
Abstract
LiFePO4/C composites were prepared via liquid phase method by using sodium ligninsulfonate (SL) as carbon source and surfactant. Effects of SL on the morphology and properties of LiFePO4/C composite were investigated, indicating that SL plays an important role in the formation of porous structures by self-assembly of carbon chains. The results illustrated that the LFP/Cs-2 (SL 30%) is well confined in the nano-porous of the carbon sheet, exhibiting a stable and high reversible capacity of 161 mAhg−1 at 0.1 C and excellent high rate capacity of 135 mAhg−1 at 8 C in CR2032 cells. In addition, the LFP/Cs-2 with a compaction density of 2.4 gcm−3 shows a remarkably high rate capability of 131 mAhg−1 at 15 C in 18,650 batteries. The excellent performance of LFP/Cs-2 is attributed to the high diffusion rate of lithium ion within the nano-porous structure and outstanding electronic conductivity of the continuous carbon conductive network.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lithium ion batteries (LIBs), including stretchable or foldable Li-ion batteries, have been the research focus in the academic and industrial communities as reflected in many review literatures or articles each year [1,2,3,4,5,6,7,8,9,10,11,12]. Cathode as the most important components of LIBs and the key factor for the performance of LIBs also continues to draw attentions, emerging various cathode materials [6]; among the compounds of the olivine family (LiMPO4 with M = Fe, Mn, Ni, or Co), only the olivine-structured LiFePO4 (LFP) has become a hot research topic and considered to be one of the most promising candidates for power batteries since the pioneer work of Prof. Goodenough in 1997 [13], who was Nobel Prize Winner in Chemistry in 2019. It has been well documented that the LFP has several characteristics of stable voltage plateau of 3.5 V vs. Li/Li+, high theoretical capacity (170 mAhg−1), good cycle stability, thermal stability, low raw material cost, environmental friendliness, and safety [14,15,16]. However, although olivine-structured LFP has many abovementioned merits, and ultimately, high-quality LFP materials have been successfully developed and commercialized in energy storage and EV, it still exists some drawbacks of poor intrinsic electronic and lithium ion conductivities originating from the lack of mixed valency and the one dimensional lithium ion diffusion, influencing its high electrochemical performance, especially high rate capability, limiting the practical use of LFP in LIBs for HEVs and EVs, especially in the so-called next generation of lithium-ion batteries.
Therefore, in order to tackle these challenges and improve the performance of LFP, several strategies have been proposed, and many efforts have been devoted to improving the extremely poor ion diffusion rate and electron conductivity. The electrical conductivity and the diffusion of lithium ions can be enhanced by using novel strategies such as surface modification using graphite, metal, metal oxide, and phosphate; particle size reduction; lattice substitution (doping); composition optimization; electrolyte additives; controlling morphology; structural engineering; and cathode prelithiation additives, all of which can improve electrochemical performance of LFP [15,16,17,18,19,20,21,22,23].
Considering that the electrochemical performances can be significantly affected by morphology of LFP, the synthesis of LFP with various morphologies and structures has been receiving extensive research interest [24]. Among the various methods for improving the battery performance of LFP mentioned above, compositing the LFP with carbon nanomaterials seems to be the most promising procedure and is widely applied and studied systemically, because it is facile and efficient [25, 26]. It is well known that electrical conductivity is one of the key factors affecting the capacity, rate, and low-temperature performance of LIBs [17, 19]. For the LiFePO4/C composite, carbon nanomaterials usually serve as a conductive agent to improve the electrical conductivity while increasing the material porosity in which the solid-state diffusion distances are significantly shortened. LiFePO4/C composite can be fabricated through carbon coating by using different carbon sources, such as biomass, polymer, and graphene [27], and MOFs (metal organic frameworks) [28,29,30] via ex situ or in situ method, such as solid state reaction method, liquid phase method, sol–gel method, hydrothermal method, and spray pyrolysis method [6]. However, during carbon coating treatment, there are two kinds of carbon attached to LFP surface, namely, graphitic carbon (sp2-hybridized carbon) and amorphous carbon [25, 26]. It is known that the conductivity of amorphous carbon is very poor, the carbon introduced in LiFePO4/C composites is usually also amorphous being formed at the sintering temperature of pure olivine phase, and the graphitization capacity of precursors is often difficult to achieve at high levels, resulting in the difficulty of increasing the rate performance of LFP cathode material to an ultra-high level. Therefore, the utilization of new graphite materials (sp2-hybridized carbon) as carbon sources to modify LFP with a higher degree of graphitization is the mainstream of current research on carbon coating on LFP. Therefore, theoretically, organic compounds with a higher degree of sp2-hybridized carbon atoms can be selected and considered as carbon sources, as long as these organic compounds can be transformed to a carbon shell with a suitable degree of graphitization. Of course, for the liquid phase method, the solubility of the carbon precursor is crucial.
