014), Solanum tuberosum (potato) shoot tips (Yamamoto et al. 2012), Diospyros kaki (persimmon) shoot tips from dormant buds (Matsumoto et al. 2015), and Saccharum officinarum (sugarcane) shoot tips (Rafique et al. 2016).
The objective of this work was to establish an efficient cryopreservation protocol for SEs derived from P. alliacea roots grown in vitro. The effect of three cryopreservation techniques—vitrification, encapsulation-dehydration and D cryo-plate, were compared on the viability and proliferation recovery of P. alliacea SEs.
Material and methods
Plant materials
Mother plants of Petiveria alliacea L. were established in vitro according to Soares et al. (2013) from seeds collected in the Niterói region, RJ, Brazil (22° 53′ 55.95″ S and 43° 05′ 09.37” O; 54 m above sea level). Specimens from each sample were deposited in the Herbarium of Rio de Janeiro (HRJ 11.710).
Plantlets were subcultured every 2 mo on MS0 medium (Murashige and Skoog 1962) at 30 ± 2 °C under a 16-h photoperiod and a light intensity of 46 μmol m−2 s−1 provided by cool white fluorescent tubes (GE lighting, São Paulo, Brazil). To produce the SEs used in cryopreservation experiments, roots from these in vitro plantlets were cultured according to Soares (2016) for 120 d in liquid MS medium supplemented with 5 mg L−1 Indole-3-butyric acid (IBA) (Sigma-Aldrich®, Steinheim, Germany) on a rotary shaker (110 rpm). For all culture media and solutions, the pH was adjusted to 5.8 and then autoclaved at 121 °C for 15 min.
Cryopreservation methods-vitrification
After pretreatment for 24 h on MS0 semi-solid medium with 0.5 M sucrose (Pettinelli et al. 2015), solidified with 2% (w/v) Phytagel™ (Sigma-Aldrich®). SEs were distributed in 2-ml sterile polypropylene cryotubes (Nalgene®, Rio de Janeiro, Brazil) (10 SEs/cryotube) and treated with vitrification solution (PVS2) or not (controls) (Sakai et al. 1990) for 15 and 30 min at 20 to 22 °C. The PVS2 solution was replaced with fresh PVS2 solution 2 min before immersion of the cryotubes in liquid nitrogen (LN), where they were kept for 24 h.
Cryotubes containing the SEs were rewarmed for 1 min in a water-bath at 40 °C. The PVS2 solution was drained from the cryotubes and replaced by 1 ml unloading solution (ULS) composed of (MS medium supplemented with 1.2 M sucrose), at 20 to 22 °C. Every 5 min, 0.5 ml of the solution contained in the cryotubes was drained and replaced with 0.5 ml fresh liquid MS0 medium containing 3% (w/v) sucrose. This procedure was repeated six times (Gagliardi et al. 2002). The SEs were then transferred to recovery medium composed of MS medium supplemented with 0.6 μM Indole-3-acetic acid (IAA) (Sigma-Aldrich®) and incubated in the dark for 1 wk. prior to being transferred to MS0 medium under standard light (16 h photoperiod and a light intensity of 46 μmol m−2 s−1) and standard temperature conditions (30 ± 2 °C).
Encapsulation-dehydration
SEs were mixed in 3% (w/v) calcium alginate, and beads encapsulating them were made by dripping the SE-alginate mixture into calcium chloride solution (1 M) (10 SEs/bead of approximately 5 mm in diameter), which were pretreated for 24 h in liquid MS medium with 0.5 M sucrose. Beads were immersed in loading solution (LS) composed of MS medium supplemented with 2.0 M glycerol and 0.4 M sucrose for 20 min at 20 to 22 °C, and then dehydrated in a laminar air flow cabinet (Veco, São Paulo, Brazil) for up to 15 min. After transfer of the beads to 2 ml cryotubes (10 beads/cryotube), the beads were immersed in LN and kept for 24 h.
Cryotubes containing the beads were rewarmed for 1 min in a water-bath at 40 °C. The beads were then immersed in ULS for 20 min at 20 to 22 °C. They were then placed on recovery medium and incubated in the dark for 1 wk., prior to being transferred to MS0 medium in standard light and temperature conditions described above.
