
Abstract
Endothelial cell apoptosis and renin-angiotensin-aldosterone system (RAAS) activation are the major pathological mechanisms for cardiovascular disease and heart failure; however, the interaction and mechanism between them remain unclear. Investigating the role of PTP1B in angiotensin II (Ang II)–induced apoptosis of primary cardiac microvascular endothelial cells (CMECs) may provide direct evidence of the link between endothelial cell apoptosis and RAAS. Isolated rat CMECs were treated with different concentrations of Ang II to induce apoptosis, and an Ang II concentration of 4 nM was selected as the effective dose for the subsequent studies. The CMECs were cultured for 48 h with or without Ang II (4 nM) in the absence or presence of the PTP1B inhibitor TCS 401 (8 μM) and the PI3K inhibitor LY294002 (10 μM). The level of CMEC apoptosis was assessed by TUNEL staining and caspase-3 activity. The protein expressions of PTP1B, PI3K, Akt, p-Akt, Bcl-2, Bax, caspase-3, and cleaved caspase-3 were determined by Western blot (WB). The results showed that Ang II increased apoptosis of CMECs, upregulated PTP1B expression, and inhibited the PI3K/Akt pathway. Furthermore, cotreatment with PTP1B inhibitor significantly decreased the number of apoptotic CMECs induced by Ang II, along with increased PI3K expression, phosphorylation of Akt and the ratio of Bcl-2/Bax, decreased caspase-3 activity, and a cleaved caspase-3/caspase-3 ratio, while treatment with LY294002 partly inhibited the anti-apoptotic effect of the PTP1B inhibitor. Ang II induces apoptosis of primary rat CMECs via regulating the PTP1B/PI3K/Akt pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disease (CVD) and heart failure (HF) result in an enormous burden in terms of morbidity and mortality (van Riet et al. 2016). Although the etiologies of CVD and HF are diverse (Suthahar et al. 2017), one of the major common pathological mechanisms of them is the endothelial cell apoptosis (Davignon and Ganz 2004; Peng et al. 2014). The cardiac coronary microvascular system is located at the end part of the coronary circulation system. It determines the level of myocardial perfusion and oxygen supply and plays a key role in maintaining normal tissue metabolism and cell growth (Bulluck et al. 2016). Cardiac microvascular endothelial cells (CMECs) comprise up to one-third of the total cardiac cells and play a critical role in maintaining normal cardiomyocyte function (Li et al. 2001). It is the main components of the cardiac coronary microvascular system and directly mediates the exchange of substances between tissue cells and circulation (Bulluck et al. 2016). Removing CMECs has significantly detrimental effects on the contractile function of the adjacent cardiac myocytes (Hedhli et al. 2011). A mismatching between angiogenesis and apoptosis of CMECs may be a critical process mediating the transition from adaptive hypertrophy to heart failure (Li et al. 2015). Clinical and experimental studies have shown that the rarefaction of cardiac capillaries observed in chronic cardiac hypertrophy subjects could promote tissue hypoxia, cell death, and replacement fibrosis, as well as contribute to the progression from compensated hypertrophy to contractile dysfunction and heart failure (Shiojima et al. 2005; Moorjani et al. 2009). It is known that apoptosis is one of the main mechanisms and a key promoter of microvascular rarefaction (Ohta et al. 2007), which also reduces the proliferation, migration, and tube formation activities of CMECs (Xiao et al. 2017). Thus, reducing CMEC apoptosis might be a potentially important therapeutic strategy for HF treatment, and it is meaningful to explore the underlying mechanisms of CMEC apoptosis in the setting of HF.
HF is characterized by the activation of the sympathetic nervous system and renin-angiotensin-aldosterone system (RAAS). Angiotensin II (Ang II), a key component of RAAS, can cause vasoconstriction, sympathetic nervous stimulation, and the release of aldosterone (Fyhrquist et al. 1995). Increased Ang II level is a common situation identified in hypertension, hypertrophy, and heart failure (Harrison et al. 2003). Previous studies revealed that Ang II participates in the pathogenesis of heart failure by inducing vascular endothelial dysfunction and endothelial injury (Smiljic 2017); Ang II also promotes apoptosis of epithelial tissue and endothelial cells as well as smooth muscle cells (SMCs) in various pathological conditions (Marshall et al. 2004; Day et al. 2011).
Protein tyrosine phosphatase 1B (PTP1B) is a known negative regulator of receptor tyrosine kinase (RTK) signaling. It could inhibit insulin signaling and promote insulin resistance, obesity, and the development of diabetes. These features make it to be a potential therapeutic target for metabolic disorders (Tsou et al. 2012). Recent studies indicate that PTP1B is widely expressed in cardiovascular tissues, especially in endothelial cells, so it may also serve as a potential therapeutic target for cardiovascular diseases (Thiebaut et al. 2016). A previous study has shown that myocardial deletion of PTP1B expression in a chronic heart failure mice model resulted in significant improvement on endothelial and cardiac dysfunction, enhancement of angiogenesis, and reduction of cardiac fibrosis. The mechanisms were found to be related to the promotion of Akt, eNOS, and ERK phosphorylation, as well as the upregulation of VEGFR2 expression (Maupoint et al. 2016). It was also observed that activated PTP1B subsequently contributed to STAT3 dephosphorylation and human glioma cell apoptosis (Akasaki et al. 2006). However, it remains unclear whether and how PTP1B involves in Ang II–induced CMEC apoptosis. The present study aims to investigate the role of PTP1B on Ang II–induced CMEC apoptosis and to explore its underlying mechanisms.
