
Abstract
Primary keratinocytes derived from human epidermis are widely used in tissue engineering and regenerative medicine. An important aspect in clinical applications is the preservation of human skin keratinocyte stem cells. However, it is difficult to expand the number of human skin keratinocyte stem cells, which are undifferentiated and highly proliferative in culture by using standard cell culture methods. It is even more difficult to identify them, since universal specific markers for human skin keratinocyte stem cells have not been identified. In this paper, we show a method to produce a large number of primary progenitor human skin keratinocytes by using our novel culture techniques. Primary human skin keratinocyte monolayers are cultured using twice the volume of medium without serum and lacking essential fatty acids. Once the cells reach 70–80% confluence, they begin to float up into the overlying medium and are called “epithelial pop-up keratinocytes (ePUKs)” allowing the cells to be passaged without the use of trypsin. We analyzed the properties of ePUKs by cell size, cell viability, immunocytofluorescence biomarker staining, and cell cycle phase distribution by fluorescence-activated cell sorting (FACS). Our results showed that these ePUKs appear to be progenitor epithelial cells, which are small in size, undifferentiated, and have a high proliferative capacity. We believe that ePUKs are suitable for use in medical applications requiring a large number of primary human progenitor skin keratinocytes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Keratinocytes derived from human epidermis, oral mucosa, and urothelium play important roles in the construction of cell-based tissue engineering devices used for soft tissue reconstruction and regeneration. In clinical applications, such as wound healing, large quantities of keratinocytes are needed at one time to manufacture tissue-engineered devices to repair burn injuries (Duncan et al. 2005). The most critical part in cell-based tissue engineering is to obtain a large numbers of stem cells. However, native keratinocyte stem cells (KSCs) represent between 1 and 10% of the basal cells and have been defined as slow cycling (Li and Kaur. 2005, Cerqueira et al. 2014) with a high proliferative potential (Jones and Watt. 1993). Thus, it becomes necessary to produce a large number of KSCs in vitro for use in clinical applications.
It is difficult to use standard tissue culture techniques to produce sufficient KSCs by continuous passaging to expand cell number because stem cells begin to lose their stem cell characteristics during the passage process (Choi et al. 2015). Additionally, specific markers for KSCs have not been identified making isolation and purification difficult (Metral et al. 2017).
In this paper, we utilized a technique developed by our laboratory to expand primary progenitor human keratinocyte and characterized these cells based on their stem cell potential. This unique technique can produce large numbers of epithelial cells containing progenitor cells derived from human epidermis, by growing monolayers under a large volume of serum-free, essential fatty acid free, low calcium medium that is replenished every 24 h. The floating cells produced by this technique are defined as epithelial pop-up keratinocytes (ePUKs) (Marcelo et al. 2012). A similar observation was identified by Chaffer et al. (2011) using human breast epithelial cells. They showed that differentiated mammary epithelial cells can convert to a stem-like state doing so in an apparent stochastic manner in vitro. These cells are basal-like human mammary epithelial cells that came from differentiated cells in culture and floated on top of existing cell monolayers. Conventionally, trypsin is used to dissociate monolayer cells for passaging and the characteristic of the cells can be effected by trypsin treatment (Huang et al. 2010, Choi et al. 2015) However, our technique of producing ePUKs does not require trypsin for subculture. ePUKs can be produced by monolayers when the cells reach 70–80% confluence. The importance of this approach is the characteristics of KSCs can be maintained without physiological change during our culture technique since we do not use trypsin to passage cells; thus, the original cell membrane phenotypes are retained. We believe it is essential to expand these progenitor/stem cells without damage, still possessing the original properties of stem cells, for clinical applications. We characterized the ePUKs using cell size, cell viability, human stem cell markers, and cell cycle phase distribution. Our results indicate differences between traditional primary human skin keratinocyte-cultured monolayer cells and ePUKs. For practical applications, the ePUK technology can be used to produce devices for wound healing and tissue engineering/regenerative medicine applications faster and in higher numbers than with other available traditional culture techniques.