Lignosulfonate is a by-product of the pulping industry, which is a type of macromolecular surfactant widely used as interfacial additive in various industrial fields, and the commercial sodium lignosulfonate (SL) is part of a by-product of sulfite pulping and is water soluble; SL was an effective chelating agent which consists of a large number of hydrocarbon chains which can be easily converted to highly conductivity carbon chains during the sintering process. SL is also an anionic surfactant with strong dispersing ability, which can make the precursor react uniformly. SL was used as the precursor for the synthesis of hierarchical porous carbons for supercapacitors with high volumetric energy density [31, 32]. However, according to literature survey and to the best of our knowledge, there is no report regarding the synthesis of LEP/C composite using SL as carbon source.
In order to achieve the excellent electrochemical performance of LiFePO4 for practical applications, it is necessary and urgent to develop a simplified and facile synthesis process for producing LiFePO4/C composites with controllable particle size and uniform carbon coating layer. We have reported a liquid phase method to synthesis LiFePO4/graphene using CH3COOLi•2H2O, Fe(NO3)3•9H2O, NH4H2PO4, and C6H8O7 as the raw materials. The prepared LiFePO4/G shows exciting performance with highly capacity retention rate after various dis-charge currents, indicating that graphene is promising conductive additives for high power Li-ion battery electrode materials [33].
Herein, the synthesis of LFP/C was carried out via an improved liquid phase method based on the abovementioned literature by using citric acid, and mixture of citric acid and sodium ligninsulfonate (SL) as carbon sources, and SL was used to modify the morphology and improve the conductivity of LFP. The effect of SL addition content on the morphology and electrochemical performance of the LiFePO4/C were investigated. LFP nanoparticles were coated by conductivity carbon layers (3–6 nm), which significantly improved the electrochemical reactivity and reversibility of LFP nanoparticles. It was found that the LFP material prepared by the improved liquid phase method using SL (30 wt%) as carbon source and surfactant would result in the formation of porous structures by self-assembly of carbon chains, greatly improving the conductivity of the material and limit the growth of the particles. It exhibited outstanding high rate performance and stable capacity retention of low temperature property.
Experimental section
Materials
LiOH·H2O, Fe(C2H3O2)2, NH4H2PO4, NH4NO3, citric acid (C6H8O7), C2H5OH, and sodium lignosulfonate (C20H24Na2O10S2, SL) were purchased from Sigma-Aldrich (Shanghai, China) and used without further purification unless stated otherwise. All chemicals were analytical grade reagents.
Synthesis of LFP/C composite
The synthesis of LFP/C was carried out via an improved liquid phase method based on the literature we published previously [33] as described below: LiOH·H2O, Fe(C2H3O2)2,, NH4H2PO4, and C6H8O7 were used as the raw materials at a molar ratio of 1.05:1:1:1. The distilled water and ethanol were used as solvent. The mixed solution was stirred at room temperature for 1 h and then dried at 80 °C. Finally, the dried precursor was heated to 700 °C at a heating rate of 5 °C·min–1 under the N2 atmosphere and maintained at this temperature for 6 h and finally was cooled slowly to obtain LFP/C.
Synthesis of LFP/CS composite using SL as a carbon source and surfactant
The synthesis method of LFP/CS was basically the same as that of LFP/C. The difference was that SL was also added as a carbon source and surfactant. The weight ratios of C6H8O7 to SL were 1:0.1 (10 wt% of the C6H8O7), 1:0.3 (30 wt% of the C6H8O7), and 1:0.5 (50 wt% of the C6H8O7); and the obtained composites were labeled as LFP/Cs-1, LFP/Cs-2, and LFP/Cs-3, respectively.
The synthesis process is schematically illustrated in Fig. 1.