D cryo-plate
After pretreatment for 24 h on MS semi-solid medium with 0.5 M sucrose (Pettinelli et al. 2015), SEs were fixed on to aluminum cryo-plates (37 mm × 7 mm × 0.5 mm with ten oval-shaped wells with a length of 2.5 mm, a width of 1.5 mm, and a depth of 0.75 mm) according to Niino et al. (2014). These plates were supplied by Dr. Takao Niino (NIAS, Tsukuba, Japan).
A volume of 2 μL of 3% (w/v) sodium alginate solution in calcium-free medium was poured in the wells of the cryo-plates. The pretreated SEs were placed in the wells and covered with 2 μL sodium alginate solution. Calcium chloride solution (100 mM) was added dropwise onto the cryo-plates until the SEs were covered. Polymerization was complete after 15 min. Cryo-plates were immersed in LS containing MS medium, 2.0 M glycerol, and 0.4 M sucrose for 20 min at 20 to 22 °C and then dehydrated in a laminar air flow cabinet for up to 140 min before immersion in LN where they were kept for 24 h.
For rewarming, the cryo-plates were immersed in ULS for 20 min at 20 to 22 °C. SEs were detached from the cryo-plates with a scalpel and placed on recovery medium and incubated in the dark for 1 wk, prior to being transferred to MS0 medium in the standard light and temperature conditions described above.
Assessment of viability and proliferation recovery
Viability of SEs was evaluated by 2.3.5-triphenyl–tetrazolium chloride (TTC) staining (Towill and Mazur 1975) after each step of the protocol. SEs were immersed in 1 ml TTC solution (0.6% w/v) and incubated at 28 ± 1 °C for12 h. SEs were considered viable when they totally stained red.
Proliferation recovery of treated SEs was evaluated by recording the number of adventitious SEs produced from the original SEs (SEs/SE) after 90 d of culture in MS0 medium.
Statistical analysis
Cryopreservation experiments were repeated two times with ten SEs per treatment. Significant differences in numbers of SEs produced per original embryo (SEs/SE) were assessed by analysis of variance (ANOVA) and comparison test of means Tukey-Kramer with the assistance of the InStat (Graphpad software, San Diego, CA), with significant values at p ≤ 0.05.
Results
Pretreatment
Pretreatment of SEs with 0.5 M sucrose did not alter their viability and proliferation capacity compared to untreated SEs (data not shown).
Vitrification
The viability of control (−LN) SEs decreased with increasing durations of exposure to PVS2, whereas proliferation recovery varied between about 6 (15 min PVS2 exposure) and 10 (30 min exposure) SEs/SE (Table 1). No viability was noted with SEs cryopreserved without PVS2 treatment. Viability of SEs cryopreserved after 15 and 30 min PVS2 treatment was similar, reaching 85%. Proliferation recovery was significantly higher after 30-min PVS2 treatment (12.6 ± 1.4 SEs/SE) compared to 15 min treatment (8.0 ± 0.1 SEs/SE). Recovery took place through the formation of friable callus, on the surface of which adventitious SEs, and plantlets could be observed 90 d after treatment, on both control (−LN, Fig. 1 a) and cryopreserved (+LN, Fig. 1 b, c) SEs.

Recovery of SEs cryopreserved by vitrification after 90 d in culture. (a) Non-cryopreserved control. (b, c) Cryopreserved SEs treated with PVS2 for 15 (b) and 30 min (c). Bar = 0.5 cm
Encapsulation-dehydration
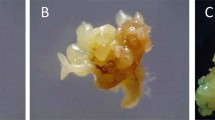
The viability of control (−LN) SEs was 100% regardless of the dehydration duration tested, while proliferation recovery was between about 6 and 13 SEs/SE after 10 and 15 min of dehydration, respectively (Table 2). The viability of cryopreserved explants was zero without dehydration and reached a maximum of 70% after 10 min of dehydration. No significant differences in proliferation recovery of cryopreserved SEs were noted between the different dehydration durations tested. After 90 d in culture, the encapsulated original SEs (Fig. 2 a) had proliferated, giving rise to callus, adventitious SEs, and young plantlets from control (Fig. 2 b) and cryopreserved (Fig. 2 c) explants.