Materials and Methods
Reagents
High-glucose Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and trypsin were purchased from Life Technologies (Carlsbad, CA). An In Situ Cell Death Detection Kit was purchased from Roche (Mannheim, Germany) and a caspase-3 activity assay kit was purchased from the Beyotime Institute of Biotechnology (Shanghai, China). Ang II (A9525) was bought from Sigma-Aldrich (St. Louis, MO). TCS 401 (PTP1B Inhibitor) and LY294002 (PI3K inhibitor) were obtained from MedChemExpress (Shanghai, China). Primary antibodies for anti-phospho-Akt (Ser473, no. 4060), PI3K p85 (19H8, no. 4257), Bax (D3R2M, no. 14796), caspase 3 (no. 9662), and cleaved caspase 3 (Asp175, no. 9661) were obtained from Cell Signaling Technology (Boston, MA). The antibodies for PTP1B (no. M1511-7) and Akt (no. ET1609-47) were purchased from Hangzhou Huaan Biotechnology Corporation (Hangzhou, China); antibody for Bcl-2 (ab59348) and HRP-conjugated monoclonal mouse anti-rat β-actin antibody (ab49900) were purchased from Abcam (Cambridge, UK).
Isolation and cultivation of rat CMECs
Rat cardiac microvascular endothelial cells were isolated and identified according to our previous work (Wang et al. 2017). Two-week-old Sprague-Dawley male rats were euthanized by cervical dislocation. Then, the hearts were rapidly excised after thoracotomy and rinsed with ice-cold phosphate-buffered saline (PBS) solution under sterile conditions. After cutting off the atrial myocardium and great vessels, the left ventricle was dissected, and the endocardium and epicardium were removed. The remaining left ventricular tissues were minced into 1-mm3 pieces and uniformly plated on 10-cm culture dishes, which were humidified with 1 ml FBS. Tissue pieces were then cultured in a humidified incubator with 5% CO2 and 95% air for 4 h. Once the pieces were firmly attached to culture dishes, 6 ml DMEM supplemented with 10% FBS was added to each dish and kept in a 5% CO2 and 95% air incubator for another 48 h, until abundant polygonal or star-like cells crept out of the myocardial tissues. After removal of tissue pieces and non-adherent cells by careful washing, 6 ml DMEM was added again to each dish for further cultures. When the cells were 80–90% confluent, they were washed with PBS, digested in 0.25% trypsin and passaged onto 6-well plates, followed by incubation at 37°C for 12 h, allowing cells to adhere and spread on the substrate.
The study was approved by the ethical committees of the Zhongshan Hospital Affiliated to Fudan University, and all the experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996).
Treatment of CMECs
The passage 2 CMECs at approximately 60–70% confluences were used in subsequent experiments. First, we treated CMECs with different concentrations of Ang II (1, 2, and 4 nM). According to the results of the apoptosis index and related proteins expressions, we choose the concentration of 4 nM as the ideal concentration for Ang II–induced apoptosis used for subsequent experiments. After screening the optimal concentration (4 nM) for Ang II–induced apoptosis, the CMECs were treated with or without Ang II (4 nM), the PTP1B inhibitor TCS 401 (8 μM), and the PI3K inhibitor LY294002 (10 μM). Then, the CMECs were cultivated in incubator chamber (at 37°C and 5% CO2) for 48 h.
Determination of apoptosis in CMECs after various treatments
Apoptotic cells were identified using a terminal transferase–mediated dUTP nick-end labeling (TUNEL) staining kit according to the manufacturer’s instructions. For TUNEL staining, CMECs were cultured on coverslips on a 24-well plate. After intervention, cells were successively fixed in 4% paraformaldehyde for 15 min, blocked with 3% H2O2 in methanol for 10 min, and incubated in a permeabilization solution (0.1% Triton X-100) for 2 min. Subsequently, a 50-μl TUNEL reaction mixture was added to each sample, followed by incubation for 1 h at 37°C in the dark. After being rinsed with PBS three times, cells were counterstained with DAPI for 10 min. Finally, TUNEL-positive nuclei were visualized using fluorescence microscopy (Leica, Germany) and sequential images were obtained from five random fields per slides. The apoptosis index was expressed as the “percentage of TUNEL-positive cells” (the number of positively stained apoptotic CMECs divided by the total number of CMECs and multiplied with 100%).
Analysis of caspase-3 activity
We further measured caspase-3 activity to detect the apoptosis level of CMECs induced by Ang II. Firstly, a standard curve was constructed using the absorbance of p-nitroanilide standards; then, caspase-3 activity in cell extracts on each sample was measured with a caspase-3 cellular activity assay kit according to the manufacturer’s instructions. Cells were then scraped off, collected by centrifugation at 600g for 5 min and lysed at 4°C. Lysates were clarified by centrifugation at 18,000g for 15 min. The reaction mixture contained 50 μl of cellular lysates, 40 μl of assay buffer, and 10 μl of caspase-3 substrate (Ac-DEVD-AMC). After incubating the obtained mixture at 37°C for 2 h in the dark, enzymatic activity was measured in a luminescence spectrophotometer (excitation = 380 nm; emission = 405 nm). Then, the protein concentration of cell lysates was determined using the BCA protein assay (Beyotime Biotechnology, Shanghai, China) and results were presented as caspase-3 activity/mg (of total protein).