Schematic illustration of the preparation process for the LFP/C composites
Material characterization
The crystal structures of the samples were characterized by X-ray diffraction (XRD) using a Bruker D8 ADVANCE diffractometer with Cu-Kα1,2 radiations at a voltage of 40 kV and a current of 40 mA. The scans were performed in the 2θ range from 10° to 90° with a scanning step width of 0.02° and 1 s time per step for each sample. Raman analysis was performed on a JY HR800 Raman spectrophotometer. Thermogravimetric analysis (TGA) was performed on a 209 F3 Thermogravimetric Analyzer (TG 209 F3 NETZSCH Germany), which was carried out from temperature to 750 ℃ under N2 atmosphere at the rate of 5 ℃min−1. Scanning electron microscopy (SEM) was performed using a Hitachi S-4800 microscope operating at 5 kV. Transmission electron microscopy (TEM) was performed with a Hitachi H-7000 microscope at an acceleration voltage of 100 kV. The accurate content of carbon in the LFP composite was measured by HCS-140 high frequency infrared carbon–sulphur analyzer. Tristar II3020 surface area analyzer was used to measure the surface area of materials. The resistivity of materials was measured by RTS-8 four-probe tester.
Electrochemical measurements
Electrochemical measurements were performed by using CR2032 coin cell with Li metal as the anode and Column 18,650 lithium ion batteries with graphite as the anode. The working electrode was prepared by mixed 85 wt% of active materials (LFP/C and LFP/Cs-2), 10 wt% of acetylene black, and 5 wt % of polyvinylidene fluoride (PVDF), using N–N-methyl-2-pyrrolidone (NMP) as the solvent. The electrolyte was a 1 mol·L−1 LiPF6 in a 1:1:1 (V/V/V) ethylene carbonate/diethyl carbonate/dimethyl carbonate ration and Celgard 2300 porous polypropylene film as the separator. The charge/discharge experiment was performed between 2.0 and 3.75 V using a Land CT2001A battery test system (Hubei LANBTS Company, Hubei China) under the condition of 25 ± 2℃ (rate charging cutoff voltage of 3.75 V; constant voltage charging cutoff voltage of 3.75 V, current 0.05 mA; rate discharge cutoff voltage of 2.00 V; charge and discharge current density: 0.1 C (nominal specific capacity: 150 mAh/g)). Cyclic voltammetry (CV) was performed by on CHI 660E electrochemical working station (Chenhua, Shanghai China) at a scanning rate of 0.5 mVs−1 between 2.0 and 4.2 V. Electrochemical impedance spectroscopy (EIS) was performed by on CHI 660E electrochemical working station (Chenhua, Shanghai China) at an amplitude of 5 mV over a frequency range from 10 kHz to 0.01 Hz.
Results and discussion
XRD and Raman spectra analyses
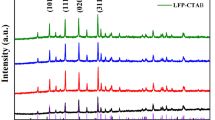
XRD measurements were carried out to examine the crystal structure of as prepared LFP/C and LFP/Cs composites. Figure 2a displayed the XRD patterns for the LFP/C and LFP/Cs samples (LFP/Cs-1, LFP/Cs-2, and LFP/Cs-3) with different SL addition content ranging from 0 to 50 wt%, matching with the standard olivine orthorhombic structure Pnma space group perfectly (LiFePO4, JCPDS card No. 83–2092) [16]. No peaks of impurity phases were observed, indicating that the obtained LFP/C samples with strong diffraction peaks were highly crystalline with high purity and good crystallization, concluding that the SL did not change the crystalline structure of the LiFePO4.

a XRD patterns and b Raman spectra of the LiFePO4/C with different SL addition content
Raman spectroscopy is a particularly useful tool for the structure characterization of the carbon coating layer on the particles surface. Figure 2b displayed the Raman spectra of the LFP/C and LFP/Cs samples. As can be seen from Fig. 2b, the four LFP samples exhibited two distinctive peaks at 1350 cm−1 and 1580 cm−1, which were characteristics of the D-band and G-band, respectively. The D-band corresponds to disorder in the sp2-hybridized carbon, and it was activated owing to existence of carbon lattice defects in carbonation process. The G-band corresponds to the breathing mode of aromatic rings. The intensity ratio of D and G band, ID/IG, was a reflection of sp2-hybridized carbon atoms [34]. The Raman results consistent with the XRD results further demonstrated the formation of LiFePO4/C and LiFePO4/Cs composites. The ID/IG values of the four samples were close to each other (1.05, 1.04, 1.07, and 1.03 for LFP/C, LFP/Cs-1, LFP/Cs-2, and LFP/Cs-3, respectively), because the carbon source and carbon coating synthesis methods were basically the same. However, we could clearly find the presence of 2D-band in the LFP/Cs-2 sample, which indicated that the carbon layer exhibited a higher degree of ordering, concluding that the LFP/Cs-2 with porous structure provided the best electronic connection in the particles.