Recovery of SEs cryopreserved by encapsulation/dehydration after 90 d in culture. (a) Encapsulated SEs before treatment. (b, c) Proliferation recovery of SEs after 90 d in culture. (b) 10 min dehydration control bead showing root formation and conversion of a SE into a plantlet. (c) cryopreserved explant showing callusing. Bar = 0.5 cm
D cryo-plate
The viability of control (−LN) SEs decreased progressively from 100% without dehydration to 50% after 140 min of dehydration (Table 3). Proliferation recovery of control SEs decreased from 45–46 SEs/SE without dehydration to 14–15 SEs/SE after 140 min of dehydration. Viability of cryopreserved SEs was zero without dehydration. It increased progressively to reach a maximum of 85% after 90 min of dehydration, and then decreased to 50% after 120 and 140 min of dehydration. No proliferation recovery of cryopreserved SEs was noted without dehydration or after 30 min of dehydration. Recovery increased to 19 SEs/SE after 90 min of dehydration, then decreased to 4–5 SEs/SE after 140 min of dehydration. After 90 d in culture, the production of callus, of adventitious SEs and of plantlets was observed on control (Fig. 3 a) and cryopreserved explants (Fig. 3 b).

Recovery of SEs cryopreserved by D cryo-plate after 90 d in culture. (a) Non-cryopreserved control. (b) Cryopreserved SEs after dehydration on a cryo-plate for 90 min. Bar = 0.5 cm
Discussion
In this study, cryopreservation of P. alliacea SEs was achieved using three different techniques, which included vitrification, encapsulation-dehydration, and D cryo-plate. The tolerance of P. alliacea somatic embryos to cryopreservation was previously evaluated through survival after the vitrification technique (Pettinelli et al. 2012). It has been reported that somatic embryos of P. alliacea, originating from leaves, when treated with PVS2 for longer exposure times than 15 min, did not survive after immersion in LN (Pettinelli et al. 2012). In the present study, for the first time, the most efficient technique was evaluated based on the recovery of somatic embryos after in vitro culture. With vitrification, high viability (85%) and intermediate proliferation recovery after LN exposure (12.6 ± 1.4 SEs/SE) were achieved. Modifying different steps of the protocol may be instrumental to improve recovery. In these experiments, the highest proliferation recovery was achieved after the 30-min PVS2 treatment, which was the longest duration tested. It has been observed that in SEs originating from roots of this species, increasing the duration of PVS2 exposure may improve recovery. Normally, longer exposure times are better as in the case of SEs of Castanea sativa (European chestnut, Corredoria et al. 2014) and embryogenic callus of Aesculus hippocastanum (Horse chestnut, Lambardi et al. 2005), for which the highest recovery was obtained after 60 and 90-min PVS2 treatments, respectively. Treating SEs with the PVS3 vitrification solution, which contains 5.4 M glycerol and 1.5 M sucrose (Nishizawa et al. 1993) and is considered less toxic than PVS2 (Sakai and Engelmann 2007) should also be tested. Finally, the use of alternative LS (Kim et al. 2009a) and different vitrification solutions (Kim et al. 2009b), which have been specially designed to treat materials sensitive to PVS2 and PVS3 solutions, should be examined.
SEs cryopreserved using the encapsulation-dehydration technique had a lower viability (70%) and very low proliferation recovery (maximum of 1.9 ± 0.2 SEs/SE) in the experimental conditions tested. Various modifications to the protocol should be tested to improve the results. In this study, a 24-h pretreatment with 0.5 M sucrose was used. Higher sucrose concentrations and/or longer pretreatment durations are usually employed (González-Arnao and Engelmann 2006) and should be tested with P. alliacea embryos. In the case of Citrus spp. SEs, pretreatment with 0.75 M sucrose for 24 h led to optimal results (González-Arnao et al. 2003), while with Olea europea L. SEs (Shibli and Al-Juboory 2000), the optimal pretreatment conditions were 0.75 to 1.25 M sucrose for 4 d.