Materials and Methods
Establishment of primary skin keratinocytes culture from human native skin
The Institutional Review Board at the University of Michigan approved the use of the skin, and donors provided informed consent for research use. Full-thickness human skin (from a breast reduction surgery) came from surgically discarded tissues obtained from five different individuals (females; 19, 34, 36, 58, 67 yr old) (N = 5). Establishment of cell strains of keratinocytes was performed as previously reported (Marcelo et al. 1992). Initially, skin was washed in a solution of Dulbecco’s phosphate-buffered saline (DPBS) containing 125 μg/mL gentamicin (ThermoFisher Scientific, Waltham, MA) and 1 μg/mL fungizone (Gibco®, Gaithersburg, MD). Then, tissue samples were digested overnight at room temperature in a 0.125% trypsin solution prepared with trypsin powder (Sigma-Aldrich, St Louis, MO) and DPBS (1×) without calcium or magnesium. After 16 h, the trypsin solution was aspirated completely and the tissue was resuspended in chemically defined medium (EpiLife®) supplemented with defined growth factors (EDGS) (Gibco®) 0.06 mmol/L calcium chloride (Gibco®), 27 μg/mL gentamicin, 0.228 μg/mL fungizone, and 10% fetal bovine serum (FBS). The epidermal layer was scraped down to the basal layer with a no. 21 scalpel. The resulting cell suspension was filtered through 200 μm filters and plated at 2.0–3.0 × 106 cells/mL with medium containing 2% FBS. Medium was changed to serum-free medium after 24 h.
Standard monolayer, ePUKs, and ePUK monolayer cell cultures
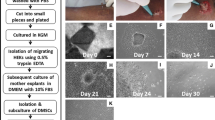
Enzymatically dissociated primary skin keratinocytes from human breast skin of 2.0–3.0 × 107 cells were seeded in T-75 flasks for primary skin keratinocytes standard monolayer (SM) and ePUKs (PP) cultures. The cells were grown in serum-free medium at 37°C, 5% CO2, and atmospheric oxygen. SM culture flasks were fed 15 mL complete medium that was changed every 48 h until 80% confluence was reached. By contrast, PP culture flasks were fed 30 mL medium every 24 h until starting to produce ePUKs. For experiments, monolayer cells of SM culture were passed with trypsin/EDTA (Sigma-Aldrich) into another T-75 flask at 80% confluence. At this point, the cell number, cell size, and cell viability were measured by Countess™ (Invitrogen, Carlsbad, CA) after staining with 0.4% trypan blue, and 1.0 × 106 cells were seeded in T-75 flasks fed with 15 mL complete medium for P1 SM culture when first passage cells of SM culture reached 80% confluence; they were harvested with enzyme-free cell dissociation solution (Gibco®) for immunocytofluorescence staining. For PP culture, monolayer cells continued to proliferate when they reach confluence and push keratinocytes upward, pop-up, into the overlying medium (Marcelo et al. 2012). The collected epithelial pop-up keratinocytes (ePUKs) were measured by the cell number, cell size, and cell viability. Collected ePUKs were split into two parts. One part of ePUKs was poured into new T-75 flasks (1.0 × 106 cells/30 mL) and forms the new culture defined as the PM culture. The second part of ePUKs was used for immunocytofluorescence staining. As for the PM culture, 30 mL medium was changed every 24 h same as PP culture. The culture protocol is shown in Fig. 1.

Experimental scheme of cell cultures and cells analyzed. SM: The monolayer cells harvested from primary skin keratinocyte standard monolayer culture, which was fed 15 mL serum-free medium every 2 d. PP: ePUKs collected from ePUK culture, which was fed 30 mL serum-free medium every single day. PM: The monolayer cells harvested from ePUK monolayer culture, which was fed 30 mL serum-free medium every single day.
Glucose Consumption
As an indicator of metabolic activity, spent and fresh medium from SM culture and the PP culture were measured for glucose consumption using a glucometer (BRS GM-100, BioReactor Sciences) every day after seeding the primary human skin keratinocytes.
Percentage of glucose consumption = the value (mg/dL) of (fresh media-spent media)/fresh media × 100. The fresh media contained 114 mg/dL of glucose which is close to the level of 110 mg/dL physiologic serum level seen in humans.
ePUK characterization
Cell suspension containing ePUKs in the spent medium and the cell suspensions containing monolayer cells from either SM culture or PM culture harvested with enzyme-free cell dissociation solution were counted and analyzed for size distribution and viability using a Countess® Automated cell counter (INVITROGEN, Carlsbad, CA) after staining with 0.4% trypan blue. The cells were then centrifuged onto slides using a Shandon Cytospin for further immunofluorescent biomarker evaluation.