It is known that the amount of carbon in LiFePO4/C composite greatly affects the surface area of LiFePO4 [35]. The carbon content of the samples was measured by HCS-140 high frequency infrared carbon–sulphur analyzer. Carbon content was 1.08 wt%, 1.12 wt%, 1.16 wt%, and 2.01 wt% for LFP/C, LFP/Cs-1, LFP/Cs-2, and LFP/Cs-3, respectively (Table 2), which was consistent with the amount of added carbon source.
TG/DTG analyses
Safety and thermal stability have become a necessity for LIBs. The TG analyses were performed to determine the process of the reaction process of lattice formation under N2 atmosphere at the rate of 5 ℃min−1. In order to further explore the effect of SL on the formation of LFP lattice, LFP/C and LFP/Cs-2 were chosen for the comparison, as illustrated in Fig. 3. The weight loss started at room temperature and finished at 450 ℃. Three distinct weight loss rate peaks at 100 ℃, 160 ℃, and 350 ℃ were assigned by DTG. Table 1 listed weight loss at each stage. From the Fig. 3 and Table 1, it can be found that the TG curves and weight loss of LFP/C and LFP/Cs-2 samples were basically the same. Weight loss at 100 ℃ corresponds to the volatilization of water in precursors, weight loss at 160 ℃ corresponds to volatiles in precursors, and weight loss at 350 ℃ corresponds to the decomposition of carbon sources and organic matter. Additionally, there was no peak in the DTG curve after 450 ℃ without weight loss in the TG profile, which might be attributed to the crystal formation and growth of LFP particle. Therefore, the addition of SL has no effect on the formation of LFP lattice and the growth temperature of LFP lattice.

TG/DTG curves of the LFP/C and LFP/Cs-2 precursors
SEM and TEM analyses
As we all know that the performance of LFP greatly depends on the morphology [16, 24,25,26, 35], SEM and TEM were carried out to investigate the morphology and microstructure of all LFP/C samples, as illustrated in Figs. 4 and 5. Figure 4 displayed the SEM images of LFP/C samples with different SL addition content, showing that the morphology and particle size of LFP nanocrystals could be readily controlled by tuning the addition content of SL in the precursor solution. As presented in Fig. 4a, agglomerated particles were obtained at the LFP/C (SL 0%). The particles exhibited spheroid with an average particle size of 150 nm. Adding different amounts of SL leaded to a great change in particle morphology. As shown in Fig. 4b, when the addition content of SL was 10%, the micrographs of the LFP/Cs-1 presented well-dispersed spherical that were approximately 100 nm, demonstrating that SL was beneficial to particle dispersion. Interestingly, when the SL addition content was 30%, most of LFP/Cs-2 (SL 30%) nanoparticles were well confined in the nanopores of the carbon sheet shown in Fig. 4c. As can be seen from the Fig. 4e and f for LFP/Cs-2, nanoparticles showed a porous structure, which could provide rapid diffusion and transfer channels for lithium ions and electrons, increasing the interface area of the active material/electrolyte and reducing the inert zone in the material. In the electrode materials of lithium ion batteries, carbon with grid structure has many advantages, such as multiple and continuous conductive paths, controllable mesopore size, large pore volume, and the most importantly was short diffusion distance [17, 26, 36]. But when the SL addition content was 50%, the LFP/Cs-3 particles were strip shaped and agglomerated in a high magnitude as illustrated in Fig. 4d. Therefore, the structure, particle size, and particle size distribution of LFP could be controlled by the addition content of SL, demonstrating that SL was a key factor in obtaining high quality and uniform LFP particles.

SEM images of the LiFePO4/C with different SL additions: a LFP/C (SL 0%), b LFP/Cs-1(SL 10%), c LFP/Cs-2 (SL 30%), d LFP/Cs-3(SL 50%), e LFP/Cs-2 (SL 30%), f LFP/Cs-2 (SL 30%)

TEM images of LFP/C (a) and LFP/Cs-2 (b)
Table 2 listed the physical and chemical properties of LFP with different SL addition content; the carbon content in LFP increased gradually with the increase of SL. However, the specific surface area and powder resistivity were not proportional to SL addition content. It has well been documented that the amount of carbon in LiFePO4/C composite affects the surface area of LiFePO4 greatly; the higher the amount of carbon the lower the surface area [26, 37]. This may be due to the carbon content in the composite interfering with the 3D porous network, blocking some paths and thus isolating the large surface from the total accessible surface area of LiFePO4. It can be seen that the specific surface area of LFP/Cs-2 was the largest and its powder resistivity was the smallest, which was closely related to the morphology of LFP/Cs-2 and the structure of conductive carbon. The results showed that the LFP structure of the holes enlarged the surface area of the material and the lattice conductive carbon had excellent conductivity, which was consistent with the results of Raman and SEM.