High viability (85%) and very high proliferation recovery (19.0 ± 0.9 SEs/SE) were obtained using the D cryo-plate technique. These results may be explained by the achievement of very high cooling and warming rates, because explants are in direct contact with LN during cooling and with the ULS during warming (Engelmann 2014). Moreover, no potentially toxic vitrification solutions are used in the D cryo-plate technique, as in the cases of V cryo-plate (Yamamoto et al. 2011) and droplet-vitrification (Panis et al. 2005) techniques. Potential improvement of the current results may be achieved by modifying the pretreatment, by increasing the sucrose concentration of the medium and/or its duration (González-Arnao and Engelmann 2006), and by experimenting with loading solution alternatives, which have been specially designed to treat sensitive materials (Kim et al. 2009a).
Despite the high recovery rates of cryopreserved somatic embryos obtained in this study, regeneration was always through callus. This indicates the need for monitoring genetic and phytochemical stability, whose techniques will be standardized depending on the conservation objective of these structures.
Conclusions
In the experimental conditions tested, the D cryo-plate technique was more efficient than vitrification and encapsulation-dehydration for cryopreserving P. alliacea SEs. The protocol developed led to a high proliferation recovery of cryopreserved samples, and various options for further improvement have been identified. Once the protocol is optimized, it will have to be tested with additional accesses for routine use in the long-term conservation of P. alliacea germplasm.
References
Cantelmo L, Soares BO, Rocha LP, Pettinelli JA, Callado CH, Mansur E, Castellar A, Gagliardi RF (2013) Repetitive somatic embryogenesis from leaves of the medicinal plant Petiveria alliacea L. Plant Cell Tiss Org 115:385–393
Corredoria E, San-José MC, Ballester A, Vieitez AM (2014) Cryopreservation of zygotic embryo axes and somatic embryos of european chestnut. Cryo-Lett 25:33–42
De Sousa JR, Demuner AJ, Pinheiro JA, Breitmaier E, Cassels BK (1990) Dibenzyltrisulphide and trans-N-methyl-4-methoxyproline from Petiveria alliacea. Phytochemistry 29:3653–3655
Engelmann F (2011) Cryopreservation of plant biodiversity conservation. In vitro Cell Dev-Pl 47:5–16
Engelmann F (2014) Cryopreservation of clonal crops: a review of key parameters. Acta Hortic 1039:31–39
Engelmann F, Gonzalez-Arnao MT, Wu Y, Escobar R (2008) Development of encapsulation dehydration. In: Reed BM (ed) In: Plant cryopreservation: a practical guide. Springer-Verlag, New York, pp 59–75
Fahy GM, MacFarlane DR, Angell CA, Meryman HT (1984) Vitrification as an approach to cryopreservation. Cryobiology 21:407–426
Gagliardi RF, Pacheco GP, Valls JFM, Mansur E (2002) Cryopreservation of cultivated and wild Arachis species embryonic axes using desiccation and vitrification methods. Cryo-Lett 23:61–68
Germano DHP, Caldeira TTO, Mazella AAG, Sertie JAA, Bacchi EM (1993) Topical anti-inflammatory activity and toxicity of Petiveriaalliacea. Fitoterapia 64:459–467
González-Arnao MT, Engelmann F (2006) Cryopreservation of plant germplasm using the encapsulation-dehydration technique: review and case study on sugarcane. Cryo-Lett 27:155–168
González-Arnao MT, Ortega JJ, Navarro L, Duran-Vila N (2003) Cryopreservation of ovules and somatic embryos of citrus using the encapsulation-dehydration technique. Cryo-Lett 24:85–94