Slide preparation (cytospin)
ePUKs from PP culture, the monolayer cells harvested by enzyme-free solution from SM culture, and PM culture were analyzed with immunofluorescence at cell density of 1.0 × 105 cells/mL. The slides for immunofluorescence were prepared by using cytospin 4® (ThermoShandon, Pittsburgh, PA). The cell suspensions were spun at 750 rpm for 5 min onto Shandon Cytoslide® using Thermo Shandon double cytology Funnel® (Fisher Scientific, Pittsburgh, PA). Four slides were prepared for each group.
Immunocytofluorescence
The cells were fixed in 4% paraformaldehyde (TED PELA, INC., Redding, CA) without methanol in PBS pH 7.4 for 15 min at room temperature and washed three times with 4°C PBS. After fixation, the slides were incubated for 10 min with PBS containing 0.25% Triton X-100 (Sigma-Aldrich) (PBST) and then, the cells were washed in PBS three times for 5 min. The cells were incubated in the diluted antibodies which are shown in Table 1 (cytokeratin 1, Ki67, involucrin (IVL), filaggrin (FLG), CD71, p63, Integrin alpa6, Lgr5) in 1% BSA in PBST inside a humidified chamber with buffer overnight at 4°C. The samples were then blocked with 1% BSA in PBST for 30 min at room temperature and were washed three times in PBS for 5 min each at room temperature. Then, slides were incubated with secondary antibody (Gout-Anti-Rabbit IgG Secondary Antibody, Alexa Fluor®488 and Goat-anti-Mouse IgG Secondary antibody, Alexa Fluor®594) in 1% BSA for 1 h at room temperature in the dark. After incubation, the secondary antibody solution was decanted and the slides were washed three times with PBS for 5 min each in dark. Counter staining was VECTASHIELD® Mounting Medium with DAPI (VECTOR LABORATORIES, Burlingame, CA). Pictures were taken of four sections on each slide using a Leica CTR 4000 (Leica Microsystems, Buffalo Grove, IL) and Lumen 200 (Prior Scientific, Rockland, MA). Each section was analyzed by software LAS V4.0 (Leica Microsystems).
The measurements of the secreted KGF/FGF-7 and FGF basic proteins by skin keratinocytes
The KGF/FGF-7 (KGF) and FGF basic (b-FGF) protein levels in the spent medium of SM culture and PP culture were determined by using a double-ligand enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN) per the manufacturer’s protocol. In brief, 100 μL of sample or standard in 100 μL of diluent was added to each well coated with each monoclonal antibody. Substrate solution was added and incubated with shield after washes. Absorbance was measured on a microplate reader at 450 nm after addition of stop solution. The concentration was determined in each sample by comparison to a standard curve. Each sample was tested in quadruplicate, and the final concentration was calculated per well.
Cell cycle analysis by FACS
For analysis of cell cycle phase distribution, SM and PP were harvested and washed twice with ice cold PBS. Cell pellets were fixed in 70% ethanol, treated with 100 μg/ml RNAse (Sigma-Aldrich), and stained with 50 μg/ml propidium iodide (Sigma-Aldrich) for 30 min at room temperature in the dark. Samples were put on ice with covered foil until analyzed. Cellular DNA was analyzed by flow cytometry (LSRFORTESSA; Becton Dickinson, San Jose, CA).
Statistical analysis
All data are presented as the mean ± standard deviation (SD). Paired t test was used for the results of ELISA. All other results were evaluated using one-way analysis of variance (ANOVA) followed by post hoc Tukey-Kramer test. A P value less than 0.05 was considered statistically significant.
Results
Glucose Consumption
The trend of glucose consumption between SM culture and PP culture was different (Fig. 2). Glucose consumption of PP culture started to be higher than that of SM culture after 3 d (~ 50% confluent) until day 7. In addition, at day 5 (~ 85% confluent), there were significant differences between SM culture and PP culture. At day 7, glucose consumption of PP culture was maximum but that of culture was still 58%. In this experiment, 72% was the maximum number we could calculate with the glucometer (BRS GM-100).

Glucose consumption of primary skin keratinocyte standard monolayer (SM) culture medium (white circle) and ePUK (PP) culture medium (black circle). Glucose consumption of PP culture was higher than SM culture 3 d after, and there was difference significantly at day 5 (around 85% confluent). Data are represented as means ± SD from five experiments when the same time point was compared. *P < 0.05.