Transmission electron microscope (TEM) images of LFP/C and LFP/Cs-2 were performed for further investigation of the carbon coated on LFP particles. The images of Fig. 5a and b clearly showed the lattice fringes of such crystalline LFP particles, which were different from the surrounding amorphous carbon phase. It showed well-crystallized and a conductive carbon layer on the edge of the LFP/C and LFP/Cs-2, which were approximately 2-nm and 4-nm thick, respectively. The results showed that SL can promote the formation of uniform and highly ordered carbon coatings on the surface of LFP particles, which significantly improved the electrochemical properties of LFP, confirmed by the following cell test.
Electrochemical characterization
In order to characterize the electrochemical performance of the LFP, the capacity and rate performance of LFP/C and LFP/Cs-2 were performed with CR2032 coin cells.
Figure 6a showed initial charge/discharge voltage profiles at 0.1 C, which was consistent with the typical flat discharge curve of LFP indicative of two-phase reaction of LiFePO4/ FePO4 [37]. It was found that the capacity of the LFP/Cs-2 was much higher than that of the LFP/C even though the two samples were prepared by the same synthetic method. The specific discharge capacity of LFP/CS-2 was 161 mAhg−1, which was equivalent to 95% of the theoretical capacity of 170 mAhg−1 [13]. The coulomb efficiency was close to 100%. Moreover, as can be seen from Fig. 6b, LFP/CS-2 showed excellent rate performance; when the current densities were 0.1 C, 0.5 C, 1 C, 2 C, 3 C, 5 C, and 8 C, the corresponding discharge capacities were 160, 157, 153, 146, 140, 138, and 135 mAhg−1, respectively. Upon returning the current back to 0.1 C, a capacity of 160 mAhg−1, which was equal to that of the initial capacity of 0.1 C. Furthermore, the storage capacity at different C-rates was found to be stable. However, LFP/C without SL showed relatively poor rate capability, just as 154, 151, 146, 139, 134, 125, and 114 mAhg−1 at 0.1 C, 0.5 C, 1 C, 2 C, 3 C, 5 C, and 8 C, respectively, which was obviously worse than the LFP/Cs-2, especially in high rate discharge.

Electrochemical performance characterizations of LFP/C and LFP/Cs-2 in CR2032 cells: a charge/discharge voltage profiles at 0.1 C, b the cycle performance at different rates, c cyclic voltammetry curves at a scan rate of 0.5 mVs−1, d Nyquist plots
Cyclic voltammetry (CV) is a powerful technique for gathering kinetic information on reversible and irreversible electrode reactions. The symmetry of CV curve and redox peak area are important references of electrode materials cycle performance and the reversible capacity. CV tests were carried out using CR2032 coin cells at a scanning rate of 0.5 mVS−1 between 2.0 and 4.2 V, as illustrated in Fig. 6c. The results showed that the cathode (discharge) peak and anode (charge) peak appeared with the increase of charge–discharge reaction time in the charge–discharge reaction process. In general, the shorter the distance between oxidation peak and reduction peak, the higher the reversibility of the process. Figure 6c demonstrated that the redox peak intensity of LFP/CS-2 was higher than that of LFP/C without SL addition and the difference of peak potential was small, indicating that LFP/CS-2 electrode had excellent high rate performance and reversibility in charge–discharge reaction [38]. This was closely related to the multi-channel porous conductive grid carbon and short lithium ion diffusion path in LFP/Cs-2 structure.
Electrochemical impedance spectroscopy (EIS) is one of the most utilized methods to characterize electrodes in the context of energy applications [39] and was used to further analyze the performance of LFP/C and LFP/CS-2 electrodes, as illustrated in Fig. 6d. The Nyquist plots of LFP/C and LFP/CS-2 showed a depressed semicircle in the intermediate frequency region and a straight line in the low frequency region, which were related to the charge transfer process and the Warburg diffusion process, respectively. As we all know, the electrolytic resistance Rel was the first intercept of the X-axis semicircle, while the second intercept minus the electrolytic resistance Rel represents the charge transfer resistance Rct, which was related to the electrochemical reaction at the electrode–electrolyte interface and particle–particle contact [40,41,42]. Therefore, it was possible to state that the LFP/C and LFP/Cs-2 Rct were about 35 Ω and 21 Ω, respectively. Compared with LFP/C, the charge transfer impedance of the smallest diameter semicircle of LFP/CS-2 was the lowest, mainly because the conductive carbon chain around LFP/Cs-2 can promote electron transfer and lithium ion exchange between interfaces [26]. The direct contact of LFP particles with conductive carbon forms an excellent conductive network, which improves the charge transfer efficiency and electronic conductivity of LFP/Cs-2. Therefore, the charge transfer resistance can be reduced by the method, revealing that the role of conductive carbon is related not only to the optimal electronic path but also to the ionic path toward the electrode active material [43].