Kim HH, Lee YG, Park S, Lee S, Baek H, Cho E, Engelmann F (2009a) Development of alternative loading solutions in droplet-vitrification procedures. Cryo-Lett 30:291–299
Kim HH, Lee YG, Shin DJ, Kim T, Cho EG, Engelmann F (2009b) Development of alternative plant vitrification solutions in droplet-vitrification procedures. Cryo-Lett 30:320–334
Kubec R, Musah R (2001) Cysteine sulfoxide derivatives in Petiveria alliacea. Phytochemistry 58:981–985
Lambardi M, De Carlo A, Capuana M (2005) Cryopreservation of embryogenic callus of Aesculus hippocastanum L. by vitrification one-steps freezing. Cryo-Lett 26:185–192
Lopes-Martins RAB, Pegoraro DH, Woisky R, Penna SC, JAA S (2002) The anti-inflammatory and analgesic effects of a crude extract of Petiveria alliacea L. (Phytolaccaceae). Phytomedicine 9:245–248
Luz DA, Pinheiro AM, Silva ML, Monteiro MC, Prediger RD, Maia CSF, Fontes-Junior EA (2016) Ethnobotany, phytochemistry and neuropharmacological effects of Petiveria alliacea L. (Phytolaccaceae): a review. J Ethnopharmacol 185:182–201
Mata-Greenwood E, Ito A, Westenburg H, Cui B, Mehta RG, Kinghorn AD, Pezzuto JM (2001) Discovery of novel inducers of cellular differentiation using HL-60 promyelocytic cells. Anticancer Res 21:1763–1770
Matsumoto T, Sakai A, Yamada K (1994) Cryopreservation of in vitro grown apical meristems of wasabi (Wasabi japonica) by vitrification and subsequent high plant regeneration. Plant Cell Rep 13:442–446
Matsumoto T, Yamamoto S, Fukui K, Rafique T, Engelmann F, Niino T (2015) Cryopreservation of persimmon shoot tips from dormant buds using the d-cryoplate technique. Hort J 84:106–110
Murashige T, Skoog FA (1962) A revised medium for rapid growth and bioassays with tobacco tissues cultures. Plant Physiol 15:473–479
Niino T, Wunna WK, Nohara N, Rafique T, Yamamoto S, Fukui K, Valle M, Arizaga M, Martinez CR, Matsumoto T, Engelmann F (2014) Cryopreservation of mat rush lateral buds by air dehydration using aluminumcryo-plates. Plant Biotechnol 31:281–287
Niino T, Yamamoto S, Fukui K, Martinez C, Roman C, Arizag M, Matsumoto T, Engelmann F (2013) Dehydration improves cryopreservation of Mat Rush (Juncus decipiens Nakai) basal stem buds on Cryo-plates. Cryo-Lett 4:549–560
Nishizawa S, Sakai A, Amano Y, Matsuzawa T (1993) Cryopreservation of asparagus (Asparagus officinalis L.) embryogenic suspension cells and subsequent plant regeneration by vitrification. Plant Sci 91:67–73
Panis B, Piette B, Swennen R (2005) Droplet vitrification of apical meristems: a cryopreservation protocol applicable to all Musaceae. Plant Sci 168:45–55
Pettinelli JA, Pimenta M, Soares BO, Garcia RO, Mansur E, Engelmann F, Gagliardi RF (2015) Avaliação do protocolo de vitrificação na criopreservação de embriões somáticos de Petiveria alliacea L. Aproximando 1:1–5
Pettinelli JA, Soares BO, Cantelmo L, Pimenta M, Cochofel J, Garcia RO, Mansur E, Gagliardi RF (2012) Evaluation of the survival of somatic embryos of Petiveria alliacea L. cryopreserved after different pretreatments with sucrose and PVS2. Cryobiology 65:363
Rafique T, Shin-ichi Y, Kuniak F, Tanaka D, Arizaga MV, Abbas M, Matsumoto T, Niino T (2016) Cryopreservation of shoot-tips from different sugarcane varieties using d-cryoplate technique. Pak J Agr Sci 53:151–158
Reed BM (2008) Plant cryopreservation: a practical guide. Springer-Verlag, New York
Rosner H, Williams LAD, Jung A, Kraus W (2001) Disassembly of microtubules and inhibition of neurite outgrowth, neuroblastoma cell proliferation and MAP kinase tyrosine dephosphorylation by dibenzyltrisulphide. Biochim Biophys Acta 40:166–177
