
Abstract
Malignant gliomas are characterized by their high level of resistance to chemo- and radiotherapy and new treatment options are urgently required. We previously demonstrated that brefelamide, an aromatic amide isolated from methanol extracts of cellular slime molds Dictyostelium brefeldianum and D. giganteum, had antiproliferative effects on 1321N1 human astrocytoma cells, a model of glioma. In this study, we investigated the mechanisms by which brefelamide inhibited 1321N1 and PC12 rat pheochromocytoma cell proliferation. When cells were cultured in serum-free medium, hepatocyte growth factor (HGF) increased survival of 1321N1 cells but not PC12 cells. HGF receptor, c-MET, was strongly expressed in 1321N1 cells, but not in PC12 cells. Pretreatment of 1321N1 cells with brefelamide inhibited both HGF-induced cell survival and expression of c-MET. Phosphorylation of extracellular signal-regulated kinase (ERK) and AKT was increased by HGF, but these changes were inhibited by brefelamide pretreatment. Moreover, HGF mRNA levels and secretion were reduced by brefelamide. These results suggest that brefelamide reduces survival of 1321N1 cells via multiple effects including suppression of HGF receptor expression and HGF secretion and inhibition of ERK and AKT phosphorylation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glial cells are non-neuronal cells that provide myelin and physical support to neurons through the release of neurotrophic factors such as neurotrophin family members (Althaus and Richter-Landsberg 2000), and also express a number of neurotransmitter receptors for neuron–glia and/or glia–glia interactions (Fields et al. 2002). Gliomas originate from glial cells and are common tumors of the brain and central nervous system, expressing a number of growth factors and corresponding receptors that contribute to their malignancy by inhibiting apoptosis and stimulating angiogenesis (Abounader and Laterra 2005). Among them, hepatocyte growth factor (HGF), also known as scatter factor, and its transmembrane tyrosine kinase receptor, c-MET, play an important role in the development and progression of primary tumors and secondary metastases (Tomiya et al. 2007).
HGF was originally identified as a potent mitogenic protein for hepatocytes, and subsequent studies revealed that HGF has multiple biological effects on various cell types. HGF and c-MET are present in the developing and adult mammalian nervous system, where they have a neurotrophic function, stimulating cell proliferation, motility, invasion, angiogenesis, and protection from apoptosis (Corso et al. 2005). A wide variety of human tumors including gliomas express both HGF and c-MET, and their expression correlates with glioma grade (Birchmeier et al. 2003). Importantly, endogenous CMET and HGF gene expression knockdown in human glioblastoma cells inhibits c-MET-dependent signal transduction, affecting cell proliferation, tumorigenicity, and chemoresistance (Abounader et al. 1999).
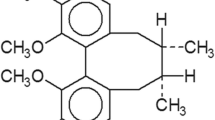
Malignant gliomas are highly refractory to current treatment modalities including radiotherapy and chemotherapy, despite significant attempts to harness the wealth of genomic data generated over the past decades to produce targeted therapies. While likely target candidates have been identified, such as epidermal growth factor (EGF) receptor, members of the RAS/RAF/extracellular signal-regulated kinase (ERK) and PI3K/AKT/mTOR pathways (Kesari et al. 2005), successful molecularly targeted therapies remain elusive. Brefelamide (Fig. 1) is an aromatic amide that was originally isolated from Dictyostelium cellular slime molds. In a previous study, we revealed that brefelamide treatment inhibited the proliferation of 1321N1 human astrocytoma cells (Kikuchi et al. 2005). Subsequently, we showed that brefelamide inhibits ERK phosphorylation by downregulating EGF receptor activity (Honma et al. 2009). In the present study, we investigated the impact of brefelamide treatment on HGF-induced survival of 1321N1 cells. We show that brefelamide reduced the survival of 1321N1 cells via multiple mechanisms including suppression of HGF receptor expression and HGF secretion and inhibition of ERK and AKT phosphorylation.

Structure of brefelamide.
Materials and Methods
Brefelamide Synthesis
N-(3-(2-Amino-3-(4-hydroxyphenoxy)phenyl)-3-oxopropyl)-4-hydroxybenzamide (brefelamide) was synthetized as previously described (Kikuchi et al. 2005). All spectral data for the synthetic products were identical to that of products originally isolated from methanol extracts of the fruiting bodies of Dictyostelium brefeldianum and D. giganteum (Kikuchi et al. 2005). 1H NMR spectrum showed that it was more than 99% pure. Brefelamide was dissolved in dimethyl sulfoxide (DMSO) to generate a 50 mM stock solution. The stock was diluted to the required concentration with complete growth medium before use (final DMSO concentration was less than 0.1%).
