
Abstract
Garnet-type solid-state electrolytes such as Ta-doped Li7La3Zr2O12 (LLZTO) are promising ceramic electrolytes for all-solid-state batteries because of their high (electro)chemical stability and ionic conductivity. LLZTO is typically synthesized from oxide precursors via solid-state reaction, but use of other precursors such as pyrochlores has also received recent interest. Here, a Ta-doped pyrochlore was prepared using non-aqueous sol–gel methods to serve as precursor for LLZTO. The dense LLZTO ceramic was directly obtained via a reactive sintering method with ionic conductivity ~ 0.4 mS/cm, which is comparable to LLZTO prepared by solid-state reaction, using only 2 h sintering at 1100 ℃. Investigation of the synthesis parameters shows that liquid phase sintering is important for achieving high pellet densities and phase purity. The grain size of LLZTO is less than 2 μm, indicating that fine-grained garnet ceramics are accessible with this approach. This work reveals an alternate method for the synthesis and processing of LLZTO materials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Solid-state ceramic electrolytes have received great attention due to the interest in high energy density batteries using lithium anodes and the desire to mitigate the safety issues related to lithium dendrite growth in conventional liquid electrolytes [1]. Among the various types of solid electrolytes, the garnet-type Li7La3Zr2O12 (LLZO) material is one of the most studied due to its high ionic conductivity at room temperature (> 10−4 S/cm) and chemical stability against lithium metal [1, 2]. Aliovalent cation doping of LLZO stabilizes the high conductivity cubic phase at room temperature [3,4,5], with Ta5+-doped LLZO (LLZTO) reported with good ionic conductivity up to 1 mS cm−1 at room temperature [2, 6, 7]. However, a drawback to LLZTO is that it is conventionally prepared using solid-state reaction (SSR) methods that require long sintering times at high temperatures (Scheme 1a). A typical SSR synthesis of LLZTO powders starts with ball milling of the Li2CO3, La2O3, ZrO2, and Ta2O5 precursors and calcination at temperatures > 900 ℃ for longer than 24 h [8], then followed by sintering above 1100 ℃ for many hours to achieve ceramic electrolytes with high relative density [9].

Overall process of synthesizing LLZTO using different methods. a Solid-state reaction (SSR) method. b Molten salt (MSS) pyrochlore to garnet (P2G) method. c Non-aqueous sol–gel (NASG) P2G method
To reduce the temperature and time required for both synthesis and sintering of LLZTO, alternate synthetic approaches have been investigated by our group and others from pyrochlore starting materials [10,11,12,13,14]. The La2Zr2O7 pyrochlore is commonly seen as a secondary phase in LLZO syntheses [15], and its presence can signify an incomplete reaction of the simple oxide precursors or decomposition of the formed garnet due to lithium loss (e.g., from excessive calcination or sintering times) [4, 16,17,18,19].
Our group has recently explored molten salt flux methods for preparing submicron particles of cubic LLZTO at lower temperatures [20, 21] and adapted the process to prepare Ta5+-doped pyrochlore with nominal composition La2.4Zr1.12Ta0.48O7.04 from La(NO3)3·6H2O, ZrOCl2·8H2O, and Ta2O5 reactants in a NaOH-KOH eutectic mixture [13]. We found that not only could the pyrochlore be added as a quasi-single-source precursor to a LiNO3-Li2O2-LiOH molten salt mixture (serving as both solvent and Li+ source) to prepare LLZTO at temperatures as low as 400–550℃, but the pyrochlore could also be converted to the garnet via reactive sintering (Scheme 1b). In the latter process, which we refer to as pyrochlore-to-garnet (P2G), the pyrochlore is mixed with LiOH (10 mol% excess relative to the target garnet stoichiometry of Li6.4La3Zr1.4Ta0.6O12) with ball milling and then pressed and sintered. At temperatures close to the melting point of LiOH (462 ℃), the liquid LiOH will start to decompose into Li2O as shown in Eq. 1 [22], which then reacts with the pyrochlore to form LLZTO following Eq. 2 [13]:
The reactive sintering of the pyrochlore to form LLZTO, aided by the liquid phase LiOH, is expected to increase the interdiffusion of various species and reaction speed. We found that dense LLZTO pellets could be obtained after sintering at 1200 °C for as little as 2 h and that the grain size of LLZTO pellets resulting from this P2G method (2–5 μm) was smaller than typical grain sizes observed with conventionally sintered LLZTO (~ 20 μm), even if the LLZTO starting powders were submicron in size (e.g., when prepared using molten salt or sol–gel based methods) [13].
Although this P2G method was effective for preparing LLZTO with good ionic conductivity and relative density, the use of corrosive molten salts to prepare the pyrochlore precursor is a limitation of the approach. Not only is it a potential safety hazard, but the NaOH-KOH salt fluxes are corrosive to alumina crucibles, necessitating more expensive zirconia or platinum crucibles. In our studies, we found that the zirconia crucible used to carry out the pyrochlore synthesis could easily crack during cooling due to thermal shock, while alumina crucibles could be etched by the highly basic molten salt, introducing Al contamination to the pyrochlore precursors and the resultant LLZTO (Fig. S1). Another concern is the disposal of the highly basic aqueous waste generated during recovery and washing of the pyrochlore from the salt medium.