Cell Size
The averaged cell sizes from all groups at all collections showed that the averaged cell size of ePUKs (PP) was the smallest among three groups with statistic difference. The averaged cell size of PM culture was smaller than SM culture even though the difference was not significant (Fig. 3a).


The characterization of ePUKs. Data are represented as means ± SD from five experiments. *P < 0.05. (a) the comparison of cell size between primary skin keratinocyte standard monolayer (SM), ePUKs (PP) and ePUK monolayer (PM) before using a Shandon Cytospin. Cell sizes of each group were the average of all data from each group. The cell size of PP was the smallest significantly. PM was still tended to be smaller than SM. (b) The average numbers of viable cells and cell size of ePUKs produced from ePUK (PP) culture each d from five cell strains. X-axis of each graph show days after seeding primary skin keratinocytes. ePUKs number of Y-axis was shown as each T75 flask. The ePUK size was the smallest when the number and viability were the highest (around day 5), when was around 85% confluent. (c, d, and e) (c) A complete monolayer of ePUK (PP) culture is shown with PP in overlying medium. (scale bar 200 μm). (d) PPwith spent medium just after seeding into new flask. (Scale bar 400 μm). (e) The monolayer of ePUKs monolayer (PM) culture 4 d after seeding. (Scale bar 400 μm).
ePUK characterization (Number, Viability, and Size)
The ePUK number and viability were the highest at day 5 (85% confluent). Approximately 3.8 × 106 ePUKs were produced from a T-75 flask. The cell size was the smallest at day 5 indicative of a high production number of small, highly viable ePUKs at 85% confluence. Viability and size seem to be related to confluence (Fig. 3b). Figure 3c showed PP culture. A large number of ePUKs could be seen on top of a complete monolayer. Cells of these ePUKs (1.0 × 106) were seeded with spent medium into new flask without the use of trypsin (Fig. 3d). Figure 3e showed the development of monolayer cells after ePUK seeding.
Growth Factor
Figure 4 showed the averaged concentration of growth factors per well. The level of keratinocyte growth factor (KGF) in the PP culture was significantly higher than SM culture at every stage from day 1 to day 7 (Fig. 4a). Level of b-FGF in the PP culture was significantly higher than SM culture at day 7, around 100% confluent of PP culture (Fig. 4b).

(a) The concentration of KGF at days 1, 4, and 7 after seeding P0 cells per well. (b) The concentration of b-FGF at days 1, 4, and 7 after seeding P0 cells per well. Data are represented as means ± SD from five experiments when two groups were compared at the same time point. *P < 0.05.
Immunocytofluorescence
Cytokeratin 1 (KRT 1) is a keratinocyte marker specifically expressed in the spinous and granular layers of the epidermis with family member keratin 10. All three culture methods exhibit the same level of KRT 1 (Fig. 5a). This suggested that PP and PM cells were identified as keratinocytes. Ki67 detects a nuclear antigen that is present only in proliferating cells (Gerdes et al. 1984). Ki67 has been reported as a specific marker of multiplying keratinocytes in human epidermis (Ando et al. 1990, Miyauchi et al. 1990, Hennen et al. 1998). From our results, all groups were almost 80% Ki67 positive. However, PPs were significantly lower than the other two groups (Fig. 5b).

The positive percentage (positive cell number/live cell number × 100) of each protein analyzed by immunofluorescence. Data is represented as means ± SD from five experiments. *P < 0.05. (a) Keratinocyte marker KRT1. (b) Proliferation marker Ki 67. (c and d) differentiation marker IVL and FLG. PP was the smallest significantly. (e–h) Stem cell markers CD71, ITGA6, Lgr5, and p63. PP was the highest significantly.
Involucrin (IVL) and filaggrin (FLG) are differentiation markers. IVL is an early differentiation marker which is present in the upper layers of spinous cells and is maintained in keratinocytes of the granular layer (Eckert et al. 2004). FLG is a terminal differentiation marker which is a structural protein specifically found in the granular and stratum corneum layers (Uchida et al. 2017). In this study, both of IVL and FLG of PP were significantly lower than the other two monolayer groups. In addition, PM culture of these differentiation markers tended to be lower than SM culture (Fig. 5c, d).