Cell Culture
1321N1 human astrocytoma cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Nissui Pharmaceutical Co., Tokyo, Japan) supplemented with 5% fetal bovine serum (FBS; HyClone Laboratories, Logan, UT, USA), 50 units/ml penicillin, and 50 g/ml streptomycin in a humidified incubator in an atmosphere of 95% air and 5% CO2 (Honma et al. 1999). PC12 rat pheochromocytoma cells were grown in DMEM supplemented with 10% FBS, 5% horse serum (Life Technologies, Rockville, MD), 50 unit/ml of penicillin and 50 mg/ml of streptomycin.
MTT Assay
1321N1 cells and PC12 cells were seeded into 96-well plates at a density of 2.5 × 104 cells/ml. Two days after seeding, the medium was replaced with serum-free DMEM, and cells were incubated with or without 10 M brefelamide for 24 h, and then treated with HGF (R&D Systems, Minneapolis, MN, USA). Then, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Wako Pure Chemicals, Tokyo, Japan) diluted to 0.1 mg in phosphate-buffered saline was added to each well, and the cells were incubated for a further 4-h period at 37 °C. After removal of the medium, the cells were lysed with DMSO to dissolve the formazan product. Absorbance at 590 nm was then measured with a plate reader (Sunrise, Tecan, Switzerland).
Western Blotting
Cells were seeded into 60-mm dishes at a density of 5 × 104 cells/ml. After 48 h from seeding, the medium was replaced with serum-free DMEM, and cells were incubated with or without brefelamide for 24 h, followed by addition of HGF. At the end of the incubation period, the medium was aspirated and cells were lysed by the addition of Laemmli sample buffer. Western blotting was performed as described previously (Honma et al. 2006). Protein concentration was quantified by BCA assay (Thermo Fisher Scientific) and samples were run on SDS-PAGE gels before transfer to polyvinylidene difluoride membranes. Membranes were incubated for 2 h with 0.5% skim milk in Tris-buffered saline with Tween 20 and then incubated with anti-ERK, anti-phospho-ERK, anti-AKT, anti-phospho-AKT, anti-c-MET (all Cell Signaling Technology, Beverly, MA), or anti-β-actin (Sigma–Aldrich, St. Louis, MO) antibodies (all at 1:1000 dilution). Membranes were then incubated for 2 h at 25 °C with a 1:3000 dilution of an anti-rabbit horseradish peroxidase (HRP)-linked immunoglobulin G (IgG) or an anti-mouse HRP-linked IgG (Cell Signaling Technology, Beverly, MA) at 25 °C for 2 h. Immunoreactivity was determined using an enhanced chemiluminescence assay kit (GE Healthcare, Buckinghamshire, UK), and immunopositive bands were detected using a LAS-3000 luminescent image analyzer (Fujifilm, Tokyo, Japan). Band density was analyzed using Image Gauge software (Fujifilm). Data are expressed as the ratio of phosphorylated to total ERK, AKT, or -actin.
Real-Time RT-PCR
Total RNA was extracted using the GenElute Mammalian Total RNA Miniprep Kit (Sigma–Aldrich), according to the manufacturer’s protocol. First-strand cDNA was prepared from total RNA (1 μg) using oligo(dT) primer and ReverTra Ace reverse transcriptase (Toyobo Co. Ltd., Osaka, Japan). Human CMET cDNA consisting of 85 base pairs (bp) and rat Cmet cDNA consisting of 310 bp were amplified using the following protocol: 30 cycles of 98 °C for 10 s, 60 °C for 30 s, and 68 °C for 60 s. Samples were then run out on 2% agarose gels by electrophoresis to confirm the PCR products. Real-time PCR analysis was performed using an Mx3000P analyzer (Stratagene, La Jolla, CA). SYBR Premix Ex Taq (TaKaRa Bio, Otsu, Shiga, Japan) was used for cDNA amplification. The sequences of the primer pairs were as follows: human CMET, 5′-TGCAAAGCTGCCAGTGAAGT-3 (sense) and 5′-GCCAAAGGACCACACATCTGA-3′ (antisense); rat Cmet, 5′-CTGGGAGCTCATGACGAGAGG-3′ (sense) and 5′-GCTAATGTTGTCTTGGGATGGC-3′ (antisense) (Lim et al. 2008); human HGF, 5′-AGAAATGCAGCCAGCATCATC-3′ (sense) and 5′-CACATGGTCCTGATCCAATCTTT-3′ (antisense) (Saghizadeh et al. 2005); human G3PDH, 5′-ACCACAGTCCATGCCATCAC-3′ (sense) and 5′-TCCACCACCCTGTTGCTGTA-3′ (antisense). The real-time PCR conditions were as follows: 95 °C for 2 min, followed by 30 cycles of 15 s at 95 °C, 30 s at 56 °C, and 30 s at 72 °C. Gene expression was normalized to G3PDH gene expression.