Western blot analysis
After 48 h of culture, cells were lysed using a RIPA lysis buffer (Beyotime Biotechnology) containing 1 mmol/l PMSF protease and phosphatase inhibitors (Beyotime Biotechnology) in an ice bath. Lysates were then centrifuged at 12,000g for 10 min to remove the cell debris. After protein concentrations were determined with a BCA protein assay kit (Beyotime Biotechnology), equivalent amounts of cell extracts (30 μg) were subjected to SDS-PAGE gel and transferred to nitrocellulose membranes (Thermo Scientific, Waltham, MA). The membranes were blocked with 5% non-fat milk for 2 h, then incubated with primary antibodies against PTP1B (dilution 1:500), Bax (1:1000), Bcl-2 (dilution 1:1000), caspase-3 (dilution 1:1000) and cleaved caspase-3 (dilution 1:1000), phospho-Akt (Ser473, dilution 1:1000), Akt (dilution 1:500), and PI3K p85 (dilution 1:1000) overnight at 4°C. This was followed by the incubation of the appropriate HRP-conjugated secondary antibody or HRP-conjugated monoclonal mouse anti-rat β-actin antibody (1:10,000), which was used to detect β-actin levels for 2 h at room temperature. Then, blots were visualized with an ECL detection kit (Thermo Scientific) and images were acquired using a Gel Doc™ XR+ System (Bio-Rad Laboratories, Inc., California, USA). At last, images were analyzed using Image Lab Analysis Software, and the results were expressed as density values that were normalized to β-actin.
Statistical analysis
All quantitative variables were expressed as mean ± standard error of the mean (SEM). The data statistical analyses were performed using GraphPad Prism software (GraphPad Software Inc., version 6.0) (California, USA). Data differences between multiple groups were measured using one-way ANOVA, followed by Tukey’s multiple comparison test to determine the differences between two groups. P values of < 0.05 was statistically significant.
Results
Ang II–induced apoptosis of primary rat CMECs
We used TUNEL staining to detect the apoptosis index of CMECs induced by Ang II. The results showed that Ang II significantly increased the apoptosis of CMECs in a concentration-dependent manner (Fig. 1A, B). Western blot analysis reviewed that Ang II promoted the protein expressions of pro-apoptotic proteins Bax and cleaved caspase-3 and the cleaved caspase-3/caspase-3 ratio, as well as inhibited the expression of anti-apoptotic protein Bcl-2 and the Bcl-2/Bax ratio in a concentration-dependent manner (Fig. 1C, D). The above results indicate that Ang II can induce CMEC apoptosis in a concentration-dependent manner.

Ang II induced the apoptosis of primary rat CMECs in a dose-dependent manner. After CMECs were treated with different concentrations of Ang II (0, 1, 2, and 4 nM) for 48 h, we used TUNEL staining to detect the apoptosis index and Western blot analysis to measure the expression of apoptosis-related proteins. (A, B) TUNEL staining showed that Ang II increased the apoptosis index of CMECs in a concentration-dependent manner. (C, D) Western blot results indicated that Ang II decreased the Bcl-2/Bax ratio and increased the cleaved caspase-3/caspase-3 ratio. Values are mean ± SEM (n = 4–6 per group). Statistical analysis was evaluated by one-way ANOVA with Tukey’s multiple comparison test. *P < 0.05, **P < 0.01. Scale bars: A = 100 μm.
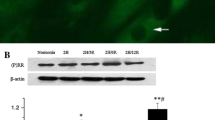
Ang II upregulated the PTP1B protein expression of CMECs and inhibited PI3K/Akt signaling
To explore the role of PTP1B in Ang II–induced CMEC apoptosis, we detected the protein expression changes of PTP1B using WB. The results showed that PTP1B protein expression was significantly upregulated in Ang II–treated CMECs in a dose-dependent manner (Fig. 2A). In addition, protein expressions of p-Akt and PI3K were significantly downregulated in a dose-dependent manner after Ang II stimulation (Fig. 2B, C). Finally, we chose the concentration (4 nM) as an ideal concentration for Ang II–induced apoptosis used for subsequent experiments.

Ang II induced PTP1B protein expressions of CMECs and inhibited the PI3K/Akt pathway. CMECs were treated with different doses of Ang II (0, 1, 2, and 4 nM) for 48 h. Western blot analysis was used to examine the related protein expressions. (A) Ang II induced the protein expression of PTP1B in CMECs. (B) Effects of Ang II on the protein expression of PI3K. (C) Expression of p-Akt/Akt in response to Ang II treatment. Values are mean ± SEM (n = 5 per group). Statistical analysis was evaluated by one-way ANOVA with Tukey’s multiple comparison test. *P < 0.05, **P < 0.01.
Inhibition of PTP1B decreased CMEC apoptosis induced by Ang II
TCS 401 (8 μM) was used to inhibit PTP1B expression. The results showed that, compared with the control group, Ang II significantly increased PTP1B protein expression, the apoptosis index, and the caspase-3 activity, while these effects were partly reversed through inhibiting the expression of PTP1B (Fig. 3A–D). Moreover, the anti-apoptotic factor Bcl-2/Bax ratio was decreased and the pro-apoptotic factor cleaved caspase-3/caspase-3 ratio was increased after Ang II stimulation, while TCS 401 treatment significantly attenuated these effects (Fig. 3E, F). These results indicated that Ang II induces the apoptosis of CMECs in a PTP1B-dependent manner.

