
Abstract
Neuroblastoma cells are neural crest derivatives that can differentiate into neuron-like cells in response to exogenous agents, and are known to be particularly sensitive to retinoic acid. The spectrum of neuroblastoma responses, ranging from proliferation, migration, differentiation, or apoptosis, is difficult to predict due to the heterogeneity of these tumors and to the broad effective range of retinoic acid. Our study focused on the effects of nanomolar concentrations of retinoic acid on neuroblastoma differentiation in two cell lines cells: SK-N-SH (HTB-11) and IMR-32. Each cell line was treated with retinoic acid from 1 to 100 nM for up to 6 d. Morphological changes were quantified; immunocytochemistry was used to observe changes in neuronal protein expression and localization, while live-cell calcium imaging utilizing pharmacological agents was conducted to identify neuron-like activity. Retinoic acid-treated HTB-11 but not IMR-32 cells developed specific neuronal phenotypes: acquisition of long neurite-like processes, expression of neurofilament-200, increased responsiveness to acetylcholine, and decreased responsiveness to nicotine and epinephrine. In addition, nanomolar levels of retinoic acid elicited increased nuclear trafficking of the CRABP2, which is traditionally associated with gene expression of cellular pathways related to neuronal differentiation. Collectively, these results show that nanomolar concentrations of retinoic acid are capable of inducing both structural and functional neuron-like features in HTB-11 cells using CRABP2, suggesting differentiation in neuroblastoma cells into neuronal phenotypes. These have important implications for both chemotherapeutic design and for the use of neuroblastomas as in vitro models for neuron differentiation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuroblastoma is a type of tumor that originates from nervous tissue; it is the most common solid tumor in infants, accounting for 6% of all childhood cancers and 15% of all tumor deaths in children (Voigt et al. 2000). These childhood tumors migrate aggressively (Holmquist-Mengelbier et al. 2006) and thus often clinically manifest as swellings, somatic distensions, or localized pain at the site of metastasis rather than at the site of origin (Brodeur et al. 1993), making early and accurate diagnosis difficult. While the genetic basis underlying neuroblastoma is not well understood, a majority of cases are non-familial and sporadic, with only 1–2% of cases linked to specific familial gene mutations (Pugh et al. 2013). Among the known gene mutations associated with neuroblastomas involve PHOX2A (Trochet et al. 2004), KIF1B anaplastic lymphoma kinase (ALK) (Mossé et al. 2008), MycN (Brodeur et al. 1984), LMO1 (Wang et al. 2011), and NBPF10 (Li et al. 2014); however, the full spectrum of gene mutations is difficult to determine due to the wide sites in which neuroblastomas are found (Pugh et al. 2013).
While a large variety of subtypes of neuroblastomas can be found, etiological studies indicate that all these subtypes originate from the multipotent cells of the neural crest and specifically from neural crest derivatives such as the sympathetic nerves or the adrenal glands (Ciccarone et al. 1989; Huang and Saint-Jeannet 2004; Jiang et al. 2011), suggesting their potential for acquisition of neuronal phenotypes (Miloso et al. 2004). Thus, the study of cell differentiation in neuroblastomas has been useful for both understanding nerve cell differentiation (Edsjö et al. 2007) as well as for the development of chemotherapeutic agents (Joshi et al. 2006).
As with normal somatic neuronal cells and neural crest derivatives (Takahashi et al. 1999; Maden 2007), neuroblastomas exhibit sensitivity to retinoids; vitamin A-related compounds such as retinoic acid (RA) elicit growth (Benson et al. 2007), differentiation (Jones-Villeneuve et al. 1982), or apoptosis (Altucci et al. 2001). Because micromolar concentrations of RA inhibit metastasis and promote cytotoxicity in neuroblastoma tumors in vitro, phase I trials of RA chemotherapies have adopted 10−5 M as the standard oral dose (Voigt et al. 2000; Voigt and Zintl 2003). This dose of RA has been shown to increase the 3-yr event-free survival rate of patients, but also produces a number of side effects due to non-specific cytotoxicity (Matthay et al. 2009). Few studies, however, have explored the effects of the full range of clinical concentrations of RA effective for neuroblastoma treatment.
RA is known to bind two families of nuclear receptors: the retinoic acid receptors (RARs), which include RAR α, β, and γ and which are activated by both all-trans-retinoic acid and 9-cis-retinoic acid, and the retinoid X receptors (RXRs), which are cellular retinoic acid-binding proteins that include RXR α, β, and γ and which are only activated by 9-cis-retinoic acid (Gupta et al. 2003). Several proteins, including cellular retinol-binding protein (CRABP), help regulate the availability of RA to nuclear receptors (Redfern et al. 1994). When RA binds an RAR, the activated receptor dimerizes with an RXR (Gupta et al. 2003), and the RAR-RXR heterodimer then associates with specific DNA-binding sequences present in the promoters of RA-responsive genes called RA response elements (Gupta et al. 2003) to trigger gene transcription (Kambhampati et al. 2003). The binding of RA to its receptors alters the transcription of a variety of genes related to differentiation, proliferation, and apoptosis. For example, it is thought that RA upsets the balance between differentiation-inhibiting and differentiation-promoting pathways such as the basic helix-loop-helix (bHLH) pathway and the phosphatidylinositol 3-kinase (PI3K)/AKT pathway (Lopez-Carballo et al. 2002; Akkuratov et al. 2015). RA binding has been shown to downregulate mRNA products of the differentiation-inhibiting genes in the bHLH family (Lopez-Carballo et al. 2002), but activate the PI3K/AKT pathway (Akkuratov et al. 2015), which promotes cell survival, differentiation, and neurite outgrowth (Qiao et al. 2012). Activation of a number of other signaling cascades including the anti-proliferative protein kinase C (PKC) pathway and the mitogenic-activated protein kinase (MAP)/extracellular signal-regulated kinase (ERK) pathway are necessary for RA-induced differentiation of neuroblastomas (Kambhampati et al. 2003).
The effects of RA stimulation can include migration, apoptosis, differentiation, adhesion, or increased survival depending upon the tumor precursor type, the stage of tumor development, and the length of the treatment exposure. While some cell types typically adopt axonic or dendritic morphologies after exposure to RA (Gudas 1994), other cell types detach from substratum and undergo apoptosis (Rozzo et al. 1997). Studies show that micromolar concentrations of RA lead to greater cell adhesion, migration inhibition, decreased invasiveness, and the adoption of neuronal phenotypes in some cell lines (Preis et al. 1988; Messi et al. 2008) and have thus been the focus of chemotherapeutic doses (Voigt and Zintl 2003); these doses of RA have been shown to increase the 3-yr event-free survival rate of patients, but also produces a number of side effects due to non-specific cytotoxicity (Matthay et al. 2009).