Enzyme-Linked Immunosorbent Assay
HGF Human ELISA Kit (Life Technologies) was used to quantify HGF secreted from cultured 1321N1 cells. Assays were performed according to the manufacturer’s instructions. All samples were measured in duplicate.
Statistical Analysis
Results are expressed as the mean ± S.E.M. Student’s t test for two-sample comparisons was used to calculate the significance of differences between values, while for multiple comparisons, one-way analysis of variance with Dunnett’s test was used. P values of <0.05 were considered statistically significant.
Results
MTT assays were performed to determine the effect of HGF on 131N1 human astrocytoma and PC12 rat pheochromocytoma cell proliferation following treatment with 0.1 or 1 ng/ml HGF for 1 or 3 d. HGF treatment increased the survival rate of 1321N1 cells but not PC12 cells in a concentration-dependent manner (Fig. 2).

Effect of HGF on survival rate of 1321N1 human astrocytoma cells and PC12 rat pheochromocytoma cells. Two days after seeding, the medium was replaced with serum-free DMEM for 24 h, and then 1321N1 human astrocytoma cells (a) and PC12 rat pheochromocytoma cells (b) were treated with vehicle (circles), 0.1 ng/ml HGF (triangles), 1 ng/ml HGF (squares), or serum as positive control (rhombuses) for 1 or 3 d. MTT assays were performed to determine cell viability. Each point represents the mean ± S.E.M. from at least three independent experiments. Asterisks indicate significant differences compared with the corresponding vehicle group (P < 0.05).
We next examined whether c-MET, the HGF receptor, was expressed in these cell lines. c-MET mRNA and protein were strongly expressed in 1321N1 cells, in contrast with PC12 cells, in which the expression levels were much lower (Fig. 3).

Expression of c-MET mRNA and protein in 1321N1 and PC12 cells. Two days after seeding, the medium was replaced with serum-free DMEM for 24 h, and then mRNA or protein was extracted. (a) Ethidium bromide staining of PCR products separated on 2% agarose gels. Arrows indicate the 85-bp fragment of human CMET cDNA (left) and the 310-bp fragment of rat Cmet cDNA (right). (b) c-MET protein from 1321N1 and PC12 cells.
Because HGF is reported to stimulate phosphorylation of ERK and AKT in many cells (Menakongka and Suthiphongchai 2010; Tang et al. 2010), we investigated these effects in 1321N1 and PC12 cells. HGF significantly increased phosphorylation of ERK and AKT at 5 min and 30 min, respectively, in 1321N1 cells (Fig. 4). However, ERK and AKT phosphorylation was not induced by HGF in PC12 cells (data not shown). To assess the effect of brefelamide on HGF-induced cell survival, we performed MTT assays following treatment of 1321N1 cells with 10 μM brefelamide for 24 h prior to the addition of 0.1 or 1 mg/ml HGF. Our data showed that brefelamide inhibited HGF-induced cell viability in 1321N1 cells (Fig. 5a).

Effect of HGF on ERK and AKT phosphorylation in 1321N1 cells. Cell lysates were separated by SDS-PAGE and then immunoblotting was performed using antibodies against (a) ERK and phospho-ERK, and (b) AKT and phospho-AKT. Each column represents the mean ± S.E.M. from at least three independent experiments. Phosphorylation levels were normalized to total ERK or total AKT levels and are shown as a percentage of the control. Asterisks indicate significant differences compared with the vehicle group (P < 0.05).