Inhibition of PTP1B decreased CMEC apoptosis induced by Ang II. After CMECs were treated with or without Ang II (4 nM) and TCS 401 (8 μM) for 48 h, we used TUNEL staining to detect the apoptosis index and Western blot analysis to measure the expression of PTP1B and apoptosis-related proteins. (A, B) The apoptosis index was measured by TUNEL staining. (C) Caspase-3 activity quantitative analysis. (D) PTP1B protein expression in CMECs was measured by Western blot analysis. (E) The ratio of anti-apoptotic factor Bcl-2/Bax was detected by Western blot analysis. (F) The pro-apoptotic factor cleaved caspase-3/caspase-3 ratio was measured by Western blot analysis. Values are mean ± SEM (n = 4–6 per group). Statistical analysis was evaluated using one-way ANOVA with Tukey’s multiple comparison test. *P < 0.05, **P < 0.01. Scale bars: A = 100 μm.
The PI3K/Akt pathway was activated by a PTP1B inhibitor
Above data revealed that the expressions of p-Akt and PI3K were downregulated after Ang II stimulation, while the expression of PTP1B protein was enhanced. We further explored whether the PI3K/Akt pathway was involved in the PTP1B-mediated apoptosis of CMECs after Ang II stimulation. The Western blot results showed that PI3K expression and Akt phosphorylation levels were decreased in Ang II–treated cells, while these effects could be partly reversed by cotreatment with a PTP1B inhibitor (Fig. 4A, B). This data indicates that PI3K/Akt pathway might be the downstream signaling pathway of activated PTP1B after Ang II treatment, and Ang II can induce CMEC apoptosis via regulating the PTP1B/PI3K/Akt pathway.

The PI3K/Akt pathway was activated by a PTP1B inhibitor. After CMECs were treated with or without Ang II (4 nM) and TCS 401 (8 μM) for 48 h, Western blot analysis was used to measure the protein expression of (A) PI3K and (B) Akt phosphorylation. Values are mean ± SEM (n = 5 per group). Statistical analysis was evaluated by one-way ANOVA with Tukey’s multiple comparison test. *P < 0.05, **P < 0.01.
LY294002, an inhibitor of the PI3K/Akt pathway, partly reduced the anti-apoptotic effect of the PTP1B inhibitor
To further detect the relationship between PTP1B and the PI3K/Akt pathway, we used a LY294002 inhibitor to suppress the PI3K/Akt pathway. The results showed that, compared with the Ang II + TCS 401 group, further LY294002 treatment significantly increased the apoptosis index and reduced PI3K expression and Akt phosphorylation (Fig. 5A–D). It also promoted CMEC apoptosis by decreasing the Bcl-2/Bax ratio and increasing the cleaved caspase-3/caspase-3 ratio (Fig. 5E, F). This data further suggests that PI3K/Akt pathway might be the downstream signaling of activated PTP1B induced by Ang II, and Ang II might induce CMEC apoptosis via regulating the PTP1B/PI3K/Akt pathway.

LY294002, an inhibitor of the PI3K/Akt pathway, partly reduced the anti-apoptotic effect of the PTP1B inhibitor. CMECs were treated with or without Ang II (4 nM), TCS 401 (8 μM), and LY294002 (10 μM) for 48 h. (A, B) The apoptosis index was measured by TUNEL staining. (C, D) The protein expressions of PI3K and Akt phosphorylation were measured by Western blot analysis. (E, F) Western blot analysis was also used to measure the protein expression of apoptosis family members Bcl-2, Bax, cleaved caspase-3, and caspase-3. Values are mean ± SEM (n = 5 per group). Statistical analysis was evaluated by one-way ANOVA with Tukey’s multiple comparison test. *P < 0.05, **P < 0.01.
Discussion
In this study, we showed that Ang II triggered the apoptotic response in primary rat CMECs. Our results further showed that Ang II induced PTP1B expression and the subsequent increase on the expression of pro-apoptosis signaling was responsible for the enhanced apoptosis after Ang II treatment. These results are consistent with the previous finding that PTP1B inhibitors possess beneficial effects on improving endothelial dysfunction (Thiebaut et al. 2018). In addition, our data extended the previous findings and suggests that the vascular protective effects of PTP1B deletion might be mediated and dependent on the effect on endothelial cell apoptosis via activating the PI3K/Akt pathway.