In order to further study its high rate and low temperature performance of LFP/Cs-2, the electrochemical performance of 18,650 batteries was tested, as illustrated in Fig. 7. It can be seen in Fig. 7b that the LFP/Cs-2 exhibited an exceptionally superior capacity of 137 mAhg−1 at 5 C, which was much higher than that of LFP/C in Fig. 7a. The capacities of 135 and 131 mAhg−1 can be maintained even at high rate of 10 C and 15 C, and the capacity retention rate can reach 93% at 15 C. However, it was clearly shown that the capacity of LFP/C decreases from 127 to 107 mAhg−1 with the increase of C-rate from 5 to 15 C, indicating that the diffusion impedance of Li ions on the surface of LFP and between particles was larger. However, LFP/Cs-2 exhibited excellent rate performance, which was mainly ascribed to the short particle size and stable crystal structure of the nano LiFePO4 and the good conductive channel between the particles provided by the conductive carbon structure.

Electrochemical performance characterizations of LFP/C and LFP/Cs-2 in 18,650 batteries. a, b. Discharge curves at different rates, c, d discharge curves at − 20 ℃
The low-temperature performance of LIBs is a critical factor restricting the practical application, and the low-temperature capacity of cathode and anode materials plays a key role in LIBs application [17, 19]. Therefore, the low temperature performance for the LFP/C and LFP/Cs-2 were also investigated, as shown in Fig. 7c and d. The discharge performance was tested at temperature of − 20 ℃ in the voltage window of 2.0–3.75 V. It was seen obviously that both electrodes of LFP/C and LFP/Cs-2 displayed a higher capacity retention rate and satisfactory discharge potential plateau at 0.2 C. However, as the test current was increased to 0.5 C, the corresponding capacity retention rate of LFP reduced dramatically from 81 to 60% and which of LFP/Cs-2 decreased from 85 to 71%. It was considered that the decrease of discharge capacity was due to the variation of electronic conductivity at high current and low temperature [17, 19]. But the low-temperature performance of LFP/Cs-2 was much better than that of LFP/C.
Conclusions
In summary, LFP/C composites with novel porous microstructures were successfully synthesized via liquid phase method by controlling the addition content of SL. LFP/Cs-2 prepared with SL (30 wt%) as additive possesses characteristics of the highest specific surface area, optimum rate performance, and low temperature discharge. The results demonstrated that the LFP/Cs-2 (SL 30%) is well confined in the nano-porous of the carbon sheet and show a remarkable effect on the electrochemical behavior, exhibiting a stable and high reversible capacity of 161 mAhg−1 at 0.1 C and good high rate capacity of 135 mAhg−1 at 8 C in CR2032 cells. Furthermore, the LFP/Cs-2 with a compaction density of 2.4 gcm−3 shows a higher rate capability of 131 mAhg−1 at 15 C in 18,650 batteries. The results of CV and EIS indicate that SL as additive can change the structure of coated carbon, reduce charge transfer resistance, improve the electronic/ionic conductivity and lithium ion diffusion coefficient of LFP cathode, and ultimately improve the rate performance and low temperature performance of LFP cathode. The outstanding performance can be attributed to the uniform dispersed nanoparticles and highly conductive carbon structure, which was conducive to the rapid diffusion and electron transfer of lithium ions in the charge–discharge process. Therefore, SL was very effective in improving the electrochemical performance of LFP cathode material for LIBs, having important application value as a high performance energy storage and high power cathode material.