All of the stem cell markers CD71, ITGA6, Lgr5, and p63 were significantly higher in PP than the two monolayer cultures (Fig. 5e–h). In particular, our data showed that the CD71 level in PP was about eight times higher than that of SM culture (Fig. 5e), and ITGA6 in PP was more than three times higher than that of SM culture (Fig. 5f). In addition, these two stem cell markers, CD71 and ITG6, PM was significantly higher than SM (Fig. 5e, f).
Cell Cycle Analysis by FACS
G1 and G2 phase of SM were 36.56 and 34.06% (Fig. 6a). In contrast, these of PP were 91.81 and 1.62% (Fig. 6b).

Cell phase distribution of the monolayer of primary skin keratinocytes standard monolayer (SM) culture (a) and ePUKs (PP) (b). Dip G1 36.56%, dip G3 34.06%, dip S 29.38% (a). Dip G1 91.81%, dip G2 1.62%, dip S 6.57% (b).
Discussion
Epithelial cells, including those isolated from the epidermis consist of a family of cells: stem cells, progenitor or transitional cells, and differentiated cells. Stem cells can be defined as cells that have the potential to divide and to produce a replica cell as well as differentiated progeny and are thought to last the lifetime of the organism (Barrandon and Green, 1985). In the interfollicular epidermis (non-hair follicle) and in the oral mucosa and other epithelia, these cells are reported to have specific expression patterns of several cell surface markers (Webb et al. 2004), and in vitro are reported to be “small” in size, less than 11 μm (Barrandon and Green, 1985). Progenitor or transitional cells are dividing cells committed to differentiation, are larger (12–20 μm), and the differentiated cells are greater than 20 μm and have a distinctive “differentiated” appearance in culture.
We report a novel culture technique with epithelial cell strains that produce a high percentage of small diameter cells. The cells have increased proliferative potential and grow in a coordinated monolayer/suspension. The technology has a number of unusual manipulations: the cells are fed once a day, with 2× the amount of medium, which is serum and fatty acid free and contains low calcium levels. The monolayer started to push or pop, “floated” the progeny cells into the overlying medium when the confluence reached 85%. The spent medium cell suspension containing about 1,000,000 cells/T75 is poured into a new flask, by passing the need for trypsin, and cells then attach and form a new monolayer. Once the cells in the new flask reach 85% confluence, they produce ePUK cells, which can form a new monolayer upon transfer to another flask, expanding the cell strain over 1–2 mo time. Our novel technique appears to allow the cells to acquire an increase in plasticity and exhibit a wider differentiation potential.
Our observations show that PP cultures consumed higher glucose than SM culture after day 3 (50% confluence) (Fig. 2). The monolayer cells of PM culture reached confluence 3 d earlier than that of standard monolayer culture at day 7. As for PP culture, it was 85% confluent at day 5 with maximum glucose consumption which was a significant difference than seen in the SM culture. In contrast, it was still around 60% confluent for SM culture at day 5. These results emphasized that constant feeding (daily medium changes), which allows maintaining a near-constant level of physiologic glucose concentration, permitting the PM cultures to produce ePUK cells that are smaller and more undifferentiated than seen in SM cultures. This could be results of the low amounts of glucose in the medium that is limiting cell growth and encouraging cell differentiation resulting in an increase in cell size. Our data shows that the small cells consumed more glucose than larger cells which was consistent with the paper of Marcelo et al. 2012. In addition, cell size of PP was significantly smaller among the three groups and was smaller than SM culture even after attachment (PM culture) when we compared skin keratinocyte of SM culture to PP and PM culture (Fig. 3a). We consider this small cell size of PP share one of the common characteristic of KSCs. Small cell populations can give rise to clones with high frequency (Barrandon and Green. 1987, Li et al. 2008, Fujimori et al. 2009). This is consistent with our data that PM grew faster than skin keratinocyte in SM cultures. Higher growth potential of a small cell population is important for the regeneration of tissues which undergo self-renewal such as skin. Many researchers have tried to isolate small cell populations from various tissues using FACS (Golipoor et al. 2016; Zhang et al. 2016), nylon filters (Dutra and Bernard 1994), and density gradient centrifugation (Sun and Green 1976, Banks-Schlegel and Green 1981). However, these methods have been known to traumatize cells. Our technique can obtain small cells, without the use of trypsin thus keeping the cell membrane components intact.