To address these issues, herein we report a new method to prepare the pyrochlore precursor that can be used to obtain LLZTO via the P2G process. A non-aqueous sol–gel (NASG) combustion synthesis method is employed to form the pyrochlore precursors that generates no liquid waste or Al contamination. The NASG pyrochlore is subsequently transformed to LLZTO using the P2G reactive sintering method (Scheme 1c). The synthesized pyrochlore is characterized, and different temperatures and lithium sources for the reactive sintering are investigated to elucidate the best parameters to give dense garnet electrolytes with high ionic conductivity and relative density. These results show that with LiOH as the Li source, a dense (> 90% relative density) LLZTO pellet with high conductivity (~ 0.4 mS/cm) can be sintered in as little as 2 h at 1100 °C.
Materials and methods
Chemicals
Lanthanum (III) 2,4-pentanedionate (“La(acac)3”), zirconium (IV) 2,4-pentanedionate (“Zr(acac)4”), lithium hydroxide (“LiOH”), and propionic acid (“PA”) were procured from Alfa Aesar and were of ACS grade purity or higher. Tantalum (V) ethoxide (Ta(OEt)5, 99% +), ACS grade anhydrous dichloromethane (“DCM”), and lithium peroxide (“Li2O2”) were procured from Sigma-Aldrich. Due to the moisture sensitivity of Ta(OEt)5 [23], it was stored in an Ar-filled glove box until use. Once taken out of the glove box, Ta(OEt)5 was added to PA immediately to prevent formation of hydrolysis products. Considering the strong odor of PA, all preparation steps should be conducted in a fume hood. Neoprene gloves should also be used when handling DCM.
Preparation of NASG pyrochlore precursor
The NASG precursor for the pyrochlore was adapted from the precursor used in the combustion synthesis of LLZTO powders reported in our previous study [24], which consisted of metal acetylacetonates dissolved in a mixture of PA and DCM with addition of a polymer to serve as a fuel and promote fine particle formation. The main difference between this NASG pyrochlore precursor and the aforementioned LLZTO precursor is the absence of a Li source and polymer. Also, similar to the MSS method we used for preparing the pyrochlore in our previous study [13], the mole ratio of La:Zr:Ta in the pyrochlore precursor was 3:1.4:0.6 so that the synthesized pyrochlore would have a nominal composition of La2.4Zr1.12Ta0.48O7.04. This would enable formation of LLZTO with nominal composition of Li6.4La3Zr1.4Ta0.6O12 after reactive sintering. In a typical synthesis (Scheme 2), 887 μL Ta(OEt)5 liquid was added to 100 mL PA, followed by 6.543 g La(acac)3 and 3.413 g Zr(acac)4 and then mixing with magnetic stirring. Then, 100 mL of DCM was added, and the mixture was stirred until the liquid became a transparent yellow solution. The NASG pyrochlore precursor can be stored in a capped bottle in air for months without degradation.

Flow chart of non-aqueous sol–gel synthesis of pyrochlore
Synthesis of pyrochlore through combustion of NASG pyrochlore precursor
To obtain the Ta-doped pyrochlore, a typical synthesis involved pouring ~ 70 mL of NASG pyrochlore precursor into a 100 mL Al2O3 cup crucible. Then, the crucible was transferred into a muffle furnace (Carbolite) and heated in air to combust the precursor. The optimized pyrochlore synthesis was carried out at 850 °C with a heating rate of 5 °C/min and hold time of 8 h. Then, the furnace was naturally cooled to room temperature. After synthesis, the product (~ 1 g white powder) was ground by hand using an agate mortar and pestle prior to use in the preparation of LLZTO.
Preparation of LLZTO pellets by reactive sintering of pyrochlore
In a typical LLZTO synthesis using the P2G approach, 1 g of pyrochlore prepared from combustion of the NASG precursor and 0.25 g LiOH to serve as lithium source (25% excess Li was added) were first ball-milled at 8000 rpm for 90 min using a SPEX 8000 M vibratory mill using a plastic jar with tungsten carbide ball. Then, about 100–150 mg of the ball-milled powder was placed in a 7-mm stainless steel die and pressed at ~ 300 MPa using a SpecAc manual hydraulic press to prepare each green pellet. The green pellet was then sintered in a lidded MgO crucible containing LLZTO and Li2O2 powders to create a Li-rich atmosphere [16, 25] (see more details in Figure S2). The crucible was placed in an Al2O3 tube and sintered in a Lindberg/Blue M TF55030A tube furnace in air. The relative density of pellets compared to the theoretical density of the LLZTO of 5.4 g/cm3 [26] was determined by the mass, thickness, and cross-sectional area of the pellet. Different sintering conditions and Li sources were also investigated, with use of 1100 °C sintering temperature and LiOH resulting in LLZTO with best phase purity and relative density.