Effect of brefelamide on HGF-induced cell survival, c-MET expression, and ERK and AKT phosphorylation in 1321N1 cells. (a) Two days after seeding, the medium was replaced with serum-free DMEM and cultured in the presence of 10 μM brefelamide for 24 h. 1321N1 cells was treated with vehicle (circles), 0.1 ng/ml HGF (triangles), 1 ng/ml HGF (squares), or serum as a positive control (rhombuses) for 1 or 3 d. MTT assays were performed to determine cell viability. Each point represents the mean ± S.E.M. from three independent experiments. Asterisks indicate significant differences compared with the corresponding vehicle group (P < 0.05). (b) Real-time PCR analysis of the expression of CMET mRNA. Each column represents the mean ± S.E.M. from six independent experiments. Asterisk indicates a significant difference compared with the group without brefelamide treatment (P < 0.05). (c) Western blotting analysis of c-MET protein expression. Cell lysates were separated by SDS-PAGE and then western blotting was performed using an anti-c-MET antibody. Each column represents the mean ± S.E.M. from three independent experiments. Data are normalized to the corresponding -actin level and are shown as a percentage of the control. Asterisk indicates a significant difference compared with the group without brefelamide treatment (P < 0.05). Cell lysates were separated by SDS-PAGE and then immunoblotting was performed using antibodies against (d) ERK and phospho-ERK and (e) AKT or phospho-AKT. Each column represents the mean ± S.E.M. from at least three independent experiments. Phosphorylation levels were normalized to total ERK or total AKT levels and are shown as a percentage of the control.
Next, we examined whether brefelamide affected the expression of c-MET in 1321N1 cells. Dosing of the cells with brefelamide for 24 h significantly inhibited the expression of c-MET mRNA and protein (Fig. 5b and c). Moreover, brefelamide treatment affected HGF-induced ERK and AKT phosphorylation in 1321N1 cells (Fig. 5d and e).
We next performed real-time PCR and ELISA to quantify the levels of HGF mRNA and HGF protein secretion following incubation of 1321N cells with brefelamide. Both HGF mRNA levels and HGF protein secretion were significantly reduced by brefelamide treatment (Fig. 6).

Effect of brefelamide on HGF secretion in 1321N1 cells. Two days after seeding, the medium was replaced with serum-free DMEM, and cultured in the presence or absence of 10 μM brefelamide for 24 h. (a) Real-time PCR analysis of HGF mRNA expression. Each column represents the mean ± S.E.M. from six independent experiments. Asterisk indicates a significant difference compared with the group without brefelamide application (P < 0.05). (b) ELISA analysis of HGF protein expression. Each column represents the mean ± S.E.M. from four independent experiments. Asterisk indicates a significant difference compared with the group without brefelamide application (P < 0.05).
Discussion
In this study, we demonstrated for the first time that brefelamide reduced HGF-induced cell survival in 1321N1 human astrocytoma cells, a model of glioma.
Brefelamide is assumed to exert its effects through multiple mechanisms including inhibition of ERK phosphorylation via downregulation of EGFR, and also reduction of HGF receptor expression and HGF secretion in parallel with inhibition of ERK and AKT phosphorylation in 1321N1 cells.
HGF exerts mitogenic, morphogenic, and motogenic activities in various types of cells (Brinkmann et al. 1995; Matsumoto and Nakamura 1996). In the present study, vehicle treatment in serum-free conditions decreased 1321N1 cell number because the growth factors contained in serum were necessary for the survival of 1321N1 cells. However, HGF treatment increased cell survival and stimulated phosphorylation of ERK and AKT in 1321N1 cells but not PC12 cells. The reason for the difference is most likely because c-MET, the HGF receptor, is strongly expressed in 1321N1 cells. It has been shown that ERK plays a pivotal role in cell proliferation, while AKT regulates multiple cellular functions such as cell proliferation and survival, angiogenesis, glycogen synthesis, protein synthesis, and transcription (Brazil and Hemmings 2001). Thus, it is possible that HGF-induced survival of 1321N1 cells was a consequence of ERK and AKT phosphorylation.