However, RA is known to exert cellular effects at a broad range of concentrations, and few studies have explored the effects of the full range of concentrations of RA on neuroblastoma cell regulation. We focused our study on the effects of nanomolar concentrations of RA on neuroblastomas. We employed two cell lines; the SK-N-SH (HTB-11) is a neuroblastic (N-type) line that contains the enzymes required for noradrenergic biosynthesis, the structures required for norepinephrine uptake, and opioid, muscarinic, and nerve growth factor receptors (Ciccarone et al. 1989). For contrast, we used the IMR-32, which is a substrate adherent (S-type) line that consists of Schwann cell precursor-like cells that have larger morphologies, stronger substrate adherence, and that lack most neuronal properties, including neurotransmitter biosynthesis enzymes, catecholamine uptake, and receptor proteins (Ciccarone et al. 1989). In this study, neuroblastoma cells of the HTB-11 and the IMR-32 cell lines were cultured and incubated in varying nanomolar RA concentrations for up to 6 days. The extent of differentiation was determined by observing morphological changes, immunostaining for neuronal proteins, and calcium imaging. The proteins and time points identified by immunocytochemical staining served as indicators of the process of neuroblastoma differentiation without the activation of apoptotic pathways. Live-cell calcium imaging was then conducted to observe changes in functional response after RA differentiation. Altered responses to a series of stimuli indicated the extent of changes induced by low-concentration RA differentiation. Together, these studies provide information regarding the complicated mechanisms of cancer proliferation as well as the pathways involved in neuronal development.
Methods and materials
Unless otherwise noted, all materials were purchased from Sigma-Aldrich Chemical Company, St. Louis, MO.
Cell cultures
SK-N-SH (HTB-11) and IMR-32 neuroblastoma cell lines were obtained from the American Type Culture Collection (ATCC; Manassas, VA); each batch was thawed from a frozen stock, used for a maximum of five passages. Cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM). Media was supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cells were cultured in 75-cm2 cell culture-treated flasks (Thermo Scientific, Waltham, MA) incubated at 37°C in a humidified incubator at 5% CO2 for regular passaging and cell line maintenance. As the cells approached confluence, adherent cells were split following treatment with 0.5% porcine trypsin supplemented with 2 mM ethylenediaminetetraacetic acid, resuspended in culture medium, and re-plated with fresh media to 15–20% original density. Prior to treatment and experimentation, cells were seeded onto tissue culture-treated 6-well plates (Becton, Dickinson and Company Biosciences, San Jose, CA) containing 22 × 22-mm no. 1 premium glass coverslips (Fisher Scientific, St. Louis, MO).
Possible mycoplasma contamination was detected using 4′,6-diamidino-2-phenylindole (DAPI) in conjunction with cell viability assays (see below) as described previously (Battaglia et al. 1994); cells that showed signs of possible mycoplasma contamination and abnormal growth properties were discarded and not included in the study. To quantify cell growth, cells were viewed under a phase contrast-inverted cell culture microscope, and a number of cells in five separate microscope fields at ×200 magnification were counted. Since cells that died typically rounded up, detached from the culture, and floated, we counted the number of floating cells in the culture using a hemocytometer and verified their viability using 0.4% trypan blue. To quantify neurite-like process growth, photomicrographs were taken under phase contrast illumination, processes were visually identified, and lengths were digitally measured using ImageJ. Processes were counted if they originated directly from the cell body and if their length was at least 40 μm (twice the diameter of the cell body). Data for cell counts and process measurements within each treatment were validated with ANOVA, and compared against controls or the other treatments with the Student’s t test using a p value of 0.05 to determine significance. Statistical comparisons for this and all subsequent analyses were made using GraphPad Prism (La Jolla, CA).
Retinoic acid treatment
Cells were seeded onto 6-well plates containing glass coverslips then incubated overnight. For each experiment, 11-cis retinoic acid (RA; Sigma Chemical Co., St. Louis, MO) was diluted using culture media from a stock solution stored at −20°C and added directly to each well for a final concentration of 1 nM, 3.1 nm, 10 nm, 31 nm, or 100 nM. The day of RA treatment was designated as day 0. Untreated control cells were maintained for later comparison. Cells were monitored under an Olympus CX-41 tissue culture microscope throughout the study and quantified as described above.
Immunocytochemistry and immunofluorescence microscopy
Coverslips with cells were fixed in methanol for 5 min, subjected to three 10-min washes with phosphate-buffered saline (PBS), and blocked for 30 min in a solution of 10% normal goat serum and 0.3% Triton X-100 detergent in PBS. Cells were subjected to three more 10-min rinses in PBS and incubated overnight at 4°C with one of the following antibodies (see Table 1). For controls, antibodies were substituted with non-immune serum. After the overnight incubation, cells were subjected to three 10-min rinses with PBS and incubated for 1.5 h in the dark with the appropriate secondary antibody conjugated to either fluorescein isothiocyanate (FITC) or tetramethylrhodamine (TRITC) diluted 1:100 in PBS. Following incubation, cells were subjected to two 15-min PBS washes, incubated for 5 min in 1 μM DAPI, and then subjected to three additional 10-min washes in deionized water. Slides were mounted using Vectashield Mounting Medium (Vector Laboratories Inc., Burlingame, CA) and stored in a light-tight box at 4°C.
Slides were viewed at ×100 under oil immersion using a Nikon Eclipse E400 epifluorescence microscope using the appropriate filters and photographed as TIFF images using a cooled Orca CCD camera (12-bit resolution; Hammamatsu Corp., Bridgewater, NJ) set at a constant 500-ms exposure time. Ten fields of view per slide were photographed under oil immersion at ×100 unless otherwise noted. The digital TIFF images were analyzed using ImageJ (National Institute of Health, Bethesda, MD) to identify differentially expressed or co-localized proteins in RA-treated neuroblastoma cells. For each image, the number of DAPI-stained nuclei and immunostained cells were counted. Specific cellular locations of protein aggregation were identified and photos were superimposed to analyze co-localization of doubly labeled proteins. Changes in cellular morphologies were observed and any discrete neurite-like processes longer than the length of the cell body were tabulated.