Material characterization
Thermogravimetric analysis (TGA) of the pyrochlore precursor was performed using a Labsys EVO from Setaram. The NASG precursor was dried to remove the solvents by heating on a hotplate in the fume hood at 120 °C for 2 h. The dried sample (~ 20 mg) was put in an Al2O3 crucible and heated to 900 °C in air with a heating rate of 5 °C/min. The pyrochlore powders and LLZTO-sintered pellets were characterized by X-ray diffraction (XRD) using a Malvern PANalytical Aeris research edition powder diffractometer with CuKα source. Raman spectroscopy was performed at room temperature on Ta-doped pyrochlore synthesized at 600 and 850 °C as well as on stoichiometric pyrochlore synthesized at 850 °C using the same process. The as-prepared powders were hand ground and then cold-pressed into pellets for the Raman measurement. Raman spectra were measured in a FER-SCI-1024B X-VR spectrometer (Teledyne Princeton Instruments) equipped with a charge-coupled device (CCD) detector (cooled to -55 °C). Excitation using the 532 nm line from a CW diode (100 mW) was carried out with a Samba 100 (Hübner Photonics) laser source. The scanning time was 10 min for each sample. The morphology and elemental composition of the samples were characterized using scanning electron microscopy (SEM, FEI Nova 200 and Thermo Scientific Helios 5 UX). Transmission electron microscopy (TEM) was performed using a NION UltraSTEM 100.
Ionic conductivity evaluation
The LLZTO pellets after the P2G reactive sintering process were polished in ambient atmosphere using 2000 grit SiC sandpaper to make a clean and smooth surface and then transferred into an Ar-filled glove box. Electrical contacts were made using Li/Sn alloy as non-blocking electrodes due to its good wetting ability to the surface of LLZTO [24, 27]. The Li/Sn alloy (20 wt% or ~ 1.5 mol% Sn) was prepared and melted at 230 °C on a hotplate inside the glove box. The LLZTO pellet was pressed onto the molten Li/Sn alloy until both surfaces were coated. Then, any excess Li/Sn alloy contacting the edge of the pellet was removed using sandpaper to prevent shorting of the two sides, and two pieces of Cu foil were pressed onto the Li/Sn alloy with tweezers. This pellet assembly was placed in a foil poly bag and sealed with an impulse sealer with the pieces of Cu foil extending out of the seal to serve as electrical connections to the potentiostat.
The assembled pouch cell was taken out of the glove box, and electrochemical impedance spectroscopy (EIS) was performed with a frequency range of 7 MHz–1 Hz and a 50 mV stimulus voltage at room temperature with a Biologic SP-200 potentiostat. To fit the impedance data, a (Rbulk/Qbulk)(Rgb/Qgb)(Ri/Qi) equivalent circuit was used, where “Rgb” refers to the grain boundary resistance and “Ri” is the interface resistance. Qbulk, Qgb, and Qi are constant phase elements (CPEs) to account for nonideal capacitance. The ionic conductivity of the pellet σ (mS/cm) was determined using Eq. 3:
where t is the thickness of the pellet, A is the cross-sectional area, and Rtotal is the total resistance, which is the sum of Rbulk and Rgb.
Lithium stripping/plating
The Li stripping/plating characteristics were evaluated in LLZTO symmetric cells with Li/Sn contacts using galvanostatic cycling. The initial current density was 0.05 mA/cm2, which was then increased to 0.1, 0.2 mA/cm2, etc. until the cell was short-circuited. At each current density, the cell was tested for 100 h or 50 cycles; i.e., in each cycle, the cell was polarized in one direction for 1 h, and then, the direction was reversed for another hour.
Results and discussion
The NASG pyrochlore precursor was calcined between 600 and 850 °C to understand the effect of combustion temperature on the doped pyrochlore product. For comparison, a stoichiometric La2Zr2O7 (LZO) sample was prepared at 850 °C using the same NASG method but without Ta dopant or excess La. XRD characterization showed that crystallized doped pyrochlore without secondary phases could be obtained at 850 °C (Fig. 1a) and the XRD patterns were similar to those for the stoichiometric LZO. When a synthesis temperature lower than 850 °C was used, the XRD reflections of the doped pyrochlore were wider and less defined, indicating less crystallinity or smaller crystallite size. The small shoulder at higher angles to the highest intensity reflection for the pyrochlore at 2θ = 29° could originate from La2O3 (corresponding to the [011] planes) (PDF card number 01–083-4961) or La2O2CO3 (corresponding to the [103] planes) (PDF card number 00–023-0320). It has been reported that La(acac)3 will first decompose to La2O2CO3 and then to La2O3 [28]. Additionally, the powders obtained at lower temperature were gray in color, instead of white as observed for the sample synthesized at 850 °C (Figure S3), which could be from defects from the low crystallinity of La2O2CO3 (the synthesis temperature in this work is lower than the typical synthesis temperature reported by other groups [29]) or from residual carbon [30].