Disruption of coordinated tissue growth and elimination may contribute to the progression from adaptive left ventricular (LV) hypertrophy to heart failure and the increased workload accompanied by ventricular remodeling (Hein et al. 2003). Recently, it has been shown that a reduction in cardiac capillary density (microvascular rarefaction) significantly promoted contractile dysfunction in transgenic mice (Shiojima et al. 2005). Endothelial cell apoptosis is found to be the main mechanism of microvascular rarefaction. RAAS, traditionally viewed as an endocrine system, functions as a regulator of apoptosis in a variety of cell types through both paracrine and autocrine mechanisms (Wu et al. 2016; Chang et al. 2018). RAAS was activated in the setting of heart failure and contributed to the progression of the heart failure process in experimental and clinical studies (Brilla et al. 1995; Felder et al. 2001; Mentz et al. 2015; Nijst et al. 2017). Furthermore, numerous studies showed that the inhibition of RAAS was beneficial in improving cardiac function and attenuating cardiac remodeling in animal heart failure models and in patients with heart failure (Aird 2007; Gao et al. 2014; Gordon et al. 2014; Ponikowski et al. 2016; Suematsu et al. 2016). Until now, the effect of Ang II on the apoptosis of well-differentiated cardiac microvascular endothelial cells has not yet been determined. Human umbilical vein endothelial cell and other kinds of endothelial cell lines respond differently to Ang II in vivo (Aird 2007). Under normal conditions, the concentration for induction of apoptosis in these cell lines is far above the plasma Ang II concentration (Filippatos et al. 2001). In a preliminary experiment, we tried higher concentrations of Ang II (1, 2, and 4 μM) reported in other literature to observe the expression of protein PTP1B during the process of apoptosis(Liu et al. 2018; Yang et al. 2019), but the results were unsatisfactory. Subsequently, we continued to search the relevant literature and found that the concentration of Ang II (> 100 pg/ml, i.e., > 0.1 nM) in HF patients was much less than what we used (Dzau et al. 1984; Hisatake et al. 2017), so we decided to try lower concentration. What is exciting is that we have reaped exciting results, as expected, after decreasing the order of magnitude. In agreement with the previous hypothesis, stimulation with Ang II (4 nM) for 48 h induced an obvious increase in the apoptotic effect of CMECs (i.e., increased apoptosis index, caspase-3 activity, and cleaved caspase-3/caspase-3 ratio, as well as decreased Bcl-2/Bax ratio) and the protein expression of PTP1B.
PTP1B is a ubiquitously expressed protein present in all vascular cell types (Vercauteren et al. 2006). Studies demonstrated that PTP1B is highly expressed in endothelial cells, and elevated levels of PTP1B were evidenced in endothelial progenitor cells isolated from obese individuals (Heida et al. 2010). Reports demonstrated that endothelial PTP1B controls vascular tree formation through the regulation of VEGFR2 signaling (Lanahan et al. 2014). Pharmacological inhibition or systemic gene deletion of PTP1B has been shown to protect against endothelial dysfunction associated with type 1 diabetes (Herren et al. 2015). Similarly, endothelial PTP1B has also been reported to contribute to adhesion molecule signaling and to regulate vascular inflammation and eosinophil recruitment (Berdnikovs et al. 2013). Additionally, previous studies have implicated the role of PTP1B in apoptosis. Apoptosis was found to be reduced in mice lacking endothelial PTP1B (Maupoint et al. 2016). PTP1B deficiency in hepatocytes was shown to confer resistance to apoptosis by altering the balance of pro-apoptotic and anti-apoptotic members of the Bcl-2 family (Gonzalez-Rodriguez et al. 2007), and small interference RNA against PTP1B reduced FasR-induced caspase-3 and caspase-8 activation following the exposure of neonatal rat cardiac myocytes to hypoxia re-oxygenation (Song et al. 2008). However, it remains largely unknown whether endothelial PTP1B expression changed along with the apoptosis induced by Ang II and whether the change in PTP1B expression could affect heart function. Our data indicates that PTP1B protein levels and CMEC apoptosis are increased after Ang II stimulation, which is consistent with recent reports that elevated PTP1B expression was observed in apoptotic cardiomyocytes induced by hypoxia/re-oxygenation (Song et al. 2008). This data collectively suggests that the increase of PTP1B might be associated with the increased apoptosis of CMECs and cardiomyocytes. Additionally, PTP1B is a known major molecule in insulin signal transduction (Stull et al. 2012) and is reported to negatively regulate insulin transduction by dephosphorylating insulin receptors (IR) and insulin receptor substrates (IRS) (Salmeen and Barford 2005). PI3K is a member of the insulin signaling system (Lungkaphin et al. 2014), along with IR and IRS, and PI3K was also modulated by them. Given this, we first examined the association of PTP1B with the PI3K/Akt signaling pathway. Our results demonstrate that the increased PTP1B expression is associated with a decreased expression of PI3K p85 in the Ang II group; while cotreatment with the PTP1B inhibitor could downregulate PTP1B expression and increase PI3K p85 expression and the phosphorylation of Akt after Ang II stimulation. These data suggest that PTP1B can regulate the PI3K/Akt pathway.
The PI3K/Akt signaling pathway participates in various cellular processes, such as cell survival, proliferation, apoptosis, and tube formation (Jung et al. 2012; Matson et al. 2017). A previous study showed that isoflurane promoted PI3K/Akt activation, upregulated B cell lymphoma 2 (Bcl-2)–associated X protein and Bcl-2 expression levels, and reduced the expression levels of caspase-3 and caspase-8 in myocardial cells (Pi et al. 2018). Caspase-3 is a crucial enzyme of the apoptotic pathway, and its activation is a central aspect of caspase-dependent apoptosis (Chan et al. 2010). In addition, the Bcl-2 family can regulate the mitochondrial apoptotic pathway and determine whether mitochondria could start apoptosis or not (Asmarinah et al. 2014). Bcl-2 is an anti-apoptotic protein, while Bax is a pro-apoptotic protein; therefore, they constitute a critical intracellular checkpoint for apoptosis (Wallgren et al. 2013). In the present study, we observed a significant increase of caspase-3 activity and decrease of Bcl-2/Bax ratio in CMECs subjected to Ang II, along with upregulated PTP1B expression and a downregulated PI3K/Akt pathway. While treated with PTP1B inhibitor significantly reversed the pro-apoptotic effect of Ang II in CMECs and activated the PI3K/Akt pathway. Based on the above results, we speculate that Ang II could induce CMEC apoptosis via activating PTP1B while downregulating PI3K/Akt pathway. In this study, we further used the LY294002 (PI3K inhibitor)-inhibited PI3K/Akt pathway; LY294002 is a potent inhibitor of PI3K p85 (Jiang et al. 2018; Pei et al. 2018) and it can decrease the expression of phosphorylated Akt (Ser473), inhibit growth, and induce apoptosis. And the results showed that the anti-apoptotic effect of the PTP1B inhibitor was attenuated by the PI3K inhibitor.