References
Li M, Lu J, Chen ZW, Amine K (2018) 30 years of lithium-ion batteries. Adv Mater 30:1800561
Miao Y, Hynan P, Jouanne AV, Yokochi A (2019) Current Li-ion battery technologies in electric vehicles and opportunities for advancements. Energies 12:1074
Ding YL, Cano ZP, Yu AP, Lu J, Chen ZW (2019) Automotive Li-ion batteries: current status and future perspectives. Electrochem Energy Rev 2:1–28
Kim T, Song WT, Son DY, Ono LK, Qi YB (2019) Lithium-ion batteries: outlook on present, future, and hybridized technologies. J Mater Chem A 7:2942
Xie J, Lu YC (2020) A retrospective on lithium-ion batteries. Nat Comm 11:2499
Sharma DK, Prabhakar A (2021) A review on air cooled and air centric hybrid thermal management techniques for Li-ion battery packs in electric vehicles. J Energ Storage 41:102885
Weiss M, Ruess R, Kasnatscheew J, Levartovsky Y, Levy NR, Minnmann P, Stolz L, Waldmann T, Wohlfahrt-Mehrens M, Aurbach D, Winter M, Ein-Eli Y, Janek J (2021) Fast charging of lithium-ion batteries: a review of materials aspects. Adv Energy Mater 11:2101126
Zhang XH, Li Z, Luo LG, Fan YL, Du ZY (2022) A review on thermal management of lithium-ion batteries for electric vehicles. Energy 238:121652
Li JL, Fleetwood J, Hawley WB, Kays W (2022) From materials to cell: state-of-the-art and prospective technologies for lithium-ion battery electrode processing. Chem Rev 122:903–956
Lv C, Zhou X, Zhong LX, Yan CS, Srinivasan M, Seh ZW, Liu CT, Pan HG, Li SZ, Wen YG, Yan QY (2022) Machine learning: an advanced platform for materials, development and state prediction in lithium-ion batteries. Adv Mater 34:2101474
Shi CM, Wang TY, Liao XB, Qie BY, Yang PF, Chen MJ, Wang X, Srinivasan A, Cheng Q, Ye Q, Li AC, Chen X, Yang Y (2019) Accordion-like stretchable Li-ion batteries with high energy density. Energ Storage Mater 17:136–142
Liao XB, Shi CM, Wang TY, Qie BY, Chen YL, Yang PF, Cheng Q, Zhai HW, Chen MJ, Wang X, Chen X, Yang Y (2019) High-energy-density foldable battery enabled by zigzag-like design. Adv Energy Mater 9:1802998
Padhi AK, Nanjundaswamy KS, Masquelier C, Okada S, Goodenough JB (1997) Phospho olivines as positive electrode material for rechargeable lithium batteries. J Electrochem Soc 144:1188–1194
Yuan LX, Wang ZH, Zhang WX, Hu XL, Chen JT, Huang YH, Goodenough JB (2011) Development and challenges of LiFePO4 cathode material for lithium-ion batteries. Energy Environ Sci 4:269–284
Zhang HH, Zou ZG, Zhang SC, Liu J, Zhong SL (2020) A review of the doping modification of LiFePO4 as a cathode material for lithium ion batteries. Int J Electrochem Sci 15:12041–12067
Mauger A, Julien CM (2018) Olivine positive electrodes for Li-ion batteries: status and perspectives. Batteries 4(39):1–32
Meng FB, Xiong XY, Tan L, Yuan B, Hu RZ (2022) Strategies for improving electrochemical reaction kinetics of cathode materials for subzero-temperature Li-ion batteries: a review. Energ Stor Mater 44:390–407
Ahsan Z, Ding B, Cai Z, Wen C, Yang W, Ma Y, Zhang S, Song G, Javed MS (2021) Recent progress in capacity enhancement of LiFePO4 cathode for Li-ion batteries. ASME J Electrochem En Conv Stor 18(1):010801
Na Y, Sun XH, Fan AR, Cai S, Zheng CM (2021) Methods for enhancing the capacity of electrode materials in low-temperature lithium-ion batteries. Chin Chem Letters 32:973–982
Li L, Wu L, Wu F, Song SP, Zhang XQ, Fu C, Yuan DD, Xiang Y (2017) Review—recent research progress in surface modification of LiFePO4 cathode materials. J Electrochem Soc 164(9):A2138–A2150
An Qi Sun XH, Guo JZ, Cai S, Zheng CM (2020) Review—key strategies to increase the rate capacity of cathode materials for high power lithium-ion batteries. J Electrocheml Soc 167:140528
Wani TA, Suresh G (2021) A comprehensive review of LiMnPO4 based cathode materials for lithium-ion batteries: current strategies to improve its performance. J Energ Stor 44:103307
Zhang N, Deng T (2022) Shuoqing Zhang, Changhong Wang, Lixin Chen, Chunsheng Wang, and Xiulin Fan, Critical review on low-temperature Li-ion/metal batteries. Adv Mater 34:2107899