Since the brightness of the staining is proportional to the amount of protein in the cells, the differential amount of protein expression was quantified from the digital photographs using available software (ImageJ, National Institutes of Health). For each trial, five images from separate microscope fields of each coverslip were randomly selected. The pixel intensity threshold value of the staining in each image was determined by visual inspection of the control images; this threshold value was subsequently kept constant throughout the data analysis. The protein expression and distribution were quantified based on standard methods (Masliah et al. 1990; Kokolakis et al. 2008). To quantify protein staining in a specific area of the cell (i.e., the nucleus), a region of interest (ROI) was manually identified in the image using ImageJ, and measurements were restricted to this area. The values for each of the images taken from one slide were grouped and compared to those from other slides that were subjected to the same experimental treatment using a paired sample t test. If the data were not significantly different, the data from these slides were combined for subsequent analyses. The effect of different experimental treatments was determined using a paired sample t test. Data from the experimental treatments were normalized to their corresponding controls (=100%). For all statistical measures, significance level was set at p = 0.05. Values were normalized so that trends among data sets could be most clearly compared with each other and control groups. Error bars for all figures show the standard error of the mean.
Calcium imaging
Cells cultured on glass coverslips in 6-well plates were prepared for imaging as follows: culture medium was replaced with warm culture medium supplemented with 1 μM Fura-2 and 20 μg/ml Pluronic F-127 (both from Molecular Probes/Invitrogen, Carlsbad, CA). Plates were returned to the incubator for 1 h prior to imaging. Coverslips with Fura-2-loaded cells were situated in a recording chamber, continuously bathed with a Ringer’s solution superfusion (145 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 1 mM Na-pyruvate, 20 mM HEPES-Na), and secured to the stage of an inverted fluorescence microscope (Nikon Eclipse TE2000-U). Stimulus solutions, each mixed at 100 μM in Ringer’s and prepared immediately prior to use, were held in a stimulus bank, and included ATP, acetylcholine (ACH), epinephrine (Epi), nicotine (Nic), and high K+ Ringer’s solution (Ringer’s with 100 mM Na+ equimolarly substituted with K+). Stimuli were applied by switching the gravity-fed superfusion for 2 to 3 min. The recording chamber configuration and the stimulus application system allowed the superfusion solutions to be completely exchanged within 5 s without interrupting fluid level or flow.
Intracellular calcium concentrations ([Ca2+]i) were quantified and analyzed as follows: cells were illuminated at excitation wavelengths of 340 and 380 nm and images were acquired at exposure times of 200–300 ms every 5 s. Light emission from the Fura-2 within the cells was filtered at 510 nm and recorded with a cooled Hammamatsu Orca CCD camera. Images from the camera were digitized and analyzed using IPLabs software (Scanalytics Inc./BD Biosciences, Bethesda, MD). Responses to stimuli were monitored in real time and quantified using standard ratiometric calcium imaging techniques, where relative calcium levels were estimated by calculating the ratio of the intensity of the emitted light at 340- and 380-nm excitation. Individual cells were visually identified and selected by drawing an ROI to restrict calcium measurements within the individual ROIs. Immediately prior to the application of a stimulus solution, the baseline [Ca2+]i was noted. If a distinct and sustained change in [Ca2+]i following stimulation (within 5–20 s) and a return of [Ca2+]i toward the original baseline following solution removal (within 50 s) were noted, this counted as a response. Average data for individual cells within each treatment were validated with ANOVA, and compared against controls or the other treatments with the Student’s t test using a p value of 0.05 to determine significance. Statistical comparisons were made using GraphPad Prism (La Jolla, CA).
Western blotting
Western blots were conducted using standard immunoblotting techniques. Briefly, cells were cultured in 150-cm2 flasks and treated with RA as described above. Cells were harvested using cell scrapers, lysed, fractionated into the nuclear and cytosolic fractions, homogenized in Laemmli buffer, and separated on a 15% SDS-polyacrylamide electrophoresis gel with 15 μg total protein per lane. Protein concentration was determined using a Coomassie protein assay (Thermo Scientific, Rockford, IL). Proteins were transferred to a nitrocellulose membrane, incubated in 10% goat serum for 1 h, and reacted with anti-RARα (1:1000), anti-FABP5 (1:500), or anti-CRABP2 (1:1000) overnight at 4°C. Uniform gel loading and appropriate protein localization was confirmed using nuclear lamin (1:5000) and β-tubulin III (1:1000) for nuclear and cytosolic localization, respectively. Immunoreactive proteins were visualized with Vectastain ABC Elite Kit (Vector Labs, Burlingame, CA) using peroxidase-conjugated secondary antibodies and diaminobenzidine (DAB) following the manufacturer’s instructions.
Results
Retinoic acid and cell growth in two neuroblastoma cell lines
Untreated HTB-11 cells appeared as triangular- or spindle-shaped cells with long fibrous processes (Fig. 1 A). After treatment with RA, cultures consisted of smaller, circular cells, many of which had neurite-like processes (Fig. 1 B). These morphologies typically appeared within 2 days of treatment with 1 nM RA, and were more pronounced at higher concentrations. No rounded or detached cells were visible.

Changes in cell morphology and growth rates resulting from a 6-d treatment with retinoic acid in HTB-11 or IMR-32 cells. HTB-11 (A, B) and IMR-32 (C, D) cell lines were prepared in 6-well plates as described above and left untreated or subjected to 6 d of exposure to 31 nM retinoic acid. Cells were photographed under phase contrast on the sixth day of treatment. (A) Prior to treatment (day 1), HTB-11 cells exhibited triangular- and spindle-shaped cell bodies terminating in short, fibrous processes. (B) Treatment with 31 nM retinoic acid for 6 d resulted in cells that developed more pronounced neurite-like processes with rounder cell bodies. (C) IMR-32 cells at day 6 exhibited spindle-like, triangular, and stout rectangular cell bodies with few processes. (D) Treatment with 31 nM retinoic acid for 6 d resulted in the detachment and death of many cells; cells remaining exhibited rounded cell bodies with some short processes, with no observable difference from the untreated cells. Scale bar 50 μm.
Untreated IMR-32 cell cultures consisted of spindle-like, triangular, and stout rectangular cells with short fibrous processes (Fig. 1 C). Treatment with RA induced cells bodies to appear more round, as well as induced detachment of cells (seen as small, phase bright bodies that moved when the plate was gently agitated). After 6 days of incubation in 31 nM RA, the majority of IMR-32 cells had detached (Fig. 1 D).
To quantify the effects of RA on cell growth and cell differentiation, we conducted five separate experiments on the two different cell lines, where cells were counted after 1, 3, and 6 days in vitro following RA treatment at different concentrations; we also measured cell viability at each time point. In Fig. 2 A, HTB-11 cells treated with RA after 1 day generally showed a concentration-dependent reduction in the overall number of cells when compared to controls. After 3 through 6 days of treatment, the reduction in these numbers was consistent (approximately 70% compared to control) but no longer concentration-dependent. The reduction in numbers was due to reduction in cell proliferation and not due to cell death and/or detachment; we did not see a marked increase in the number of trypan blue-stained rounded or floating cells present in the cultures (T 15 = 2.956; p < 0.005). In contrast, IMR-32 cells treated with RA (Fig. 2 B) showed a strong concentration-dependent reduction in overall number after 1 day in vitro. This effect was exacerbated after 3 through 6 days of treatment. The reduction in this cell number was due to an increase in cell death; at each time point, we quantified a high number of trypan blue-stained floating cells that accounted for the reduction in the cell number of each treatment compared to controls. Thus, for this cell line, the observed reduction in numbers was due to decreased cell viability and not due to decreased cell proliferation.

