
Abstract
Methanol oxidation in fuel cells is an effective guarantor of renewable energy. So, this article investigates the electrochemical oxidation of methanol at various nickel-modified electrodes in NaOH solution using cyclic voltammetry and chronoamperometry techniques. Herein, a new electrochemical electrode was characterized by means of the scanning electron microscope (SEM), energy dispersive spectroscope (EDS), thermogravimetric analysis (TGA) and voltammetry method using a redox probe (ferricyanide). The catalytic activity of a new reduced p-nitroaniline/Ni(II) polymer transformed graphite electrode (poly-p-PD/Ni(II)/GE) for the oxidation of methanol is higher than that of the others. Quasi-reversible redox couple peaks were observed at + 0.25 and + 0.117 V and belong to the redox of Ni2+/Ni3+ cations. The surface coverage (Γ) of electrochemical activity of the adsorbed redox cations at the surface of the poly-p-PD/Ni(II)/GE was estimated to be 4.1 × 10−6 mmol/cm2. The effect of scan rate (30–90 mV/s) for methanol in NaOH reveals the oxidation reaction of methanol under a diffusion-controlled process at the fabricated electrode. The values of electro-active surface area (A), transfer coefficient (α), number of electron transfers (n) and diffusion coefficient (D) have been calculated to be 0.562 cm2, 0.183, 1 and 1.59 × 10−4 cm2/s, respectively. The developed electrode used to be eventually displayed high stability while stored in the lab.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
A fuel cell is a battery that converts a chemical reaction into electrical energy. There are various types of fuel cells such as phosphoric acid, alkaline, regenerative, solid oxide and direct alcohol fuel cells. The importance of fuel cells stems from their multiple applications in transport, computers and portable and stationary power generation [1]. We can say that the only two near-defective problems of fuel cells are high cost and the availability of hydrogen as a fuel source. As long as hydrogen is available to the fuel cell, it will be able to operate. So, some fuel cells use solutions such as hydrocarbons and alcohols as a source of hydrogen. Alcohol such as methanol and ethanol are used as fuel because they can store a high energy density [2]. In a direct methanol fuel cell, methanol can be used as fuel due to its high electrochemical activity and liquid state at room temperature, thus facilitating its storage [3]. Moreover, methanol can be produced from more than one source such as coal, oil and natural gas [2].
In recent years, the electrooxidation of methanol has garnered great attention due to several applications of the direct alcohol fuel cells. Unfortunately, the complete oxidation of methanol to hydrogen is slow and difficult and furthermore is accompanied with relatively high overpotential so a catalyst must be used [3]. Many efforts were made to develop a modified electrode with high catalytic activity for the oxidation of methanol [4,5,6,7]. The most active catalyst for the electrooxidation of methanol is Pt [8]. However, carbon monoxide species is produced as an intermediate during the oxidation of methanol. It is strongly adsorbed on the surface of Pt, poisoning the Pt electrode. Additionally, its supply due to its cost is limited [9].
However, the addition of Ni component to a catalyst can significantly improve its electro-catalytic activity and CO tolerance. Therefore, numerous scientific studies have focused on the application of this transition metal or its alloys. Rostami et al. [1] made binary or ternary alloys of Ni, Co and Cu on the surfaces of graphite electrodes for the electrocatalytic oxidation of methanol. Meanwhile, Abdel Hameed RM and El-Khatib KM [3] used commercial carbon electrodes modified with Ni–P and Ni–Cu–P catalysts for the same purpose. Mousavi et al. [10] modified a carbon paste electrode with bis(salicylaldehyde)-nickel(II)-dihydrate complex and reduced graphene oxide. They noted that the electrooxidation of methanol occurred by NiOOH groups through further electrochemical oxidation of Ni(OH)2. In the same way, methanol oxidised at the surface of nickel hydroxide nanoparticles modified glassy carbon electrode (GCE) as prepared by Raoof et al. [11]. Liu et al. [12] developed a biosensor for electrooxidation of methanol in an alkaline medium based on DNA-Ni(II)/MWNTs/Chitosan complex membrane/GCE. Jeon et al. [13] used a cold plasma process to fabricate a polymeric nickel complex-modified indium tin oxide (ITO) electrode. They estimated that the electrochemical oxidation of methanol can occur on Ni-modified electrodes, while not observed on the surface of bare ITO.
The presence of Ni as a catalyst clearly enhances the electrochemical oxidation of methanol. However, for more catalytic activity, sensitivity and stability, a conducting polymer film could be added as a modifier. Polymers have been the subject of great interest in recent years because of their high surface area, tunable conductivities and strong adherence to the electrode surface [14, 15]. Thus, a polymer film of reduced p-nitroaniline seems a good analytical tool for Ni catalyst because of charge migration within the p-nitroanaline leading to low-energy electronic transitions [16]. This specific effect results from an electron-donating amino group in a para position with an electron-withdrawing nitro group in its aromatic system [17]. Hence, this work aims to develop a new reduced p-nitroaniline/Ni(II) polymer-modified graphite electrode (poly-p-PD/Ni(II)/GE) for electrocatalytic oxidation of methanol in an alkaline medium.
Experimental
Nickel sulphate hexahydrate, potassium ferricyanide, sodium hydroxide, sulphuric acid, p-nitroanaline (p-NA) and methanol were of the analytical grade of Merck and used without further purification. All solutions were freshly prepared at room temperature (25 ± 2 °C) using double-distilled water.
The electrochemical measurements were performed with a three-electrode cell attached to an EG&G model 263 potentiostat/galvanostat [18, 19]. Working electrodes were commercial graphite rods and Pt-modified electrodes, while counter and reference electrodes were pure platinum wire and the saturated calomel electrode (SCE), respectively. Scanning electron microscopy (SEM, JEOL JSM 6510 lv) was used to be employed to study the surface morphology, and energy dispersive spectroscopy (EDS) to provide the Ni(II) content of the modified electrode. The thermal stability was carried out using thermogravimetric analyser (TGA, TA Instruments Q500).
The modified electrodes were prepared firstly by polishing the graphite rods (r = 2 mm)with various grades of emery papers until they had a mirror-like surface. The Pt electrode (r = 1.5 mm)was polished carefully with Al2O3 slurry (0.05 μm), then washing with acetone and redistilled water. Secondly, each electrode was immersed in a solution of 0.1 M H2SO4 containing 0.1 M of NiSO4.6H2O and 5.0 mM of p-NA and was conditioned by cyclic sweeping between − 1.2 and 1.0 V (vs. SCE) for 20 cycles as displayed in Scheme 1. Finally, the physically adsorbed material could be removed by carefully rinsing the two modified electrodes with distilled water.