a XRD patterns of Ta-doped pyrochlore synthesized from combustion of NASG precursor at sintering temperatures of 600–850 °C compared to stoichiometric pyrochlore synthesized at 850 °C from the stoichiometric NASG precursor. Unless otherwise specified, the sintering time is 8 h for all samples. The La2Zr2O7 reference pattern is for the stoichiometric, undoped pyrochlore (PDF card number 01–085-6855), La2O2CO3 reference is from PDF card 00–023-0320, and La2O3 reference is from PDF card 01–083-4961. b Raman spectra of Ta-doped pyrochlore synthesized at 600 or 850 °C for 8 h and stoichiometric pyrochlore synthesized at 850 °C for 8 h
To further analyze the structure of the Ta-doped pyrochlore, the Raman spectra were obtained from doped pyrochlore synthesized at 600 °C for 8 h and 850 °C for 8 h (Fig. 1b), along with the undoped, stoichiometric LZO prepared at 850 °C. The features at around 200 cm−1 and 900–1100 cm−1 are likely from La2O2CO3 [31]. The stoichiometric LZO sample displayed broadened vibrational modes at around 311, 403 and 511, and 525 cm−1, which are consistent with the 4F2g Raman modes (Zr-O stretching or bending) expected for the pyrochlore (Fd-3 m, no. 227) structure [32, 33]. The broadening of the modes is consistent with the low synthesis temperature used and previous reports [34, 35]. The Raman spectra for the doped pyrochlores were similar to each other except for the much higher intensity in the sample prepared at 850 °C (see Figure S4); both spectra showed slight shifts in the 4F2g modes to higher wavenumbers. Notably, a new band at 803 cm−1 was also observed which was not present in the Raman spectrum of the stoichiometric LZO, which could be from Ta-O stretching since similar modes have been seen in amorphous and disordered tantalum oxides [36,37,38,39].
To analyze the thermal decomposition process of the NASG precursor, the dried precursor was characterized with thermogravimetric analysis (TGA) between room temperature and 900 °C. Figure S5a shows the sample mass as a function of temperature corresponding to three distinct weight loss segments. The first part (from ~ 250 to 300 °C) corresponds to the decomposition of Ta(OEt)5 in air (to form Ta2O5) [40]. Then, there is rapid weight loss from ~ 300 to 375 °C, which could be due to the decomposition of zirconium acetato-propionate and the formation of ZrO2 [41]. There might be a combination of reactions [28] taking place from ~ 375 to 600 °C, indicating the final transformation from La(acac)3 to La2O3. The theoretical calculated mass loss is 58.4%, while the observed mass loss was 52.4%. Considering the decomposition temperatures of La(acac)3 and intermediate La2O2CO3 are relatively high [28], the difference between the calculated and observed mass loss is likely due to the incomplete decomposition of La2O2CO3.
The XRD pattern of the sample subjected to TGA analysis was measured afterwards (Figure S5b), and broad reflections from the pyrochlore structure were observed along with some other reflections that could be assigned to La2O3, La2O2CO3, and tetragonal ZrO2. The low crystallinity and phase purity of this sample could arise from the insufficient hold time at high temperatures during the TGA analysis, which prevents full transformation of the precursor material to the pyrochlore. Interestingly, while most of the mass loss was observed by 600 °C, performing the combustion at 600 °C for 32 h (Fig. 1a) was not effective for crystallizing the pyrochlore.
Therefore, to ensure complete decomposition of the precursor and a phase-pure, crystalline product, a synthesis temperature of 850 °C and hold time of 8 h were used to prepare pyrochlore particles to be subsequently used in the P2G process to form LLZTO. SEM imaging revealed that the as-synthesized NASG pyrochlore exhibited agglomerated particles with sizes of several tens of micrometers after hand grinding (Fig. 2a). These large particles, particularly compared to the fine nanopowders resulting from the closely related non-aqueous polymer combustion synthesis of LLZTO [24], presumably form due to insufficient carbon templating since no polymer was added to the NASG precursor. However, the broad reflections in the XRD pattern (Fig. 1) (reference pattern is for stoichiometric pyrochlore La2Zr2O7, from reference [42]) suggest that the materials have low crystallinity [43] or that the crystallite size is very small from the Scherrer effect [44]. To gain further understanding, the pyrochlore particles were ball-milled for 90 min to enable TEM observation. After ball milling, the particle size of the pyrochlore could be reduced to as small as hundreds of nanometers according to SEM imaging (Fig. 2b). TEM analysis (Fig. 2c) showed that the pyrochlore particles were polycrystalline, with average grain size of 5–10 nm. The fast Fourier transform (FFT) indicates the (222) plane in the pyrochlore, which should have the strongest diffraction intensity.

a SEM image of as-synthesized pyrochlore made via NASG at 850 °C for 8 h (after hand grinding). b SEM and c TEM image and FFT of pyrochlore after ball milling
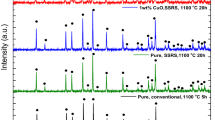
To prepare LLZTO, the as-synthesized pyrochlore particles were ball-milled with the Li source (LiOH). SEM imaging of this ball-milled mixture (Fig. S6) shows that the particle size is like that seen for the ball-milled pyrochlore without LiOH (Fig. 2c), suggesting that the particle sizes shown in Fig. 2b reflect the primary/actual particle sizes of the pyrochlore used for reactive sintering. The reactive sintering process to realize the pyrochlore-to-garnet (P2G) transformation was carried out by heating the green pellets comprising the ball-milled mixtures at different temperatures and sintering times. The XRD pattern of reactively sintered pellets (Fig. 3, top trace) shows that phase-pure LLZTO could be synthesized at 500 °C for 2 h with a good match to the structure for Li6.5La3Zr1.5Ta0.5O12 (PDF 04–018-9024) [45], indicating complete transformation of the pyrochlore to garnet without other secondary phase formation. However, the relative density of the pellet reactively sintered at 500 °C was very low (~ 50%), while higher densities > 90% could be obtained using sintering at 1100 °C for 2 h. The SEM images (Figure S7) show that the P2G pellet prepared at 500 °C has a lot of porosity. Furthermore, the dark contrast in the backscattered electron (BSE) image suggests the presence of phases with low molecular weight in the pellet, possibly unreacted LiOH, which is not observed in the XRD pattern.