Bi ZY, Zhang XD, He W, Min DD, Zhang WH (2013) Recent advances in LiFePO4 nanoparticles with different morphology for high-performance lithium-ion batteries. RSC Adv 3:19744
Xin YM, Xu HY, Ruan JH, Li DC, Wang AG, Sun DS(2021) A review on application of LiFePO4 based composites as electrode materials for lithium ion batteries. Int J Electrochem Sci 16:210655
Eftekhari A (2017) LiFePO4/C nanocomposites for lithium-ion batteries. J Power Sources 343:395–411
Geng J, Zhang SH, Hu XX, Ling WG, Peng XX, Zhong SL, Liang FG, Zou ZG (2022) A review of graphene-decorated LiFePO4 cathode materials for lithium-ion batteries. Ionics 28:4899–4922
Liu YY, Gua JJ, Zhang JL, Yu F, Dong LT, Nie N, Li W (2016) Metal organic frameworks derived porous lithium iron phosphate with continuous nitrogen-doped carbon networks for lithium ion batteries. J Power Sources 304:42–50
Zhu W, Li A, Wang ZP, Yang JX, Xu YH (2021) Metal–organic frameworks and their derivatives: designing principles and advances toward advanced cathode materials for alkali metal ion batteries. Small 7:2006424
Wang ZY, Tao HZ, Yue YZ (2019) Metal-organic-framework-based cathodes for enhancing the electrochemical performances of batteries: a review. ChemElectroChem 6(21):5358–5537
Pang J, Zhang W, Zhang J, Cao GP, Han MF, Yang YS (2017) Facile and sustainable synthesis of sodium lignosulfonate derived hierarchical porous carbons for supercapacitors with high volumetric energy density. Green Chem 19(16):3916–3926
Zhang XS, Jian WB, Zhao L, Wen FW, Chen JL, Yin J, Qin YL, Lu K, Zhang WL, Qiu XQ (2022) Direct carbonization of sodium lignosulfonate through self-template strategies for the synthesis of porous carbons toward supercapacitor applications. Colloids Surf A Physicochem Eng Asp 636:128191
Shang WL, Kong LY, Ji XW (2014) Synthesis, characterization and electrochemical performances of LiFePO4/ graphene cathode material for high power lithium-ion batteries. Solid State Sci 38:79–84
Tian Z, Liu SS, Ye F, Yao SJ, Zhou ZF, Wang SM (2014) Synthesis and characterization of LiFePO4 electrode materials coated by graphene. Appl Surf Sci 305:427–432
Seher J, Fröba M (2021) Shape matters: the effect of particle morphology on the fast-charging performance of LiFePO4/C nanoparticle composite electrodes. ACS Omega 6:24062–24069
Wu XL, Jiang LY, Cao FF, Guo YG, Wan LJ (2009) LiFePO4 nanoparticles embedded in a nanoporous carbon matrix: superior cathode material for electrochemical energy-storage devices. Adv Mater 21:2710–2714
Yim CH, Baranova EA, Abu-Lebdeh Y, Davidson I (2012) Highly ordered LiFePO4 cathode material for Li-ion batteries template by surfactant-modified polystyrene colloidal crystals. J Power Sources 205:414–419
Wang Y, Zhu B, Wang Y, Wang F (2016) Solvothermal synthesis of LiFePO4 nanorods as high-performance cathode material for lithium ion batteries. Ceram Int 42:10297–10303
Meddings N, Heinrich M, Overney F, Lee JS, Ruiz V, Napolitano E, Seitz S, Hinds G, Raccichini R, Gaberscek M, Park J (2020) Application of electrochemical impedance spectroscopy to commercial Li-ion cells: a review. J Power Sources 480:228742
Choi YM, Pyun SI (1997) Effects of intercalation-induced stress on lithium transport through porous LiCoO2 electrode. Solid State Ion 99:173–183
Wang C, Hong J (2007) Ionic/electronic conducting characteristics of LiFePO4 cathode materials. The Determining Factors for High Rate Performance. Electrochem Solid-State Lett 10:A65–A69
Park CY, Park SB, Oh SH, Jang H, Cho WI, Ion L (2011) Diffusivity and improved electrochemical performances of the carbon coated LiFePO4. Bull Korean Chem Soc 32:836–840
Itou Y, Ogihara N, Kawauchi S (2020) Role of conductive carbon in porous Li-ion battery electrodes revealed by electrochemical impedance spectroscopy using a symmetric cell. J Phys Chem C 124:5559–5564
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shang, WL., Kong, LY., Sun, Y. et al. Electrochemical performance improvement of LiFePO4/C composite cathode materials by using sodium ligninsulfonate as carbon source and surfactant for lithium-ion batteries. Ionics 29, 563–572 (2023). https://doi.org/10.1007/s11581-022-04840-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-022-04840-9