In a previous study, we demonstrated that brefelamide inhibited the proliferation of 1321N1 cells. Therefore, we examined the effect of brefelamide on HGF-induced 1321N1 cell survival. Brefelamide reduced cell survival and ERK and AKT phosphorylation in 1321N1 cells. Moreover, brefelamide inhibited the expression of c-MET mRNA and protein. Therefore, brefelamide might inhibit ERK and AKT phosphorylation by reducing HGF receptor expression in 1321N1 cells. Luteolin, an inhibitor of fatty-acid synthase, blocked HGF-induced scattering and motility in DU145 prostate cancer cells by reducing expression of c-MET protein and phosphorylation of AKT (Coleman et al. 2009). FASN, which catalyzes the synthesis of long-chain saturated fatty acids, is overexpressed in many human cancers, and this overexpression is correlated with advanced disease (Menendez and Lupu 2007). In contrast with our results, luteolin failed to inhibit ERK phosphorylation, suggesting that brefelamide may not be a FASN inhibitor. It is likely that the inhibition of AKT phosphorylation led to suppression of HGF-induced proliferation.
Xie et al. (2011) recently reported that HGF autocrine status could be the dominant determinant of c-MET signaling pathway activity. Chattopadhyay et al. (2004) showed that HGF induced proliferation of U87 human astrocytoma cells, while the proliferation of these cells was inhibited by transforming growth factor- (TGF-)-1. Furthermore, TGF--1 and other TGF- family proteins, such as bone morphogenetic proteins and activin A, inhibited HGF secretion from U87 human astrocytoma cells through transcriptional repression without inducing concomitant changes in c-MET expression. In the present study, HGF secretion was significantly reduced by brefelamide treatment. The reduction in HGF secretion from 1321N1 cells may be a critical effect of brefelamide that suppresses glioma progression. Further studies are necessary to elucidate the detailed mechanisms of brefelamide-induced inhibition of HGF secretion.
Recent findings have suggested several promising molecular therapeutics that specifically inhibit c-MET in gliomas. PHA665752, a highly selective c-MET inhibitor that competitively inhibits binding of ATP to the tyrosine kinase domain of c-MET, is capable of antagonizing HGF-induced migration and proliferation or survival of c-MET-expressing neuroblastoma cells (Crosswell et al. 2009). SGX523, a selective and orally bioavailable small-molecule inhibitor of c-MET kinase, inhibits c-MET, AKT, and ERK phosphorylation, cell proliferation, cell cycle progression, migration, and invasion in human glioblastoma cell lines, glioblastoma primary cells, glioblastoma stem cells, and medulloblastoma cell lines (Guessous et al. 2010). Consequently, inhibition of c-MET may be a feasible and promising approach for brain tumor therapy.
Conclusions
We have shown that brefelamide inhibited HGF-induced survival of 1321N1 human astrocytoma cells and reduced c-MET expression, HGF secretion, and ERK and AKT phosphorylation. Further studies are required to determine the precise mechanisms of c-MET and HGF inhibition. However, treatment of gliomas with brefelamide may be an effective therapeutic strategy.
References
Abounader R, Laterra J (2005) Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncology 7:436–451
Abounader R, Ranganathan S, Lal B, Fielding K, Book A, Dietz H, Burger P, Laterra J (1999) Reversion of human glioblastoma malignancy by U1 small nuclear RNA/ribozyme targeting of scatter factor/hepatocyte growth factor and c-met expression. J Natl Cancer Inst 91:1548–1556
Althaus HH, Richter-Landsberg C (2000) Glial cells as targets and producers of neurotrophins. Int Rev Cytol 197:203–277
Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF (2003) Met, metastasis, motility and more. Nat Rev Mol Cell Biol 4:915–925
Brazil DP, Hemmings BA (2001) Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci 26:657–664
Brinkmann V, Foroutan H, Sachs M, Weidner KM, Birchmeier W (1995) Hepatocyte growth factor/scatter factor induces a variety of tissue-specific morphogenic programs in epithelial cells. J Cell Biol 131:1573–1586
Chattopadhyay N, TFH J, Godbole MM, Brown EM (2004) Transforming growth factor beta receptor family ligands inhibit hepatocyte growth factor synthesis and secretion from astrocytoma cells. Brain Res Mol Brain Res 121:146–150