The results of ePUKs characterization show that there is a relation between ePUK number, viability, and size. ePUK size was small when ePUK number and viability were high (around day 5) (Fig. 3b). From these results, one can obtain high-quality ePUKs (more than 6000 cells/mL after 100% confluent [data not shown]) which have a high potential of proliferation and differentiation at 85% confluence (around day 5). The number and viability of cells gradually decreased overtime and cell size was bigger after day 5.
When we compared spent medium between SM culture and PP culture, we found a major difference of KGF concentration. KGF concentration in both cultures was almost the same level at every stage even though PP culture was significantly higher than SM culture just 1 day after starting culture (Fig. 4a). In contrast, b-FGF concentration of PP culture was almost the same level as SM culture until day 4. However, the level of both monolayers was increased at day 7 with the PP culture being significantly higher than SM culture at day 7 (Fig. 4b). Furthermore, these levels of PP culture were gradually increased, almost threefold over the SM culture at day 14 (data not shown). It would, thus, appear that b-FGF is also related to the proliferation of monolayer of PP culture and ePUK production since it is known that it can stimulate keratinocyte migration and promote proliferation of keratinocytes (O'Keefe et al. 1988, Shipley et al. 1989, Sogabe et al. 2006). These concentrations were per culture not per cell, because the proliferation test was not done for this study. We have recognized that the PP culture can proliferate quicker and produce more ePUKs than the SM culture. Hence, it seems that the total cell number was different depending on the group even at the same time. These two growth factors of KGF and b-FGF are not enough evidence to explain the differences between SM and PP cultures at this time.
Ki67, a nuclear protein, is exclusively expressed in proliferating cells (Bullwinkle et al. 2006). This protein is expressed during all active cell cycles excluding the quiescent stage (Bruno and Darzynkiewicz. 1992). In our data, Ki67-positive cells in PP was significantly lower number than that of SM culture. This is because PP includes more quiescent stage cells than skin keratinocyte seen in the standard monolayer. This result was consistent with our cell phase distribution analysis. There were large differences between SM and PP. PP had almost 92% of G0/G1 stage cells even though SM had 37%. As for G2 stage, PP had only 1.6% whereas SM had 34% (Fig. 6a, b).
The levels of IVL and FLG showed similar patterns as Ki67 (Fig. 5b–d). This data demonstrated that PP are more undifferentiated cells, though, they still have the capacity to differentiate if placed in the appropriate environment (Natesan et al. 2011).
PP had the highest level for all evaluated stem cell markers among analyzed samples. Stem cell markers, CD71, ITGA6, and Lgr5 of PM, were significantly higher than those of SM except for p63. Parsa et al. (1999) reported keratinocytes p63 mRNA is confined to small cells known to be capable of DNA synthesis and colony formation. This is absent from the larger cells that are undergoing terminal differentiation and consistent with our data (Parsa et al. 1999). The differentiation marker of PM and putative stem cell marker, p63 were similar to the SM culture. This indicates that PP consisted of a small cell-undifferentiated population. Immunofluorescence results also suggested that PP had a more potential to be of a stem cell lineage than SM. Several studies reported that basal keratinocytes with phenotype ITGA6 high/CD71 low have important stem cell attributes (Li et al. 1998), and stem cells can divide asymmetrically and give rise to progenitor cells called transit amplifying (TA) cells that are actively proliferating cells that migrate from the lower suprabasal layers to the upper layers (Potten. 1981; Hall and Watt. 1989; Li et al. 1998). It has been reported that small-diameter cells have increased prolific capacity (Barrandon and Green. 1985) (Fujimori et al. 2009) and have an ITGA6 high/CD71 high phenotype that has been used as a marker of TA cells (Tani et al. 2000; Metral et al. 2017). From these reports, our immunofluorescence data suggests that ePUKs are proliferating cells, such as TA cells, maintained by the serum and fatty acid-free low calcium medium which impeded differentiation. A subpopulation of ePUKs could be progenitor/stem cell keratinocyte after analysis using appropriate biomarker profiles for identifying stem cells.
To enhance ePUK production further and make it more efficient and less labor-intense process, we have advocated the design and use of a bioreactor. These bioreactors could maintain the monolayers in active form for much longer periods of time by continuous feeding of the culture to maintain a constant physiologic glucose concentration instead of a single medium change every 24 h (Marcelo et al. 2012). In addition, the bioreactors would be set up to “capture” the floating ePUKs as they are produced.