XRD patterns of LLZTO pellets prepared from P2G method. The NASG pyrochlore was reacted with different Li sources: LiOH, Li2O, Li2O2, Li2CO3, and Li2CO3 (without 25% excess Li) at 1100 °C for 2 h unless otherwise indicated
To understand the role of LiOH on the LLZTO formation process, different Li sources were used to prepare the P2G mixture, including Li2O2, Li2O, and Li2CO3. Figure 3 shows the XRD patterns of the reactively sintered LLZTO pellets obtained from mixing the pyrochlores with these other Li sources sintered with the same conditions (25% excess Li, 1100 °C, and 2 h). When using Li2O as the Li source, phase-pure LLZTO was obtained, but the relative density (less than 80%) was lower than that for the LLZTO pellet sintered with LiOH. Using Li2O2 as the Li source resulted in LLZTO but with some secondary phases (likely Li2CO3) also observed. When using Li2CO3, a high intensity reflection at 2θ ~ 30° was observed, which is most likely from unreacted Li2CO3. Another pellet was prepared using stoichiometric amounts of Li2CO3, but this reflection was still observed, likely because of the use of a closed crucible (Figure S2), which was utilized to create a Li-rich atmosphere during sintering but may also restrict the release of CO2 from Li2CO3 during decomposition and can promote its re-formation [16]. From these results, we conclude that LiOH is the best choice as lithium source for reactive sintering of the pyrochlore to form phase-pure LLZTO.
Several pellets prepared from pyrochlore/LiOH mixtures were sintered at 1100 °C for 2 or 6 h to measure the relative densities that could be obtained using these conditions. Figure 4a shows that the average relative density of the LLZTO pellet after sintering for 2 h (n = 12 pellets) is around 92%, while pellets sintered for 6 h had density around 95% (n = 42). Although there was a larger range of relative densities for pellets sintered for only 2 h, the average densities were only slightly different, indicating that the P2G process can be completed in a short time after the sintering temperature is reached.

Characterization of LLZTO pellets prepared using P2G with LiOH and reactive sintering at 1100 °C. a Relative densities and b Nyquist plots of impedance spectra from LLZTO pellets obtained using sintering time of 2 (red) or 6 h (blue). c Relationship between relative density and ionic conductivity from LLZTO obtained using sintering time of 2 (red) or 6 h (blue). SEM images of fracture surface of pellets with sintering time of 2 (d, e) or 6 h (f, g). h Lithium stripping and plating on LLZTO pellet obtained using sintering time of 2 h; insets show zoomed-in figure for each current density
The Nyquist plots obtained from performing EIS on representative pellets sintered at 1100 °C for 2 and 6 h are shown in Fig. 4b. The total ionic conductivities determined from the fitting (see Figure S8) were ~ 0.4 mS/cm for both types of LLZTO pellets (0.37 mS/cm for 2 h sintering and 0.34 mS/cm for 6 h sintering). As expected, the ionic conductivity tends to increase with the relative density for both kinds of pellets (Fig. 4c). The relationship between temperature and ionic conductivity was also investigated for pellets sintered at 1100 °C for 2 h to determine its activation energy (Figure S9). EIS measurements were performed between 30 and 100 °C, and a linear fit of the ln(σ) (S cm−1) vs 1/T (K−1) plot was taken. Based on the slopes of the linear fits, the activation energy was determined to be 0.41 eV, which corresponds well to other values (0.39–0.42 eV) in the literature and our previous work [18, 24, 46].
From SEM imaging of the pellet fracture surface (Fig. 4d, e), the average grain size for the pellet sintered for 2 h was determined to be ~ 2 μm. When the sintering time was increased to 6 h, the grain size increased on average with a nonuniform distribution (for size distribution, see Figure S10) of grain sizes (Fig. 4f, g). The lithium stripping/plating experiment performed on a P2G pellet sintered at 2 h (Fig. 4h) shows that the pellet could sustain a current density of 0.2 mA/cm2 for 100 h without requiring any externally applied stack pressure or heating and shorted at 0.3 mA/cm2. This is better than results from sintered SSR LLZTO powders also coated with Li/Sn alloy, which shorted at 0.05 mA/cm2 [27]. While the LLZTO grain size and distribution did not appear to affect the ionic conductivity of the P2G pellets, it could play a role in the Li stripping/plating properties [47, 48], which will be investigated in future studies.
The small grain sizes seen in the LLZTO P2G pellets sintered for 2 h can be explained by the nanosized dimensions of the pyrochlore particles after ball milling, which could potentially help to promote uniform nucleation of LLZTO during reactive sintering. During the reactive sintering process, the pyrochlore nanoparticles react with molten LiOH to form cubic LLZTO (Scheme 3). Considering the LLZTO grains form directly from reaction of the pyrochlore nanoparticles with LiOH/Li2O in a short time, we should also expect a relatively small grain size for LLZTO.