Schematic illustration of the electropolymerization of GE
Results and discussion
Modification process of the graphite electrode
Generally, conducting polymers have an effective role in the activity and selectivity of modified electrodes for measuring analytical species. Consequently, cyclic voltammetry (CV) within the potential range (− 1.2 to 1.0 V vs. SCE) in 0.1 M H2SO4 solution containing 5.0 mM of p-NA and 0.1 M NiSO4 at a scan rate of 100 mV/s can be utilised to develop a polymer film of poly-p-PD with Ni2+ on a graphite electrode, as seen in Fig. 1. In the first cycle, anodic peak (a1), cathodic peak (c1) and cathodic peak (c2) were observed at peak potentials + 0.282 V, + 0.063 V and − 0.729 V, respectively. The mentioned two peaks (a1 and c1) belong to the redox couple of Ni2+/Ni3+ cations [20]. Additionally, the peak-to-peak separation was obtained (ΔEp = (Epa1 − Epc1) = 0.219 V), this value indicating that the redox process of Ni(II)/Ni(III) demonstrates a quasi-reversible behaviour [10]. If we consider this, the other irreversible cathodic peak (c2) belongs to the reduction of p-nitroanaline to p-phenylenediamine (p-PD) [21]. Also, in Fig. 1, an increase in the three peaks’ current upon continuous scanning cycles reflects the continuous growth and increases the thickness of the polymer film of poly-p-PD and Ni(II) on the surface of the GE [22].