Conclusion
Using this primary culture technique, large quantities of human primary epithelial cells, including progenitor cells, a population of small-diameter and undifferentiated cells, can be generated without the use of the enzyme trypsin to passage the cultures. The cells have basal progenitor cell characteristics induced by daily feeding with larger than normal medium volumes which maintain a physiologic level of glucose concentration. The ePUK epithelial cells have the potential to be used in regenerative medicine and for basic studies of epithelial progenitor cell phenotype in the future.
References
Ando M, Kawashima T, Kobayashi H, Ohkawara A (1990) Proliferating cells in the normal and psoriatic epidermis detected by Ki67 monoclonal antibody. J Invest Dermatol. 1:441–446
Banks-Schlegel S, Green H (1981) Involucrin synthesis and tissue assembly by keratinocytes in natural and cultured human epithelia. J Cell Biol 90:732–737
Barrandon Y, Green H (1985) Cell size as a determinant of the clone-forming ability of human keratinocytes. Proc Natl Acad Sci 82:5390–5394
Barrandon Y, Green H. (1987) Three clonal types of keratinocyte with different capacities for multiplication. 84:2302–2306
Bruno S, Darzynkiewicz Z (1992) Cell cycle dependent expression and stability of the nuclear protein detected by Ki-67 antibody in HL-60 cells. Cell Prolif 25:31–40
Bullwinkel J, Baron-Lühr B, Lüdemann A, Wohlenberg C, Gerdes J, Scholzen T (2006) Ki-67 protein is associated with ribosomal RNA transcription in quiescent and proliferating cells. J Cell Physiol 206:624–635
Cerqueira MT, Frias AM, Reis RL, Marques AP (2014) Boosting and rescuing epidermal superior population from fresh keratinocyte cultures. Stem Cells Dev 23:34–43
Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, Arendt LM, Kuperwasser C, Bierie B, Weinberg RA. (2011) Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci.10;108(19):7950–7955
Choi HR, Byun SY, Kwon SH, Park KC (2015) Niche interactions in epidermal stem cells. World J Stem Cells 26:495–501
Duncan CO, Shelton RM, Navsaria H et al (2005) In vitro transfer of keratinocytes: comparison of transfer from fibrin membrane and delivery by aerosol spray. J Biomed Mater Res B Appl Biomater 73:221–228
Dutra TF, Bernard GW (1994) Size-selective comparison of fetal calvarial versus adult marrow osteogenic colony-forming entities. Anat Rec 239:1–8
Eckert RL, Crish JF, Efimova T, Dashti SR, Deucher A, Bone F, Adhikary G, Huang G, Gopalakrishnan R, Balasubramanian S (2004) Regulation of involucrin gene expression. J Invest Dermatol. 123:13–22
Fujimori Y, Izumi K, Feinberg SE, Marcelo CL (2009) Isolation of small-sized human epidermal progenitor/stem cells by gravity assisted cell sorting (GACS). J Dermatol Sci 56:181–187
Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H (1984) Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 133:1710–1705
Golipoor Z, Mehraein F, Zafari F, Alizadeh A, Ababzadeh S, Baazm M (2016) Migration of bone marrow-derived very small embryonic-like stem cells toward an injured spinal cord. Cell J 17:639–647
Hall PA, Watt FM (1989) Stem cells: the generation and maintenance of cellular. Diversity 106:619–623
Hennen M, Thiriar S, Noel JC, Galand P (1998) Ki-67 immunostaining of normal human epidermis—comparison with 3H-thymidine labelling and PCNA immunostaining. Dermatology 197:123–126
Huang HL, Hsing HW, Lai TC, Chen YW, Lee TR, Chan HT, Lyu PC, Wu CL, Lu YC, Lin ST, Lin CW, Lai CH, Chang HT, Chou HC, Chan HL (2010) Research trypsin-induced proteome alteration during cell subculture in mammalian cells. J Biomed Sci 17:36. https://doi.org/10.1186/1423-0127-17-36
Jones PH, Watt FM (1993) Separation of human epidermal stem cells from transit amplifying cells on the basis of differences in integrin function and expression. Cell 73:713–724
Li A, Kaur P (2005) FACS enrichment of human keratinocyte stem cells. Methods Mol Biol 289:87–96