Changes in overall cell abundance cell morphology and growth rates resulting from a 6-d treatment with retinoic acid in HTB-11 or IMR-32 cells. HTB-11 (A, B) and IMR-32 (C, D) cell lines were prepared in 6-well plates as described above and left untreated or subjected to 6 d of exposure to retinoic acid. Five microscope fields were photographed at each d, and cells were counted; the values were expressed as a proportion of the cells in the control treatment. For clarity, only the data for 1, 3, and 6 d are shown. (A) HTB-11 cells generally slowed their proliferation when treated with RA. The total number of cells was lower generally steady when compared to control cells. (B) IMR-32 cells showed a greater reduction in the proliferation rate when treated with RA.
RA-induced neuritogenesis and neurofilament expression in HTB-11 cells
RA-treated and untreated HTB-11 and IMR-32 cells were stained using primary antibodies as described in the “Methods and Materials” section. Figure 3 shows representative images of positive immunostaining seen in HTB-11 cells; Table 1 gives a summary of the immunostaining results for both HTB-11 and IMR-32 cells.

Immunostaining properties of HTB-11 cells. Images show representative samples of untreated and RA-treated (31 nM) HTB-11 cells immunostained with marker molecules that characterize neurons. For all images, DAPI-stained nuclei appear blue, while the antibody staining was visualized with either FITC (green) or TRITC (red). (A) GFAP staining, a glial cell marker, was diffused throughout the cytosol and was visible in approximately 50% of all cells. (B) RA treatment did not affect GFAP staining patterns. (C) Vimentin staining in untreated cells appeared generally diffused, and occasionally appeared to be organized in fibers. About 40–50% of these cells were vimentin+. (D) RA treatment generally resulted in denser and more fibrous vimentin staining; the proportion of vimentin+ cells was unaffected by RA treatment. (E) In untreated cells, NF200 appeared to be localized in the cell body. (F) In RA-treated cells, NF200 appeared to localize to the neurite-like processes (see Fig. 1 B). The proportion of NF200 staining localized to neurites increased with increasing concentrations and duration of RA treatment (see Fig. 3). (G) Cells double-labeled for vimentin (green) and NF200 (red) showed that these two proteins did not co-localize in treated or untreated cells. Images were acquired using a ×40 objective. Scale bar 50 μm.
The DAPI-stained nuclei that were visible in images served as an indicator for the individual cells. Based on our preliminary results, we focused on glial fibrillary acidic protein (GFAP) to stain non-neuronal phenotypes, neurofilament 200 (NF200) to stain neuron-like cells, and vimentin as a neutral stain for both neuronal and non-neuronal phenotypes (Table 1). Figure 3 A shows that GFAP was localized in the cytosol, but did not show any distinct subcellular pattern. In addition, some cells with visually distinct nuclei did not express this marker protein; these cells served as internal controls for immunoreactivity. RA treatment (Fig. 3 B) did not result in a change in the immunostaining pattern or in the number of GFAP+ cells. In untreated cells, vimentin staining (Fig. 3 C) appeared to be either diffused within the cytosol or organized into distinct filaments. RA treatment (Fig. 3 D) did not result in a noticeable change in the proportion of vimentin+ cells nor in the staining patterns. In untreated cells, NF200 primarily appeared as diffuse staining throughout the cytosol (Fig. 3 E). RA treatment resulted in a higher frequency of neurite-like processes (as in Fig. 1 B); in these cells, NF200 appeared to be organized in discrete filaments localized to these processes (Fig. 3 F).
Figure 3 G shows a sample of cells stained with vimentin (green) and NF200 (red); for these cells, there was no overlap in staining of vimentin and NF200. In all combinations tested (vimentin/NF200, GFAP/NF200, and GFAP/vimentin), we found no co-localization between any of these proteins, suggesting that neurofilament expression in cells that differentiate into a neuron-like phenotype occurs in a specific subpopulation of cells that is likely distinct from the glia-like cells.
To quantify the emergence of these neuronal phenotypes resulting from RA treatment, HTB-11 cells were treated with different concentrations of RA and incubated for 1 to 6 d, then immunostained for NF200 as described in the “Methods and Materials” section. We conducted five replicates for each treatment at each duration. We first determined the proportion of cells that had visible processes; Fig. 4 A shows the complete series of data for all RA treatments and durations. After 2-day treatment, the proportion of cells with processes ranged from 38 to 60%; while increasing RA concentrations resulted in a larger proportion of cells developing processes, this proportion was not significant. After 3 days of treatment, increasing RA concentrations in half-log steps resulted in significant increases in process development; each data point from 1 to 31 nM was significantly higher than the data point that preceded it (T 25 = 3.492; p < 0.001). At 31 nM, the response reached a maximum, above which increasing concentration of RA did not result in additional process development. Longer incubations (4 through 6 d) did not result in a change in this pattern.

RA treatment upregulates neurite formation. Cells were treated with varying concentrations of RA for 2 to 6 d. Cell processes (Fig. 1) and NF200 staining (Fig. 3) were quantified as described in the “Methods and Materials” section. Asterisks indicate the point at which increasing RA did not result in a further significant increase. (A) Increasing concentrations of RA resulted in increased formation of neurite-like processes. After 2 d in vitro, the effects of RA were evident but not as pronounced. By 3 d and at 10 nM RA, the maximal effects of RA treatment were observed, and longer treatment times did not result in a significant increase in process formation. (B) The total number of cells that expressed NF200 (either localized to processes or found in the cell body) was determined. For clarity, only the data for days 2, 3, and 6 are shown in the graph. Increasing concentrations of RA (up to 3.1 nM) resulted in a significant increase in cells expressing NF200; as in Fig. 4 A, this occurred within 3 d, and longer treatment times did not result in a significant increase in the proportion of NF200+ cells. C NF200 could either be diffusely distributed through the cytosol (Fig. 3 E) or localized to processes (Fig. 2 F). Increasing RA resulted in a larger proportion of processes that were NF200+. The processes that did not have NF200 were likely in earlier stages of formation.
We then determined the proportion of cells that were NF200+; for clarity, Fig. 4 B shows data from days 2, 3, and 6. At day 2, the proportion of NF200+ cells was relatively low (15–35%) and was not significantly altered by increasing RA concentrations (T 25 = 1.477; p > 0.05). At 3 days, 1 and 3.1 nM RA resulted in an increase in the proportion of NF200+ cells; higher concentrations (10 nM and above) did not result in a further increase in NF200+ expression.