Proposed reactive sintering of LLZTO using P2G process
Interestingly, despite the polycrystallinity of the pyrochlore particles prepared from the NASG precursor, the grain size, microstructure, crystallinity, and conductivity of the LLZTO were similar to those from LLZTO pellets prepared via P2G from pyrochlore nanocrystals made in the molten salt flux [13]. These differences in the pyrochlore synthesis method and microstructure apparently did not significantly affect the reactive sintering or properties of the resulting LLZTO. This shows that the NASG method for preparation of the pyrochlore could be a more facile approach for obtaining the LLZTO precursor than the molten salt synthesis method.
In summary, we have developed a new reactive sintering method based on a NASG pyrochlore precursor to synthesize the garnet-type LLZTO electrolyte. By leveraging the liquid phase formed in situ during sintering, we can significantly reduce the sintering temperature and time to obtain LLZTO. Compared to the previously reported, molten salt synthesis of the pyrochlore, the NASG synthesis also avoids corrosion-induced Al contamination. Sintered LLZTO ceramics from P2G show a high relative density (> 90%) and high ionic conductivity (~ 0.4 mS/cm). The reactive sintering method gives us an alternative way to prepare dense LLZTO material with greatly reduced sintering time compared to many other LLZTO synthesis methods at a relatively low sintering temperature. Additionally, the LLZTO grain sizes obtained using this P2G method are smaller than those from conventional sintering of LLZTO particles. Further characterization and electrochemical analysis will be applied to evaluate the potential benefits of the smaller grain sizes in future studies.
References
Zhang Z, Shao Y, Lotsch B, Hu Y-S, Li H, Janek J et al (2018) New horizons for inorganic solid state ion conductors. Energy Environ Sci 11(8):1945–1976. https://doi.org/10.1039/C8EE01053F
Murugan R, Thangadurai V, Weppner W (2007) Fast lithium ion conduction in garnet-type Li7La3Zr2O12. Angew Chem Int Ed 46(41):7778–7781. https://doi.org/10.1002/anie.200701144
Geiger CA, Alekseev E, Lazic B, Fisch M, Armbruster T, Langner R et al (2011) Crystal chemistry and stability of “Li7La3Zr2O12” garnet: a fast lithium-ion conductor. Inorg Chem 50(3):1089–1097. https://doi.org/10.1021/ic101914e
Rangasamy E, Wolfenstine J, Sakamoto J (2012) The role of Al and Li concentration on the formation of cubic garnet solid electrolyte of nominal composition Li7La3Zr2O12. Solid State Ionics 206:28–32. https://doi.org/10.1016/j.ssi.2011.10.022
Meier K, Laino T, Curioni A (2014) Solid-state electrolytes: revealing the mechanisms of Li-ion conduction in tetragonal and cubic LLZO by first-principles calculations. J Phys Chem C 118(13):6668–6679. https://doi.org/10.1021/jp5002463
Logéat A, Köhler T, Eisele U, Stiaszny B, Harzer A, Tovar M et al (2012) From order to disorder: the structure of lithium-conducting garnets Li7− xLa3TaxZr2− xO12 (x= 0–2). Solid State Ionics 206:33–38. https://doi.org/10.1016/j.ssi.2011.10.023
Wang Y, Lai W (2015) Phase transition in lithium garnet oxide ionic conductors Li7La3Zr2O12: the role of Ta substitution and H2O/CO2 exposure. J Power Sources 275:612–620. https://doi.org/10.1016/j.jpowsour.2014.11.062
Wang Y, Lai W (2012) High ionic conductivity lithium garnet oxides of Li7−xLa3Zr2−xTaxO12 compositions. Electrochem Solid State Lett 15(5):A68. https://doi.org/10.1149/2.024205ESL
Liu Q, Geng Z, Han C, Fu Y, Li S, He Y-b et al (2018) Challenges and perspectives of garnet solid electrolytes for all solid-state lithium batteries. J Power Sources 389:120–134. https://doi.org/10.1016/j.jpowsour.2018.04.019
Deviannapoorani C, Ramakumar S, Janani N, Murugan R (2015) Synthesis of lithium garnets from La2Zr2O7 pyrochlore. Solid State Ionics 283:123–130. https://doi.org/10.1016/j.ssi.2015.10.006
Kimura T, Yamada Y, Yamamoto K, Matsuda T, Nomura H, Hirayama T (2017) Rapid low-temperature synthesis of tetragonal single-phase Li7La3Zr2O12. J Am Ceram Soc 100(4):1313–1319. https://doi.org/10.1111/jace.14633
Hamao N, Hamamoto K, Taguchi N, Tanaka S, Akimoto J (2020) Synthesis and crystal structure of fluorite-type La2.4Zr1.2Ta0.4O7: a precursor oxide for low temperature formation of garnet-type Li6.5La3Zr1.5Ta0.5O12. Solid State Ionics 357: 115460. https://doi.org/10.1016/j.ssi.2020.115460.
Weller JM, Chan CK (2020) Pyrochlore nanocrystals as versatile quasi-single-source precursors to lithium conducting garnets. J Mater Chem A 8(34):17405–17410. https://doi.org/10.1039/D0TA05842D
Hamao N, Akimoto J (2019) A novel synthetic route of garnet-type Li6.5La3Zr1.5Ta0.5O12 using pyrochlore-type La2Zr2O7 and weberite-type La3TaO7 as starting materials. J Ceram Soc Jpn 127, no. 6: 374–7. https://doi.org/10.2109/jcersj2.19014.