Coleman DT, Bigelow R, Cardelli JA (2009) Inhibition of fatty acid synthase by luteolin post-transcriptionally down-regulates c-Met expression independent of proteosomal/lysosomal degradation. Mol Cancer Ther 8:214–224
Corso S, Comoglio PM, Giordano S (2005) Cancer therapy: can the challenge be MET? Trends Mol Med 11:284–292
Crosswell HE, Dasgupta A, Alvarado CS, Watt T, Christensen JG, De P, Durden DL, Findley HW (2009) PHA665752, a small-molecule inhibitor of c-Met, inhibits hepatocyte growth factor-stimulated migration and proliferation of c-Met-positive neuroblastoma cells. BMC Cancer 9:411
Fields RD, Stevens-Graham B (2002) New insights into neuron-glia communication. Science 298:556–562
Guessous F, Zhang Y, diPierro C, Marcinkiewicz L, Sarkaria J, Schiff D, Buchanan S, Abounader R (2010) An orally bioavailable c-Met kinase inhibitor potently inhibits brain tumor malignancy and growth. Anti Cancer Agents Med Chem 10:28–35
Honma S, Nakahata N, Kobayashi H, Ikeda S, Takeda N, Ohizumi Y (1999) Decrease in thromboxane A2 receptor expression by differentiation with dibutyryl cyclic AMP in 1321N1 human astrocytoma cells. Prostaglandins Other Lipid Mediat 58:51–62
Honma S, Saito M, Kikuchi H, Saito Y, Oshima Y, Nakahata N, Yoshida M (2009) A reduction of epidermal growth factor receptor is involved in brefelamide-induced inhibition of phosphorylation of ERK in human astrocytoma cells. Eur J Pharmacol 616:38–42
Honma S, Saika M, Ohkubo S, Kurose H, Nakahata N (2006) Thromboxane A2 receptor-mediated G12/13-dependent glial morphological change. Eur J Pharmacol 545:100–108
Kesari S, Ramakrishna N, Sauvageot C, Stiles CD, Wen PY (2005) Targeted molecular therapy of malignant gliomas. Curr Neurol Neurosci Rep 5:186–197
Kikuchi H, Saito Y, Sekiya J, Okano Y, Saito M, Nakahata N, Kubohara Y, Oshima Y (2005) Isolation and synthesis of a new aromatic compound, brefelamide, from dictyostelium cellular slime molds and its inhibitory effect on the proliferation of astrocytoma cells. J Org Chem 70:8854–8858
Lim CS, Walikonis RS (2008) Hepatocyte growth factor and c-Met promote dendritic maturation during hippocampal neuron differentiation via the Akt pathway. Cell Signal 20:825–835
Matsumoto K, Nakamura T (1996) Emerging multipotent aspects of hepatocyte growth factor. J Biochem 119:591–600
Menakongka A, Suthiphongchai T (2010) Involvement of PI3K and ERK1/2 pathways in hepatocyte growth factor-induced cholangiocarcinoma cell invasion. World J Gastroenterol 16:713–722
Menendez JA, Lupu R (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 7:763–777
Saghizadeh M, Kramerov AA, Tajbakhsh J, Aoki AM, Wang C, Chai NN, Ljubimova JY, Sasaki T, Sosne G, Carlson MR, Nelson SF, Ljubimov AV (2005) Proteinase and growth factor alterations revealed by gene microarray analysis of human diabetic corneas. Invest Ophthalmol Vis Sci 46:3604–3615
Tang MK, Zhou HY, Yam JW, Wong AS (2010) c-Met overexpression contributes to the acquired apoptotic resistance of nonadherent ovarian cancer cells through a cross talk mediated by phosphatidylinositol 3-kinase and extracellular signal-regulated kinase 1/2. Neoplasia 12:128–138
Tomiya T, Nishikawa T, Inoue Y, Ohtomo N, Ikeda H, Tejima K, Watanabe N, Tanoue Y, Omata M, Fujiwara K (2007) Leucine stimulates HGF production by hepatic stellate cells through mTOR pathway. Biochem Biophys Res Commun 358:176–180
Xie Q, Bradley R, Kang L, Koeman J, Ascierto ML, Worschech A, De Giorgi V, Wang E, Kefene L, Su Y, Essenburg C, Kaufman DW, DeKoning T, Enter MA, O’Rourke TJ, Marincola FM, Vande Woude GF (2011) Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc Natl Acad Sci U S A 109:570–575
Acknowledgments
We are grateful to Dr. Yoshiteru Oshima and Dr. Haruhisa Kikuchi (Tohoku University, Sendai, Japan) for providing brefelamide.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interest.
Additional information
Editor:Tetsuji Okamoto
Rights and permissions
About this article

Cite this article
Honma, S., Takasaka, S., Ishikawa, T. et al. Effect of brefelamide on HGF-induced survival of 1321N1 human astrocytoma cells. In Vitro Cell.Dev.Biol.-Animal 52, 705–711 (2016). https://doi.org/10.1007/s11626-016-0019-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-016-0019-z