We then determined the proportion of processes that had NF200 localized (as seen in Fig. 3 F). The trend seen in this data was similar to that seen in Fig. 4 B: at day 2, the effects of RA were not evident, and at day 3, NF200 localization to processes increased with increasing RA concentrations above 3.1 nM. At time periods longer than 3 d, the trend did not change (T 25 = 0.973; p > 0.1).
Based on the results shown above, we focused our functional studies (below) on 3.1- or 10-nM RA exposure for 3 d.
Retinoic acid induces neuron-like functional changes in HTB-11 cells
HTB-11 cell were seeded into 6-well plates and treated with 0 (control), 3.1 nM, or 31 nM RA for 3 d and prepared for live-cell imaging as described above. Cells were superfused with ATP, acetylcholine epinephrine, nicotine (each at 100 μM), or high K+ Ringer’s solution, and changes in [Ca2+]i in response to the stimulus application were monitored in real time. Sample calcium imaging data traces are shown in Fig. 5. In the top panel, the cell responded to ACH with an increase in [Ca2+]i but not to ATP, Epi, or Nic. While the [Ca2+]i level appears to change in response to Epi, this was not counted as a response since the change in magnitude was not at least twice greater than the fluctuation in [Ca2+]i immediately prior to the stimulus application. The lower panel shows a different cell that responded to Nic with a decrease in [Ca2+]i, but not to ATP, ACH, or K+.

Sample data traces showing typical stimulus-induced changes in [Ca2+]i measured using ratiometric imaging. HTB-11 cells were prepared as described in the “Methods and Materials” section and loaded with Fura-2 for 1 h. Cells were situated in a recording chamber, and stimuli were applied using superfusion. Stimuli used were nicotine (Nic), ATP, acetylcholine (ACH), epinephrine (Epi), or depolarization with 100 mM K+ (K+). Solid bars indicate stimulus applications, while the hatched bar indicates a time scale of 100 s. The top panel shows a single cell’s response to ACH with an increase in [Ca2+]i that is at least twice the magnitude of the baseline fluctuation in [Ca2+]i. The bottom panel shows a different cell that responded to Nic with a decrease in [Ca2+]i; this type of response was more commonly observed in these cells. While there appears to be a slight change in [Ca2+]i following ACH, the magnitude of the change was not large enough (i.e., at least twice the magnitude of the baseline fluctuation) to be considered a response.
Data from four complete runs of the manipulations were summarized, and responses from at least 100 cells per trial were tallied. Analyses of the variation within each run and between individual runs did not show a significant difference between the trials; thus, the data was pooled. Figure 6 shows the proportion of all measured cell responses. When stimulated with Nic, approximately 48% of untreated cells did not show a response, 4% responded with increases in [Ca2+]i, and 48% responded with decreases in [Ca2+]i. Treatment with 3.1 or 31 nM RA resulted in a similar significant decrease in the proportion of these responses (T 4 = 4.953; p < 0.005). A similar pattern was observed with Epi stimulation, where RA treatment resulted in significant decreases in the proportion of cells that responded (T 4 = 2.226; p < 0.05). In contrast, increasing concentrations of RA resulted in a significant increase in the proportion of cells that responded to ACH (T 4 = 2.360; p < 0.05). RA treatment did not affect the proportion of cells that responded to ATP or K+ depolarization (T 4 = 0.881; p > 0.2).

RA treatment induces neuron-like functional properties. The total number of responses for each cell tested was determined as shown in Fig. 5. The responses of all untreated and RA-treated (3.1 and 31 nM) cells were expressed as a percentage of the total. RA treatment resulted in a significant decrease in the response rate to nicotine, and a significant increase in the response rates to epinephrine and ACH. Stimuli that are general excitable cell activators (high K+ and ATP) were not affected by RA treatment, suggesting that RA treatment triggered a differentiation pathway that was specific for neurons.
Differentiation in response to nanomolar concentrations of RA involves CRABP2
To determine the likely signaling pathway involved with RA induction of cell differentiation, determine the effect of RA stimulation on the expression of proteins known to be involved with RA signaling: CRABP2, which has a high affinity for RA and shuttles it to its one nuclear receptor RARα, and ultimately stops cell division (Sidell 1982), and FABP5, a protein interacts with RA and shuttles it to the nuclear receptor PPARβ and eventually leads to cell proliferation (Tachibana et al. 2008; Morgan et al. 2010). We treated cells for 3 d with RA at a low concentration (10 nM; see Fig. 3). Figure 7 shows sample images of cells double labeled with either RARα, CRABP2, or FABP5 (green); nuclei were labeled with DAPI to enable determination of the precise boundary of the nuclei and the relative amount of protein that is localized in this region.

Immunostained images showing RA-induced nuclear localization of CRABP2 and RAR. Sample epifluorescence images showing immunostaining for the nuclear receptor proteins (CRABP2, RARα, or FABP5; each shown in green) and DAPI-stained nuclei (blue). Diagonal right arrows indicate nuclear localization, while diagonal left arrows show cytosolic localization. In untreated cells, CRABP2 and RARα did not localize to the nucleus. Following treatment with 10 nM (0.01) or 10 μM (10) RA, although much of the protein was still localized to the cytosol, some staining was evident in the nucleus, suggesting nuclear import. For FABP5, RA treatment did not appear to affect nuclear localization, suggesting that this receptor protein was not involved with RA signaling at these concentrations. All images were acquired using a ×100 objective. Scale bar 20 μm.
In untreated HTB-11 cells, all three proteins appeared to be diffuse throughout the cell cytosol, with is no evident nuclear localization for CRABP2 or RARα. When treated with 10 nM or 10 μM RA, FABP5 distribution does not visibly change; in contrast, both CRABP2 and RARα appear to localize to the nucleus.
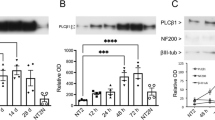
The protein expression patterns seen above were quantified using Western blotting and quantitative immunocytochemistry. Results from three replicates of the Western blotting studies (Fig. 8 A) showed that RA treatment increased the localization of RARα and CRABP2 but not FABP5, confirming the trends shown in Fig. 7. To verify these trends, the staining in the immunostained cells was quantified as described in the “Methods and Materials” section. Six replicates of the study were conducted, and immunostaining data was quantified. RA treatment resulted in a significant increase in the overall expression (Fig. 8 B) and nuclear localization (Fig. 8 C) of RARα (T 6 = 3.263; p < 0.01) and CRABP2 (T 6 = 4.941; p < 0.005) but not in FABP5 (T 6 = 1.443; p > 0.05). These results suggest that the differentiation of HTB-11 cells into neuron-like cells is initiated by transcriptional regulators that depend on CRABP2 complexing with RARα and entering the nucleus.