Conclusion
In conclusion, the present study demonstrated, for the first time that, Ang II induced apoptosis of CMECs via upregulating PTP1B and the inhibition of PTP1B might reduce Ang II–induced CMEC apoptosis via activating the PI3K/Akt pathway. This data suggests that PTP1B inhibition might be a potential novel therapeutic avenue for coronary microvascular disease and heart failure.
References
Aird WC (2007) Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res 100(2):174–190
Akasaki Y, Liu G, Matundan HH, Ng H, Yuan X, Zeng Z, Black KL, Yu JS (2006) A peroxisome proliferator-activated receptor-gamma agonist, troglitazone, facilitates caspase-8 and -9 activities by increasing the enzymatic activity of protein-tyrosine phosphatase-1B on human glioma cells. J Biol Chem 281(10):6165–6174
Asmarinah A, Paradowska-Dogan A, Kodariah R, Tanuhardja B, Waliszewski P, Mochtar CA, Weidner W, Hinsch E (2014) Expression of the Bcl-2 family genes and complexes involved in the mitochondrial transport in prostate cancer cells. Int J Oncol 45(4):1489–1496
Berdnikovs S, Abdala-Valencia H, Cook-Mills JM (2013) Endothelial cell PTP1B regulates leukocyte recruitment during allergic inflammation. Am J Physiol Lung Cell Mol Physiol 304(4):L240–L249
Brilla CG, Rupp H, Funck R, Maisch B (1995) The renin-angiotensin-aldosterone system and myocardial collagen matrix remodelling in congestive heart failure. Eur Heart J 16(Suppl O):107–109
Bulluck H, Yellon DM, Hausenloy DJ (2016) Reducing myocardial infarct size: challenges and future opportunities. Heart 102(5):341–348
Chan YY, Chang CS, Chien LH, Wu TF (2010) Apoptotic effects of a high performance liquid chromatography (HPLC) fraction of Antrodia camphorata mycelia are mediated by down-regulation of the expressions of four tumor-related genes in human non-small cell lung carcinoma A549 cell. J Ethnopharmacol 127(3):652–661
Chang RL, Chang CF, Ju DT, Ho TJ, Chang TT, Lin JW, Li JC, Cheng SM, Day CH, Viswanadha VP, Huang CY (2018) Short-term hypoxia upregulated Mas receptor expression to repress the AT1 R signaling pathway and attenuate Ang II-induced cardiomyocyte apoptosis. J Cell Biochem 119(3):2742–2749
Davignon J, Ganz P (2004) Role of endothelial dysfunction in atheerosclerosis. Circulation 109(23 Suppl 1):Iii27–Iii32
Day RM, Lee YH, Han L, Kim YC, Feng YH (2011) Angiotensin II activates AMPK for execution of apoptosis through energy-dependent and -independent mechanisms. Am J Physiol Lung Cell Mol Physiol 301(5):L772–L781
Dzau VJ, Packer M, Lilly LS, Swartz SL, Hollenberg NK, Williams GH (1984) Prostaglandins in severe congestive heart failure. Relation to activation of the renin--angiotensin system and hyponatremia. N Engl J Med 310(6):347–352
Felder RB, Francis J, Weiss RM, Zhang ZH, Wei SG, Johnson AK (2001) Neurohumoral regulation in ischemia-induced heart failure. Role of the forebrain. Ann N Y Acad Sci 940:444–453
Filippatos GS, Gangopadhyay N, Lalude O, Parameswaran N, Said SI, Spielman W, Uhal BD (2001) Regulation of apoptosis by vasoactive peptides. Am J Physiol Lung Cell Mol Physiol 281(4):L749–L761
Fyhrquist F, Metsarinne K, Tikkanen I (1995) Role of angiotensin II in blood pressure regulation and in the pathophysiology of cardiovascular disorders. J Hum Hypertens 9(Suppl 5):S19–S24
Gao Y, Gao JP, Chen CX, Wang HL, Guo J, Wu R (2014) Beneficial effects of houttuynin on ventricular remodeling induced by coronary artery ligation in rats. Eur J Pharmacol 740:200–208
Gonzalez-Rodriguez A, Escribano O, Alba J, Rondinone CM, Benito M, Valverde AM (2007) Levels of protein tyrosine phosphatase 1B determine susceptibility to apoptosis in serum-deprived hepatocytes. J Cell Physiol 212(1):76–88
Gordon O, He Z, Gilon D, Gruener S, Pietranico-Cole S, Oppenheim A, Keshet E (2014) A transgenic platform for testing drugs intended for reversal of cardiac remodeling identifies a novel 11betaHSD1 inhibitor rescuing hypertrophy independently of re-vascularization. PLoS One 9(3):e92869
Harrison DG, Cai H, Landmesser U, Griendling KK (2003) Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J Renin Angiotensin Aldosterone Syst 4(2):51–61
Hedhli N, Huang Q, Kalinowski A, Palmeri M, Hu X, Russell RR, Russell KS (2011) Endothelium-derived neuregulin protects the heart against ischemic injury. Circulation 123(20):2254–2262
Heida NM, Leifheit-Nestler M, Schroeter MR, Muller JP, Cheng IF, Henkel S, Limbourg A, Limbourg FP, Alves F, Quigley JP et al (2010) Leptin enhances the potency of circulating angiogenic cells via src kinase and integrin (alpha)vbeta5: implications for angiogenesis in human obesity. Arterioscler Thromb Vasc Biol 30(2):200–206