Huang X, Lu Y, Guo H, Song Z, Xiu T, Badding ME et al (2018) None-mother-powder method to prepare dense Li-garnet solid electrolytes with high critical current density. ACS Appl Energy Mater 1(10):5355–5365. https://doi.org/10.1021/acsaem.8b00976
Cai J, Polzin B, Fan L, Yin L, Liang Y, Li X et al (2021) Stoichiometric irreversibility of aged garnet electrolytes. Mater Today Energy 20:100669. https://doi.org/10.1016/j.mtener.2021.100669
Janani N, Ramakumar S, Dhivya L, Deviannapoorani C, Saranya K, Murugan R (2011) Synthesis of cubic Li7La3Zr2O12 by modified sol–gel process. Ionics 17:575–580. https://doi.org/10.1007/S11581-011-0611-X
Sakamoto J, Rangasamy E, Kim H, Kim Y, Wolfenstine J (2013) Synthesis of nano-scale fast ion conducting cubic Li7La3Zr2O12. Nanotechnology 24(42):424005. https://doi.org/10.1088/0957-4484/24/42/424005
Yang T, Gordon ZD, Li Y, Chan CK (2015) Nanostructured garnet-type solid electrolytes for lithium batteries: electrospinning synthesis of Li7La3Zr2O12 nanowires and particle size-dependent phase transformation. J Phys Chem C 119(27):14947–14953. https://doi.org/10.1021/acs.jpcc.5b03589
Weller JM, Chan CK (2020) Reduction in formation temperature of Ta-doped lithium lanthanum zirconate by application of Lux-Flood basic molten salt synthesis. ACS Appl Energy Mater 3(7):6466–6475. https://doi.org/10.1021/acsaem.0c00716
Weller JM, Whetten JA, Chan CK (2018) Synthesis of fine cubic Li7La3Zr2O12 powders in molten LiCl–KCl eutectic and facile densification by reversal of Li+/H+ exchange. ACS Appl Energy Mater 1(2):552–560. https://doi.org/10.1021/acsaem.7b00133
Kiat J, Boemare G, Rieu B, Aymes D (1998) Structural evolution of LiOH: evidence of a solid–solid transformation toward Li2O close to the melting temperature. Solid State Commun 108(4):241–245. https://doi.org/10.1016/S0038-1098(98)00346-9
Jones AC (1998) MOCVD of electroceramic oxides: a precursor manufacturer’s perspective. Chem Vap Deposition 4(5):169–179. https://doi.org/10.1002/(SICI)1521-3862(199810)04:05%3c169::AID-CVDE169%3e3.0.CO;2-6
Weller JM, Whetten JA, Chan CK (2020) Nonaqueous polymer combustion synthesis of cubic Li7La3Zr2O12 nanopowders. ACS Appl Mater Interfaces 12(1):953–962. https://doi.org/10.1021/acsami.9b19981
Huang X, Lu Y, Song Z, Rui K, Wang Q, Xiu T et al (2019) Manipulating Li2O atmosphere for sintering dense Li7La3Zr2O12 solid electrolyte. Energy Storage Mater 22:207–217. https://doi.org/10.1016/j.ensm.2019.01.018
Li Y, Han J-T, Wang C-A, Xie H, Goodenough JB (2012) Optimizing Li+ conductivity in a garnet framework. J Mater Chem 22(30):15357–15361. https://doi.org/10.1039/C2JM31413D
Wang C, Xie H, Zhang L, Gong Y, Pastel G, Dai J et al (2018) Universal soldering of lithium and sodium alloys on various substrates for batteries. Adv Energy Mater 8(6):1701963. https://doi.org/10.1002/aenm.201701963
Ciontea L, Nasui M, Petrisor Jr T, Mos R, Gabor M, Varga R et al (2010) Synthesis, crystal structure and thermal decomposition of [La2(CH3CH2COO)6·(H2O)3]·3.5H2O precursor for high-k La2O3 thin films deposition. Mater Res Bull 45, no. 9: 1203–8. https://doi.org/10.1016/j.materresbull.2010.05.019.