Quantification of immunostaining shows RA-induced increases in nuclear localization of CRABP2 and RA, but not FABP5. (A) Western blots of untreated cells (C) or cells treated with 10 nM (0.01) or 10 μM (10) RA. These correspond to the immunostained cells shown in Fig. 7. The top two bands show loading and staining controls for nuclear (lamin) and cytosolic (tubulin) proteins that are unaffected by RA. Note that 10 nM RA resulted in changes in nuclear localization of RARα and CRABP2 but not FABP5, supporting the visually observable results from Fig. 7. (B) Immunostaining of different proteins was quantified as described in the “Methods and Materials” section. The measured values were consistent with the trends seen in the western blots (A). (C) The total amount of protein localized to the nucleus (determined by visual determination of the location of the nucleus using DAPI staining) was expressed as the proportion of the measured total amount of protein (as measured in B). Treatment with 10 nM RA resulted in increased localization of RARα to the nucleus; increased RA did not result in a further increase in nuclear localization (not shown). The proportion of nuclear localization of CRABP2 increased with increasing RA concentrations. Nuclear localization of FABP5 was not affected by RA treatment. Asterisks indicate a significant change in the measured quantity resulting from RA treatment.
Discussion
The present study, working with IMR-32 and HTB-11 cell lines, focused on quantifying changes in both structural (cell morphology, protein expression) and functional (Ca+2-mediated cellular response to stimulatory compounds) responses elicited by nanomolar concentrations of RA. Our results indicate that nanomolar concentrations of RA elicit CRABP2-mediated differentiation pathways (Figs. 7 and 8) in HTB-11 cells that activate expression of both structural (Figs. 3 and 4) and functional (Fig. 6) neuronal phenotypes (Miloso et al. 2004). IMR-32 cells subjected to the same treatments showed increased mortality (Fig. 2), suggesting that the effects of RA were cell line-specific.
Because neuroblastomas are derived from the neural crest and arise from neural tissue, the expression of neuronal proteins during cell differentiation would be expected. The lack of expression of most of the neuronal proteins tested in this study is likely due to the heterogeneity of neuroblastoma tumors (Brodeur 1995; Maris 2005). Neuroblastoma cells lines, including HTB-11 and IMR-32, contain a mixture of the N-type, S-type, and intermediate-type (I-type) cells (Ross et al. 1995). S-type cells tend to express high levels of vimentin and not GFAP in vitro due to their Schwann cell derivation (Ciccarone et al. 1989). The immunostaining patterns seen in our study (Table 1) suggest that the IMR-32 cell line consisted of mostly S-type cells, while the expression of NF200 in HTB-11 cells (Fig. 3) suggests that this cell line consists primarily of N-type cells. In addition, the differential viability and adhesion patterns seen in our two cells lines (Figs. 1 and 2) could also be related to the heterogeneity of the different tumor cell lines. S- and N-type cells exhibit varying levels of invasiveness and substrate adherence (Corey et al. 2010); prior to differentiation, S-type cells remain tightly adhered to the substrate due to integrin upregulation while N-type cells adhere loosely (Meyer et al. 2004). This downregulation of adhesion proteins is thought to account for the highly migratory and metastatic nature of N-type cells (Voigt et al. 2000). Binding of RA to its receptor sites upregulates expression of platelet endothelial cell adhesion molecule (PECAM-1; Voigt et al. 2000), which is one of several proteins involved in tumor angiogenesis and integrin activation (Meyer et al. 2004). In N-type HTB-11 cells, retinoic-acid induced PECAM-1 expression and the formation of substrate-adhered neurite-like processes appear to inhibit migration and metastatic invasion (Voigt et al. 2000; Messi et al. 2008). In S-type IMR-32, the opposite effect was seen; cells were detached and did not survive RA treatment, suggesting a differential gene expression pathway associated with RA-induced cytotoxicity (Voigt et al. 2000; Voigt and Zintl 2003).
In addition to heterogeneity of the different tumor lines, the cells within each cell line show heterogeneity as well. Although RA treatment of HTB-11 resulted in extensive neurite outgrowth (Figs. 1 and 2), the immunostaining patterns of the cells we never uniform (Figs. 3 and 4). When tested with pharmacological agents, a majority of cells were non-responsive (Fig. 6), suggesting that these cells were either not fully differentiated or of a different cell type. This tumor heterogeneity and individual differences in cell responsiveness to RA treatment underscores the difficulty in using specific chemotherapeutic agents that are broadly applied to an entire population of cells (Skipper and Schabel 1984; Fidler and Poste 1985; Tang 2012).
The effects of RA treatment seen in our study could be elicited by several different mechanisms; inhibition of cell division, induction of terminal differentiation, or initiation of apoptosis could all result in the morphological changes seen in our study. The involvement of CRABP2 (Figs. 7 and 8) suggests that the most likely mechanism of elicited by nanomolar concentrations of RA is gene expression that elicits terminal differentiation (Didierjean et al. 1991; Houldsworth et al. 2002). To activate gene expression, RA typically binds to RA binding proteins to enable nuclear import and subsequent associating with RA receptors associated with the DNA (Mangelsdorf and Evans 1995; Balmer and Blomhoff 2002). The two major activation pathways related to RA-elicited gene activation involve either CRABP2-mediated association with nuclear RARs (Dolle et al. 1990; Pemrick et al. 1993) or FABP5-mediated activation of PPAR β/γ (Tachibana et al. 2008; Morgan et al. 2010). Studies show that CRABP2 is typically associated with growth inhibition, cell cycle arrest, or differentiation, while FABP5 is typically associated with cell proliferation and cell survival (for review, see Wolf 2008). Activation of these two pathways differs for each cell type and for different concentration ranges (Allenby et al. 1993; Schug et al. 2007). The results of our study show that for the SK-N-SH, nanomolar levels of RA are capable of modulating gene expression through the canonical pathways previously described (Sidell 1982; Allenby et al. 1993).
Neuroblastoma cells have long been used as models for nerve cell differentiation (Gomez et al. 1996; Edsjö et al. 2007; Xie et al. 2010). In our study, we showed that RA at nanomolar concentrations can trigger the emergence of a subset of structural and functional cellular features associated with neurons. Previous studies using calcium fluxes to measure acetylcholine activation of muscarinic receptors in neuroblastoma cell lines have shown that these cells possess acetylcholine-sensitive muscarinic binding sites (Forsythe et al. 1992). The RA-induced differentiation in our study resulted in an increase in muscarinic and adrenergic receptor activity, and a decrease in nicotinergic receptor activity in subpopulations of cells (Fig. 6). Thus, these subpopulation neuroblastoma cells likely differentiated into a specific neuronal phenotype, likely related to sympathetic/parasympathetic origins (Landis and Keefe 1983; McGehee and Role 1995), suggesting that RA at very low levels can affect the developmental trajectory of neuron-like cells (Henion and Weston 1994; Sucov and Evans 1995). This has implications for the study of nerve cell differentiation in vivo (Encinas et al. 2000).