Li A, Simmons PJ, Kaur P (1998) Identification and isolation of candidate human keratinocyte stem cells based on cell surface phenotype. Proc Natl Acad Sci 95:3902–3907
Li J, Miao C, Guo W, Jia L, Zhou J, Ma B, Peng S, Liu S, Cao Y, Duan E (2008) Enrichment of putative human epidermal stem cells based on cell size and collagen type IV adhesiveness. Cell Res 18:360–371
Marcelo CL, Duell EA, Rhodes LM, Dunham WR (1992) In vitro model of essential fatty acid deficiency. J Invest Dermatol 99:703–708
Marcelo CL, Peramo A, Ambati A, Feinberg SE (2012) Characterization of a unique technique for culturing primary adult human epithelial progenitor/stem cells. BMC Dermatol 12(8). https://doi.org/10.1186/1471-5945-12-8
Metral E, Bechetoille N, Demarne F, Rachidi W, Damouret O (2017) α6 integrin (α6 high)/transferrin receptor (CD71) low keratinocyte stem cells are more potent for generating reconstructed skin epidermis than rapid adherent cells. Int J Mol Sci 18(2). https://doi.org/10.3390/ijms18020282
Miyauchi S, Hashimoyo K, Miki Y (1990) The innermost cell layer of the outer root sheath is positive with Ki-67. J Invest Dermatol. 95:393–396
Natesan S, Wrice LN, Baer GD, Christy JR (2011) Debrided skin as a source of autologous stem cells for wound repair. Stem Cells 29:1219–1230
O'Keefe EJ, Chiu M L, Payne RE Jr. (1988) Stimulation of growth of keratinocytes by basic fibroblast growth factor. J Invest Dermatol. 90:767–769
Parsa R, Yang A, McKeon F, Green H (1999) Association of p63 with proliferative potential in normal and neoplastic human keratinocytes. J Invest Dermatol. 113:1099–1105
Potten CS (1981) Cell replacement in epidermis (keratopoiesis) via discrete units of proliferation. Int Rev Cytol 69:271–318
Shipley GD, Keeble WW, Hendrickson JE, Coffey RJ Jr, Pittelkow MR (1989) Growth of normal human keratinocytes and fibroblasts in serum-free medium is stimulated by acidic and basic fibroblast growth factor. J Cell Physiol 138:511–518
Sogabe Y, Abe M, Yokoyama Y, Ishikawa O (2006) Basic fibroblast growth factor stimulates human keratinocyte motility by Rac activation. Wound Repair Regen 14:457–462
Sun TT, Green H (1976) Differentiation of the epidermal keratinocyte in cell culture: formation of the cornified envelope. Cell 9:511–521
Tani H, Morris RJ, Kaur P (2000) Enrichment for murine keratinocyte stem cells based on cell surface phenotype. Proc Natl Acad Sci 97:10960–10965
Uchida R, Aoki R, Aoki YA, Tajima A, Takayama Y (2017) Promoting effect of lactoferrin on barrier function and epithelial differentiation of human keratinocytes. Biochem Cell Biol 95:64–68
Webb A, Li A, Kaur P (2004) Location and phenotype of human adult keratinocyte stem cells of the skin. Differentiation 72(8):387–395
Zhang X, Hua R, Wang X, Huang M, Gan L, Wu Z, Zhang J, Wang H, Cheng Y, Li J, Guo W (2016) Identification of stem-like cells and clinical significance of candidate stem cell markers in gastric cancer. Oncotarget 7:9815–9831
Acknowledgements
We would like to thank Dr. Cynthia Marcelo for suggestions of our experiments. We also thank Eve Bingham and James Washington for their critical editing of this manuscript. The author, A.M., would like to thank Eve for teaching her skin keratinocyte culture methodology. This study was supported by NIH grants R01 DE 019431 and R01 DE 013417 to SEF.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The Institutional Review Board at the University of Michigan approved the use of the skin, and donors provided informed consent for research use.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Editor: Tetsuji Okamoto
Rights and permissions
About this article

Cite this article
Miyazawa, A., Kuo, S. & Feinberg, S.E. Production of progenitor cells from primary human epithelial cell monolayer cultures. In Vitro Cell.Dev.Biol.-Animal 54, 413–422 (2018). https://doi.org/10.1007/s11626-018-0259-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-018-0259-1