Mu Q, Wang Y (2011) Synthesis, characterization, shape-preserved transformation, and optical properties of La(OH)3, La2O2CO3, and La2O3 nanorods. J Alloy Compd 509(2):396–401. https://doi.org/10.1016/j.jallcom.2010.09.041
Todorovsky D, Getsova M, Vasileva M (2002) Thermal decomposition of lanthanum-titanium citric complexes prepared from ethylene glycol medium. J Mater Sci 37(18):4029–4039. https://doi.org/10.1023/A:1019600815906
Orera A, Larraz G, Sanjuán M (2013) Spectroscopic study of the competition between dehydration and carbonation effects in La2O3-based materials. J Eur Ceram Soc 33(11):2103–2110. https://doi.org/10.1016/j.jeurceramsoc.2013.03.010
Subramanian M, Aravamudan G, Rao GS (1983) Oxide pyrochlores—a review. Prog Solid State Chem 15(2):55–143. https://doi.org/10.1016/0079-6786(83)90001-8
Panghal A, Kumar Y, Kulriya P, Shirage PM, Singh N (2021) Structural assessment and irradiation response of La2Zr2O7 pyrochlore: impact of irradiation temperature and ion fluence. J Alloy Compd 862:158556. https://doi.org/10.1016/j.jallcom.2020.158556
Kong L, Karatchevtseva I, Gregg DJ, Blackford MG, Holmes R, Triani G (2013) A novel chemical route to prepare La2Zr2O7 pyrochlore. J Am Ceram Soc 96(3):935–941. https://doi.org/10.1111/jace.12060
Paul B, Singh K, Jaroń T, Roy A, Chowdhury A (2016) Structural properties and the fluorite–pyrochlore phase transition in La2Zr2O7: the role of oxygen to induce local disordered states. J Alloy Compd 686:130–136. https://doi.org/10.1016/j.jallcom.2016.05.347
Krishnan RR, Vinodkumar R, Rajan G, Gopchandran K, Pillai VM (2010) Structural, optical, and morphological properties of laser ablated ZnO doped Ta2O5 films. Mater Sci Eng, B 174(1–3):150–158. https://doi.org/10.1016/j.mseb.2010.03.065
Joseph C, Bourson P, Fontana MD (2012) Amorphous to crystalline transformation in Ta2O5 studied by Raman spectroscopy. J Raman Spectrosc 43:1146–1150. https://doi.org/10.1002/jrs.3142
Cusick AB, Lang M, Zhang F, Sun K, Li W, Kluth P et al (2017) Amorphization of Ta2O5 under swift heavy ion irradiation. Nucl Instrum Methods Phys Res, Sect B 407:25–33. https://doi.org/10.1016/j.nimb.2017.05.036
Tsuchiya T, Imai H, Miyoshi S, Glans P-A, Guo J, Yamaguchi S (2011) X-ray absorption, photoemission spectroscopy, and Raman scattering analysis of amorphous tantalum oxide with a large extent of oxygen nonstoichiometry. Phys Chem Chem Phys 13(38):17013–17018. https://doi.org/10.1039/C1CP21310E
George P, Gedanken A (2008) Ta2O5 nanobars and their composites: synthesis and characterization. Journal of nanoscience nanotechnology 8(11):5801–5806. https://doi.org/10.1166/jnn.2008.230
Petit S, Morlens S, Yu Z, Luneau D, Pilet G, Soubeyroux J-L et al (2011) Synthesis and thermal decomposition of a novel zirconium acetato-propionate cluster:[Zr12]. J Solid State Sci 13(3):665–670. https://doi.org/10.1016/j.solidstatesciences.2010.12.041
Deiseroth HJ, Müller-Buschbaum H (1970) Ein beitrag zur pyrochlor struktur an La2Zr2O7. Z Anorg Allg Chem 375(2):152–156. https://doi.org/10.1002/zaac.19703750205
Guinier A (1994) X-ray diffraction in crystals, imperfect crystals, and amorphous bodies. Dover, New York
Cullity BD, Stock SR (2001) Elements of X-ray diffraction, Third Edition. New York: Prentice-Hall
El-Shinawi H, Cussen EJ, Corr SA (2017) Enhancement of the lithium ion conductivity of Ta-doped Li7La3Zr2O12 by incorporation of calcium. Dalton Trans 46(29):9415–9419. https://doi.org/10.1039/C7DT01573A
Badami P et al (2020) Highly conductive garnet-type electrolytes: access to Li6.5La3Zr1.5Ta0.5O12 prepared by molten salt and solid-state methods. ACS Appl Mater Interfaces 12(43):48580–48590. https://doi.org/10.1021/acsami.0c14056
Cheng L, Wu CH, Jarry A, Chen W, Ye Y, Zhu J et al (2015) Interrelationships among grain size, surface composition, air stability, and interfacial resistance of Al-substituted Li7La3Zr2O12 solid electrolytes. ACS Appl Mater Interfaces 7(32):17649–17655. https://doi.org/10.1021/acsami.5b02528
Sharafi A, Haslam CG, Kerns RD, Wolfenstine J, Sakamoto J (2017) Controlling and correlating the effect of grain size with the mechanical and electrochemical properties of Li7La3Zr2O12 solid-state electrolyte. J Mater Chem A 5(40):21491–21504. https://doi.org/10.1039/C7TA06790A
Acknowledgements
This work was supported by NSF CAREER award DMR-1553519 and CMMI-1929806. J.M.W. acknowledges support from an ASU Fulton Schools of Engineering Dean’s Fellowship. The authors would like to thank C. Birkel and J. Jamboretz for assistance with Raman measurements. The authors gratefully acknowledge the use of facilities within the LeRoy Eyring Center for Solid State Science at Arizona State University supported in part by NNCI-ECCS-1542160 and the Southwest Center for Aberration Corrected Electron Microscopy at ASU.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Guo, J., Weller, J.M., Yang, S. et al. Reactive sintering of garnet-type Li6.4La3Zr1.4Ta0.6O12 (LLZTO) from pyrochlore precursors prepared using a non-aqueous sol–gel method. Ionics 29, 581–590 (2023). https://doi.org/10.1007/s11581-022-04837-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-022-04837-4