References
Akkuratov EE, Wu J, Sowa D, Shah ZA, Liu L (2015) Ouabain-induced signaling and cell survival in SK-N-SH neuroblastoma cells differentiated by retinoic acid. CNS Neurol Disord Drug Targets 14:1343–1349
Allenby G, Bocquel MT, Saunders M, Kazmer S, Speck J, Rosenberger M, Lovey A, Kastner P, Grippo JF, Chambon P (1993) RA receptors and retinoid X receptors: interactions with endogenous retinoic acids. Proc Natl Acad Sci U S A 90:30–34
Altucci L, Rossin A, Raffelsberger W, Reitmair A, Chomienne C, Gronemeyer H (2001) Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat Med 7:680–686
Balmer J, Blomhoff R (2002) Gene expression regulation by retinoic acid. J Lipid Res 43:1773–1808
Battaglia M, Pozzi D, Crimaldi S, Parasassi T (1994) Hoechst 33258 staining for detecting mycoplasma contamination in cell cultures: a method for reducing fluorescence photobleaching. Biotech Histochem 69:152–156
Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ (2007) All-trans retinoic acid mediates enhanced Treg cell growth differentiation and gut homing in the face of high levels of co-stimulation. J Exp Med 204:1765–1774
Brodeur GM (1995) Molecular basis for heterogeneity in human neuroblastomas. Eur J Cancer 31:505–510
Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, De Bernardi B, Evans AE, Favrot M, Hedborg F (1993) Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11:1466–1477
Brodeur G, Seeger R, Schwab M, Varmus H, Bishop J (1984) Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 224:1121–1124
Ciccarone V, Spengler BA, Meyers MB, Biedler JL, Ross RA (1989) Phenotypic diversification in human neuroblastoma cells: expression of distinct neural crest lineages. Cancer Res 49:219–225
Corey JM, Gertz G, Sutton TJ, Chen Q, Mycek KB, Wang B-S, Martin AA, Johnson SL, Feldman EL (2010) Patterning N-type and S-type neuroblastoma cells with Pluronic F108 and ECM proteins. J Biomed Mater Res A 93:673–686
Didierjean L, Durand B, Saurat JH (1991) Cellular retinoic acid-binding protein type 2 mRNA is overexpressed in human psoriatic skin as shown by in situ hybridization. Biochem Biophys Res Commun 180:204–208
Dolle P, Ruberte E, Leroy P, Morriss-Kay G, Chambon P (1990) Retinoic acid receptors and cellular retinoid binding proteins. I A systematic study of their differential pattern of transcription during mouse organogenesis. Development 110:1133–1151
Edsjö A, Holmquist L, Påhlman S (2007) Neuroblastoma as an experimental model for neuronal differentiation and hypoxia-induced tumor cell dedifferentiation. Semin Cancer Biol 17:248–256
Encinas M, Iglesias M, Liu Y, Wang H, Muhaisen A, Cena V, Gallego C, Comella JX (2000) Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated neurotrophic factor-dependent human neuron-like cells. J Neurochem 75:991–1003
Fidler IJ, Poste G (1985) The cellular heterogeneity of malignant neoplasms: implications for adjuvant chemotherapy. Semin Oncol 12:207–221
Forsythe ID, Lambert DG, Naborski SR, Lindsdell P (1992) Elevation of cytosolic calcium by cholinoceptor agonists in SH-SY5Y human neuroblastoma cells: estimation of the contribution of voltage-dependent currents. Br J Pharmacol 107:207–214
Gomez G, Restrepo D, Rawson N, Lowry LD, Keane WM, Rothstein JL (1996) Induction of differentiation of human olfactory neuroblastoma cells into odorant-responsive cells. Neuroscience 74:567–577
Gudas LJ (1994) Retinoids and vertebrate development. J Biol Chem 269:15399–15402
Gupta RA, Sarraf P, Brockman JA, Shappell SB, Raftery LA, Willson TM, DuBois RN (2003) Peroxisome proliferator-activated receptor γ and transforming growth factor-β pathways inhibit intestinal epithelial cell growth by regulating levels of TSC-22. J Biol Chem 278:7431–7438
Henion PD, Weston JA (1994) Retinoic acid selectively promotes the survival and proliferation of neurogenic precursors in cultured neural crest cell populations. Dev Biol 161:243–250
Holmquist-Mengelbier L, Fredlund E, Löfstedt T, Noguera R, Navarro S, Nilsson H, Pietras A, Vallon-Christersson J, Borg Å, Gradin K, Poellinger L (2006) Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 10:413–423
Houldsworth J, Heath SC, Bosl GJ, Studer L, Chaganti RSK (2002) Expression profiling of lineage differentiation in pluripotential human embryonal carcinoma cells. Cell Growth Differ 13:257–264
Huang X, Saint-Jeannet JP (2004) Induction of the neural crest and the opportunities of life on the edge. Dev Biol 275:1–11
Jiang M, Stanke J, Lahti JM (2011) The connections between neural crest development and neuroblastoma. Curr Top Dev Biol 94:77–127
Jones-Villeneuve EM, McBurney MW, Rogers KA, Kalnins VI (1982) Retinoic acid induces embryonal carcinoma cells to differentiate into neurons and glial cells. J Cell Biol 94:253–262
Joshi S, Guleria R, Pan J, DiPette D, Singh US (2006) Retinoic acid receptors and tissue-transglutaminase mediate short-term effect of retinoic acid on migration and invasion of neuroblastoma SH-SY5Y cells. Oncogene 25:240–247
Kambhampati S, Li Y, Verma A, Sassano A, Majchrzak B, Deb DK, Parmar S, Giafis N, Kalvakolanu DV, Rahman A, Uddin S (2003) Activation of protein kinase Cδ by all-trans-retinoic acid. J Biol Chem 278:32544–32551
Kokolakis G, Panagis L, Stathopoulos E, Giannikaki E, Tosca A, Krüger-Krasagakis S (2008) From the protein to the graph: how to quantify immunohistochemistry staining of the skin using digital imaging. J Immunol Methods 331:140–146
Landis SC, Keefe D (1983) Evidence for neurotransmitter plasticity in vivo: developmental changes in properties of cholinergic sympathetic neurons. Dev Biol 98:349–372
Li W, Li K, Zhao L, Zou H (2014) Bioinformatics analysis reveals disturbance mechanism of MAPK signaling pathway and cell cycle in glioblastoma multiforme. Gene 547:346–350
Lopez-Carballo G, Moreno L, Masia S, Perez P, Barettino D (2002) Activation of the phosphatidylinositol 3-kinase/Akt signaling pathway by retinoic acid is required for neural differentiation of SH-SY5Y human neuroblastoma cells. J Biol Chem 227:25297–25304