Hein S, Arnon E, Kostin S, Schonburg M, Elsasser A, Polyakova V, Bauer EP, Klovekorn WP, Schaper J (2003) Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation 107(7):984–991
Herren DJ, Norman JB, Anderson R, Tremblay ML, Huby AC, Belin de Chantemele EJ (2015) Deletion of protein tyrosine phosphatase 1B (PTP1B) enhances endothelial cyclooxygenase 2 expression and protects mice from type 1 diabetes-induced endothelial dysfunction. PLoS One 10(5):e0126866
Hisatake S, Kiuchi S, Kabuki T, Oka T, Dobashi S, Ikeda T (2017) Serum angiotensin-converting enzyme 2 concentration and angiotensin-(1-7) concentration in patients with acute heart failure patients requiring emergency hospitalization. Heart Vessel 32(3):303–308
Jiang J, Xu Y, Ren H, Wudu M, Wang Q, Song X, Su H, Jiang X, Jiang L, Qiu X (2018) MKRN2 inhibits migration and invasion of non-small-cell lung cancer by negatively regulating the PI3K/Akt pathway. J Exp Clin Cancer Res 37(1):189
Jung KH, Choi MJ, Hong S, Lee H, Hong SW, Zheng HM, Lee HS, Hong S, Hong SS (2012) HS-116, a novel phosphatidylinositol 3-kinase inhibitor induces apoptosis and suppresses angiogenesis of hepatocellular carcinoma through inhibition of the PI3K/AKT/mTOR pathway. Cancer Lett 316(2):187–195
Lanahan AA, Lech D, Dubrac A, Zhang J, Zhuang ZW, Eichmann A, Simons M (2014) PTP1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation 130(11):902–909
Li F, Yuan Y, Guo Y, Liu N, Jing D, Wang H, Guo W (2015) Pulsed magnetic field accelerate proliferation and migration of cardiac microvascular endothelial cells. Bioelectromagnetics 36(1):1–9
Li JM, Mullen AM, Shah AM (2001) Phenotypic properties and characteristics of superoxide production by mouse coronary microvascular endothelial cells. J Mol Cell Cardiol 33(6):1119–1131
Liu L, Meng L, Zhang P, Lin H, Chi J, Peng F, Guo H (2018) Angiotensin II inhibits the protein expression of ZO1 in vascular endothelial cells by downregulating VE-cadherin. Mol Med Rep 18(1):429–434
Lungkaphin A, Arjinajarn P, Pongchaidecha A, Srimaroeng C, Chatsudthipong L, Chatsudthipong V (2014) Impaired insulin signaling affects renal organic anion transporter 3 (Oat3) function in streptozotocin-induced diabetic rats. PLoS One 9(5):e96236
Marshall RP, Gohlke P, Chambers RC, Howell DC, Bottoms SE, Unger T, McAnulty RJ, Laurent GJ (2004) Angiotensin II and the fibroproliferative response to acute lung injury. Am J Physiol Lung Cell Mol Physiol 286(1):L156–L164
Matson DR, Hardin H, Buehler D, Lloyd RV (2017) AKT activity is elevated in aggressive thyroid neoplasms where it promotes proliferation and invasion. Exp Mol Pathol 103(3):288–293
Maupoint J, Besnier M, Gomez E, Bouhzam N, Henry JP, Boyer O, Nicol L, Mulder P, Martinet J, Richard V (2016) Selective vascular endothelial protection reduces cardiac dysfunction in chronic heart failure. Circ Heart Fail 9(4):e002895
Mentz RJ, Stevens SR, DeVore AD, Lala A, Vader JM, AbouEzzeddine OF, Khazanie P, Redfield MM, Stevenson LW, O’Connor CM et al (2015) Decongestion strategies and renin-angiotensin-aldosterone system activation in acute heart failure. JACC Heart Fail 3(2):97–107
Moorjani N, Westaby S, Narula J, Catarino PA, Brittin R, Kemp TJ, Narula N, Sugden PH (2009) Effects of left ventricular volume overload on mitochondrial and death-receptor-mediated apoptotic pathways in the transition to heart failure. Am J Cardiol 103(9):1261–1268
Nijst P, Verbrugge FH, Martens P, Bertrand PB, Dupont M, Francis GS, Tang WW, Mullens W (2017) Plasma renin activity in patients with heart failure and reduced ejection fraction on optimal medical therapy. J Renin Angiotensin Aldosterone Syst 18(3):1470320317729919
Ohta T, Eguchi R, Suzuki A, Miyakaze S, Ayuzawa R, Kaji K (2007) Hypoxia-induced apoptosis and tube breakdown are regulated by p38 MAPK but not by caspase cascade in an in vitro capillary model composed of human endothelial cells. J Cell Physiol 211(3):673–681
Pei L, Kong Y, Shao C, Yue X, Wang Z, Zhang N (2018) Heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to pharmorubicin by promoting autophagy via PI3K/Akt pathway. J Cell Mol Med 22(11):5311–5321
Peng N, Meng N, Wang S, Zhao F, Zhao J, Su L, Zhang S, Zhang Y, Zhao B, Miao J (2014) An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E(-)/(-) mice. Sci Rep 4:5519
Pi Z, Lin H, Yang J (2018) Isoflurane reduces pain and inhibits apoptosis of myocardial cells through the phosphoinositide 3-kinase/protein kinase B signaling pathway in mice during cardiac surgery. Mol Med Rep 17(5):6497–6505
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, Gonzalez-Juanatey JR, Harjola VP, Jankowska EA et al (2016) 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 18(8):891–975