Sakai A, Engelmann F (2007) Vitrification, encapsulation-vitrification and droplet-vitrification: a review. Cryo-Lett 28:151–172
Sakai A, Hirai D, Niino T (2008) In: Plant cryopreservation: A practical guide. In: Development of PVS-based vitrification and encapsulation–vitrification protocols, pp 33–57
Sakai A, Kobayashi S, Oiyama I (1990) Cryopreservation of nucellar cells of navel orange (Citrus sinensis Osb. Var. brasiliensis Tanaka) by vitrification. Plant Cell Rep 9:30–33
Salma M, Fki L, Engelmann-Sylvestre I, Niino T, Engelmann F (2014) Comparision of droplet-vitrification and D-cryoplate for cryopreservation of date palm (Phoenix dactylifera L.) polyembryogenic masses. Sci Hortic 179:91–97
Sertié JA, Hanada S, Sudo LS, Germano DHP (1995) Petiveria alliacea-antiinflammatory effect and gastric mucous protection. J Dent Res 74:793
Shibli RA, Al-Juboory KM (2000) Cryopreservation of “Nabali” olive (Olea europea L.) somatic embryos by encapsulation-dehydration and encapsulation-vitrification. Cryo-Lett 21:657–366
Soares BO (2016) Monitoramento da estabilidade genética de plantas de Petiveria alliacea L. produzidas in vitro através de marcadores fitoquímicos e moleculares. Tese de doutorado. Universidade do Estado do Rio de Janeiro
Soares BO, Fernandes DC, Cantelmo L, Rocha LP, Pettinelli JA, Christo AG, Coelho MGP, Gagliardi RF (2013) Botanical characterization of Petiveria alliacea L. from Rio de Janeiro, Brazil: systematic and funcional implications. Plant Biosyst 147:411–417
Towill LE, Mazur P (1975) Studies on the reduction of 2.3.5-triphenyltetrazolium chloride as a viability assay for plant tissue cultures. Can J Botany 53:1097–1102
Webster S, Mitchell S, Gallimore W, Ahmad M (2008) Biosynthesis of Dibenzyl Trisulfide (DTS) from somatic embryos and rhizogenous/embryogenic callus derived from guinea hen weed (Petiveria alliacea L.) leaf explant. In vitro Cell Dev-Pl 44:112–118
Williams LAD, The TL, Gardner MT, Fletcher CK, Naravani A, Bibbs N, Fleishacker R (1997) Immunomodulatory activities of Petiveria alliacea L. Phytother Res 11:251–253
Yamamoto S, Fukui K, Rafique T, Khan NI, Martinez CR, Sekizawa K, Matsumoto T, Niino T (2012) Cryopreservation of in vitro-grown shoot tips of strawberry by the vitrification method using aluminum cryo-plates. Plant Gen Resour 10:14–19
Yamamoto S, Rafique T, Priyantha WS, Fukui K, Matsumoto T, Niino T (2011) Development of a cryopreservation procedure using aluminum cryo-plates. Cryo-Lett 32:256–265
Acknowledgments
The authors are grateful to the Coordination of Improvement of Higher Education Personnel (CAPES/Brazil) and to the National Council for Scientific and Technological Development (CNPq/Brazil) for funding an international collaborative research project (Programme Science without Borders) between UERJ (University of the State of Rio de Janeiro) and IRD (Institut de Recherche pour le Développement). This study was also supported by the Carlos Chagas Filho Foundation for Research Support of the State of Rio de Janeiro (FAPERJ).
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: Barbara Reed
Rights and permissions
About this article

Cite this article
de Almeida Pettinelli, J., de Oliveira Soares, B., Cantelmo, L. et al. Cryopreservation of somatic embryos from Petiveria alliacea L. by different techniques based on vitrification. In Vitro Cell.Dev.Biol.-Plant 53, 339–345 (2017). https://doi.org/10.1007/s11627-017-9820-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11627-017-9820-y