Maden M (2007) Retinoic acid in the development regeneration and maintenance of the nervous system. Nat Rev Neurosci 8:755–765
Mangelsdorf DJ, Evans RM (1995) The RXR heterodimers and orphan receptors. Cell 83:841–850
Maris JM (2005) The biological basis for neuroblastoma heterogeneity and risk stratification. Curr Opin Pediatr 17:7–13
Masliah E, Terry RD, Alford M, DeTeresa R (1990) Quantitative immunohistochemistry of synaptophysin in human neocortex: an alternative method to estimate density of presynaptic terminals in paraffin sections. J Histochem Cytochem 38:837–844
Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, Gerbing RB, London WB, Villablanca JG (2009) Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children's oncology group study. J Clin Oncol 27:1007–1013
McGehee DS, Role LW (1995) Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol 57:521–546
Messi E, Florian MC, Caccia C, Zanisi M, Maggi R (2008) Retinoic acid reduces human neuroblastoma cell migration and invasiveness: effects on DCX LIS1 neurofilaments-68 and vimentin expression. BMC Cancer 8:1
Meyer A, van Golen CM, Kim B, van Golen KL, Feldman EL (2004) Integrin expression regulates neuroblastoma attachment and migration. Neoplasia 6:332–342
Miloso M, Villa D, Crimi M, Galbiati S, Donzelli E, Nicolini G, Tredici G (2004) Retinoic acid-induced neuritogenesis of human neuroblastoma SH-SY5Y cells is ERK independent and PKC dependent. J Neurosci Res 75:241–252
Morgan E, Kannan-Thulasiraman P, Noy N (2010) Involvement of fatty acid binding protein 5 and PPAR/in prostate cancer cell growth. PPAR Res Article ID 234629
Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, Hou, Hakonarson HTA, Schork NJ, Brodeur GM, Tonini GP, Rappaport E, Devoto M, Maris JM (2008) Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455:930–935
Pemrick SM, Lucas DA, Grippo JF (1993) The retinoid receptors. Leukemia 8:S1–10
Preis PN, Saya H, Nádasdi L, Hochhaus G, Levin V, Sadée W (1988) Neuronal cell differentiation of human neuroblastoma cells by retinoic acid plus herbimycin A. Cancer Res 48:6530–6534
Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, Kim J (2013) The genetic landscape of high-risk neuroblastoma. Nat Genet 45:279–284
Qiao J, Pritha P, Lan Qiao SL, Josifi E, Tiao JR, Chung DH (2012) PI3K/AKT and ERK regulate retinoic acid-induced neuroblastoma cellular differentiation. Biochem Biophys Res Commun 424:421–426
Redfern CP, Lovat PE, Malcolm AJ, Pearson AD (1994) Differential effects of 9-cis and all-trans retinoic acid on the induction of retinoic acid receptor-beta and cellular retinoic acid-binding protein II in human neuroblastoma cells. J Biochem 304:147–154
Ross RA, Spengler BA, Domènech C, Porubcin M, Rettig WJ, Biedler JL (1995) Human neuroblastoma I-type cells are malignant neural crest stem cells. Cell Growth Differ 6:449–456
Rozzo CV, Chiesa Caridi G, Pagnan G, Ponzoni M (1997) Induction of apoptosis in human neuroblastoma cells by abrogation of integrin-mediated cell adhesion. Int J Cancer 70:688–698
Schug TT, Berry DC, Shaw NS, Travis SN, Noy N (2007) Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 129:723–733
Sidell N (1982) Retinoic acid-induced growth inhibition and morphologic differentiation of human neuroblastoma cells in vitro. J Natl Cancer Inst 68:589–596
Skipper HE, Schabel FM Jr (1984) Tumor stem cell heterogeneity: implications with respect to classification of cancers by chemotherapeutic effect. Cancer Treat Rep 68:43–61
Sucov HM, Evans RM (1995) Retinoic acid and retinoic acid receptors in development. Mol Neurobiol 10:169–184
Tachibana K, Yamasaki D, Ishimoto K, Doi T (2008) The role of PPARs in cancer. PPAR Res 2008:102737
Takahashi J, Palmer TD, Gage FH (1999) Retinoic acid and neurotrophins collaborate to regulate neurogenesis in adult-derived neural stem cell cultures. J Neurobiol 38:65–81
Tang DG (2012) Understanding cancer stem cell heterogeneity and plasticity. Cell Res 22:457–472
Trochet D, Bourdeaut F, Janoueix-Lerosey I, Deville A, de Pontual L, Schleiermacher G, Coze C, Philip N, Frébourg T, Munnich A, Lyonnet S (2004) Germline mutations of the paired-like homeobox 2b (phox2b) gene in neuroblastoma. Am J Hum Genet 74:761–764
Voigt A, Hartmann P, Zintl F (2000) Differentiation proliferation and adhesion of human neuroblastoma cells after treatment with retinoic acid. Cell Commun Adhes 7:423–440
Voigt A, Zintl F (2003) Effects of retinoic acid on proliferation, apoptosis, cytotoxicity, migration, and invasion of neuroblastoma cells. Pediatr Blood Cancer 40:205–213
Wang K, Diskin SJ, Zhang H, Attiyeh EF, Winter C, Hou C, Schnepp RW, Diamond M, Bosse K, Mayes PA, Glessner J (2011) Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature 469:216–220
Wolf G (2008) Retinoic acid as cause of cell proliferation or cell growth inhibition depending on activation of one of two different nuclear receptors. Nutr Rev 66:55–59
Xie HR, Hu LS, Li GY (2010) SH-SY5Y human neuroblastoma cell line: in vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin Med J 123:1086–1092
Acknowledgements
This work was supported by internal research funds from the University of Scranton (GG) and Presidential University Summer Fellowships to EH and NM. We would like to thank Ms. Devaney Wood for her assistance with the Western blotting studies.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: Tetsuji Okamoto
Rights and permissions
About this article

Cite this article
Harasym, E., McAndrew, N. & Gomez, G. Sub-micromolar concentrations of retinoic acid induce morphological and functional neuronal phenotypes in SK-N-SH neuroblastoma cells. In Vitro Cell.Dev.Biol.-Animal 53, 798–809 (2017). https://doi.org/10.1007/s11626-017-0190-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-017-0190-x