Salmeen A, Barford D (2005) Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid Redox Signal 7(5–6):560–577
Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K (2005) Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 115(8):2108–2118
Smiljic S (2017) The clinical significance of endocardial endothelial dysfunction. Medicina (Kaunas) 53(5):295–302
Song H, Zhang Z, Wang L (2008) Small interference RNA against PTP-1B reduces hypoxia/reoxygenation induced apoptosis of rat cardiomyocytes. Apoptosis 13(3):383–393
Stull AJ, Wang ZQ, Zhang XH, Yu Y, Johnson WD, Cefalu WT (2012) Skeletal muscle protein tyrosine phosphatase 1B regulates insulin sensitivity in African Americans. Diabetes 61(6):1415–1422
Suematsu Y, Miura S, Goto M, Matsuo Y, Arimura T, Kuwano T, Imaizumi S, Iwata A, Yahiro E, Saku K (2016) LCZ696, an angiotensin receptor-neprilysin inhibitor, improves cardiac function with the attenuation of fibrosis in heart failure with reduced ejection fraction in streptozotocin-induced diabetic mice. Eur J Heart Fail 18(4):386–393
Suthahar N, Meijers WC, Sillje HHW, de Boer RA (2017) From inflammation to fibrosis-molecular and cellular mechanisms of myocardial tissue remodelling and perspectives on differential treatment opportunities. Curr Heart Fail Rep 14(4):235–250
Thiebaut P-A, Besnier M, Gomez E, Richard V (2016) Role of protein tyrosine phosphatase 1B in cardiovascular diseases. J Mol Cell Cardiol 101:50–57
Thiebaut PA, Delile E, Coquerel D, Brunel JM, Renet S, Tamion F, Richard V (2018) Protein tyrosine phosphatase 1B regulates endothelial endoplasmic reticulum stress; role in endothelial dysfunction. Vasc Pharmacol 109:36–44
Tsou RC, Zimmer DJ, De Jonghe BC, Bence KK (2012) Deficiency of PTP1B in leptin receptor-expressing neurons leads to decreased body weight and adiposity in mice. Endocrinology 153(9):4227–4237
van Riet EE, Hoes AW, Wagenaar KP, Limburg A, Landman MA, Rutten FH (2016) Epidemiology of heart failure: the prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur J Heart Fail 18(3):242–252
Vercauteren M, Remy E, Devaux C, Dautreaux B, Henry JP, Bauer F, Mulder P, Hooft van Huijsduijnen R, Bombrun A, Thuillez C et al (2006) Improvement of peripheral endothelial dysfunction by protein tyrosine phosphatase inhibitors in heart failure. Circulation 114(23):2498–2507
Wallgren M, Lidman M, Pedersen A, Brannstrom K, Karlsson BG, Grobner G (2013) Reconstitution of the anti-apoptotic Bcl-2 protein into lipid membranes and biophysical evidence for its detergent-driven association with the pro-apoptotic Bax protein. PLoS One 8(4):e61452
Wang J, Zhou J, Wang Y, Yang C, Fu M, Zhang J, Han X, Li Z, Hu K, Ge J (2017) Qiliqiangxin protects against anoxic injury in cardiac microvascular endothelial cells via NRG-1/ErbB-PI3K/Akt/mTOR pathway. J Cell Mol Med 21(9):1905–1914
Wu Z, Dai F, Ren W, Liu H, Li B, Chang J (2016) Angiotensin II induces apoptosis of human pulmonary microvascular endothelial cells in acute aortic dissection complicated with lung injury patients through modulating the expression of monocyte chemoattractant protein-1. Am J Transl Res 8(1):28–36
Xiao X, Xu S, Li L, Mao M, Wang J, Li Y, Wang Z, Ye F, Huang L (2017) The effect of velvet antler proteins on cardiac microvascular endothelial cells challenged with ischemia-hypoxia. Front Pharmacol 8:601
Yang D, Xiao C, Long F, Wu W, Huang M, Qu L, Liu X, Zhu Y (2019) Fra-1 plays a critical role in angiotensin II-induced vascular senescence. FASEB J 33(6):7603–7614
Funding
This study was supported by grants from the Program for the Outstanding Academic Leaders supported by Shanghai Science and Technology Commission (16XD1400700), National Basic Research Program of China (973 Program, 2012CB518605), National Natural Science Foundation of China (81370199) and Shanghai Sailing Program (19YF1406300).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international and national guidelines for the care and use of animals were followed. The experiments with animals were approved by the ethical committees of the Zhongshan Hospital Affiliated to Fudan University.
Informed consent
Informed consent was obtained from all authors who participated in the study.
Additional information
Editor: Tetsuji Okamoto
Rights and permissions
About this article

Cite this article
Wang, Y., Fan, Y., Song, Y. et al. Angiotensin II induces apoptosis of cardiac microvascular endothelial cells via regulating PTP1B/PI3K/Akt pathway. In Vitro Cell.Dev.Biol.-Animal 55, 801–811 (2019). https://doi.org/10.1007/s11626-019-00395-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-019-00395-8