CVs of a bare GE in 0.1 M H2SO4 containing 0.1 M Ni(II) and 5.0 mM of p-NA (from (a) to (e): 1, 5, 10, 15, and 20 cycles) at a scan rate of 100 mV/s
Characterization of the poly-p-PD/Ni(II)/GE
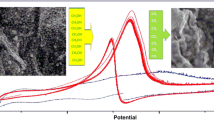
The developed electrode was once eventually characterized by means of SEM to investigate the formation and growth of the thin films of p-PD and Ni ions on the GE surface. Figure 2A and B shows the SEM photographs of the poly-p-PD/Ni(II)/GE with two magnification at accelerating potential of 20 kV. Figure 2A demonstrated almost the whole surface of the electrode at a high-magnification where a thick layer was effectively incorporated on the GE surface. The morphology of the polymer layer was once discovered to be much less uniform having clefts and creases as shown in Fig. 2B. Furthermore, in reality, a few Ni ions distributed randomly throughout the polymer layer. It's worth noting that the polymerized thin layer clings tightly to the GE surface and is mechanically stable with more active sites. Figure 2C displays the EDS spectrum corresponding to the poly-p-PD/Ni(II)/GE, which clearly shows well-defined elemental peaks of C, N, and Ni as expected. The presence of a high percent of Ni is certainly a result of the dispersion of Ni on the high surface area of the p-PD polymer layer. For further characterization, thermogravimetric curves of the poly-p-PD/GE and the poly-p-PD/Ni(II)/GE are shown in Fig. 2D. As can be seen, the TG of the poly-p-PD/GE (curve a) represented a distinct three–steps polymer degradation processes. The first step of mass loss (4.92%) within the temperature from 71.42 to 125.22 °C could be attributed to the elimination of adsorbed water, while the second and third stages of degradation within the temperature ranges of 125.22–243.7 °C and 243.7–334.03 °C, respectively, are caused by the decomposition of the polymeric backbone. As can be observed, about 78.22% weight loss occurred through these two degradation steps. On other hand, the presence of Ni(II) enhanced the thermal stability of the polymeric film, which leads to mass loss of 67.88% (curve b).

SEM images of the poly-p-PD/Ni(II)/GE with two magnifications (A, B) and its related EDS spectra (C). Thermogravimetric (D) analysis (TGA) curves of poly-p-PD/GE (curve a) and poly-p-PD/Ni(II)/GE (curve b)
Electrochemical behaviour of the poly-p-PD/Ni(II)/GE in sodium hydroxide
The proposed electrode behaviour used to be studied in sodium hydroxide in order to investigate the formation of the poly-p-PD/Ni(II) layer on the GE surface. Figure 3 compares the first and 20 cycles of the poly-p-PD/Ni(II)/GE in 0.1 M NaOH solution (pH = 12.5) obtained by CV technique. As can be seen from the first scan, the redox couple peaks appear at a potential of + 0.25 and + 0.117 V, respectively, and their current increases progressively with consecutive potential cycling [11]. These results are other proof of the modified electrode having nickel ions. The redox peaks are corresponding to the Ni(II)/Ni(III) transformation and the possible mechanism can be written as follows [23]:

CVs of the poly-p-PD/Ni(II)/GE in 0.1 M of NaOH (pH = 12.5) were (a) is first cycle and (b) is 20 cycles at scan rate 100 mV/s
Figure 4 shows the CVs of the poly-p-PD/Ni(II)/GE in a 0.1 M NaOH (pH = 12.5) solution at various scan rates (20–1000 mV/s). As can be shown, the currents of the anodic and cathodic peaks increased as the scan rate values increased. Both, the anodic and cathodic peak currents have linear dependences with the scan rate in the same range (inset Fig. 4), indicating that the redox reaction is a surface-adsorbed process independent of mass transfer [20].

CVs of the poly-p-PD/Ni(II)/GE in 0.1 M of NaOH (pH = 12.5), at different scan rates (from (a) to (k): 0.02, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, and 1 V/s). Inset, the plotted of Ip versus the same range of scan rate
The surface coverage of the electrochemical activity of the adsorbed redox cations at the surface of the poly-p-PD/Ni(II)/GE can be calculated from the slopes of the inset in Fig. 4 (Ipc = − 2.23υ–0.629 with R2 = 0.989 and Ipa = 2.007 υ + 0.193 with R2 = 0.992) using the following equation [24]:
where Ip is the peak current (mA), n is the number of electrons (n = 1), F is the Faraday constant (C/mol), A is the working electrode’s surface area (cm2), υ is the scan rate (mV/s), Γ is the surface coverage (mmol/cm2), R is the gas constant (J/mol K) and T is the absolute temperature (K). The Γ value of 4.1 × 10−6 mmol/cm2 was determined using the average of anodic and cathodic peak currents.
Estimation of the poly-p-PD/Ni(II)/GEsurface area
The electro-active surface area of the proposed electrode must be determined since it is related to electrode response rate. As a result, the CV response of 5.0 mM potassium ferricyanide was recorded as the redox probe. Figure 5 describes the CVs with different scan rates (20–500 mV/s) of 0.1 M KNO3 containing 5.0 mM K3[Fe(CN)6] at the poly-p-PD/Ni(II)/GE. It can be seen that with the scan rate increment the anodic and cathodic peaks current increased gradually. In addition, during the oxidation and reduction processes, there is a small potential change in the positive and negative directions, respectively. By plotting the peak current against the square root of the scan rate (inset Fig. 5), two straight lines were produced, suggesting that ferricyanide oxidation is a diffusion-controlled process, with slope compensation in the following Randles–Sevcik equation [25]:

CVs of the poly-p-PD/Ni(II)/GE in 0.1 M of KNO3 containing 5.0 mM K3[Fe(CN)6] at different scan rates (from (a) to (g): 20, 50, 100, 200, 300, 400, and 500 mV/s). Inset, the plotted of Ip versus the square root of scan rate
where Ip is the peak current, n is the number of electrons (for this system n = 1), F is the Faraday constant (C/mol), A is the electro-active surface area of the modified electrode (cm2), C is the concentration of the electro-active species (mM), D is the diffusion coefficient (cm2/s), ν is the scan rate (mV/s), R is the universal gas constant (J/mol.K) and T is the absolute temperature in Kelvin. With known the precise value of D which is 5.6 × 10−6 cm2/s [26], the value of A was calculated to be 0.562 cm2. The electro-active surface area of the poly-p-PD/Ni(II)/GE was large compared to the geometric area of the bare GE (0.44 cm2), which provided an effective evidence for the superior conductivity of p-PD/Ni(II) films as expected. The obtained large-surface area which caused by the polymer film of p-PD was expected to be an attractive platform for the oxidation of methanol.
The electrochemical behaviour of methanol at different modified electrodes
Herein, several efforts have been made to develop Ni-modified electrodes so that their electrocatalytic activity towards the electrooxidation of methanol in an alkaline medium was tested. Figure 6 displays CVs of 0.5 M methanol in 0.1 M NaOH (pH = 12.0) on modified Pt and various modified graphite electrode surfaces swept from − 0.25 to 1.0 V (vs. SCE) at a scan rate of 50 mV/s. In Fig. 6(a–d) curves represent bare GE, poly-p-PD/GE, poly-p-PD/Ni(II)/PtE and poly-p-PD/Ni(II)/GE, respectively. As seen from this figure, there is no significant effect at bare GE pointing towards the electrooxidation of methanol in alkaline solution (curve a). However, the addition of polymer film of poly-p-PD to the GE causes a low current density with the smallest significant effect (curve b). This is expected due to the difficulty of electrooxidation of methanol without Ni(III) according to the following proposed mechanism [27].

CVs of 0.5 M methanol in 0.1 M of NaOH (pH = 12.0) at (a) bare GE, (b) poly-p-PD/GE, (c) poly-p-PD/Ni(II)/PtE and (d) poly-p-PD/Ni(II)/GE at scan rate 50 mV/s
The presence of a thin polymer film of nickel on the poly-p-PD/Pt surface leads to the appearance of an anodic peak at + 0.45 V with a partially low current (curve c). This behaviour can be explained by the formation of poisoning species of CO in alkaline solution strongly bound to the Pt surface, leading to low surface coverage [28].
Conversely, when Pt electrode is replaced by the graphite one (curve d), the anodic peak potential obviously shifts to a more positive value (+ 0.62 V). Furthermore, its current density clearly increases almost five times with the near absence of overpotential. Thus, the presence of polymer film of nickel-base can be said to generally catalyse the electrooxidation of methanol, especially on the GE [29,30,31]. Additionally, the polymer film provides a high electronic conductivity, increases the reactive surface area and can effectively facilitate methanol transport into the modified electrode [32].
Figure 7 compares the behaviour of different electrodes towards the electrooxidation of methanol at conditions similar to Fig. 6. As can be observed, the large current density of the anodic peak for catalytic oxidation of methanol is achieved by using the poly-p-PD/Ni(II)/GE. Therefore, the poly-p-PD/Ni(II)/G modified electrode had been chosen to investigate the electrooxidation of methanol in NaOH solution as seen in Scheme 2. It can be seen that the poly-p-PD is regarded as a bridge that connects Ni(II) with the GE.

A comparison diagram of anodic peak current from Fig. 6 for bare GE, poly-p-PD/GE, poly-p-PD/Ni(II)/PtE and poly-p-PD/Ni(II)/GE

Schematic illustration of the electrochemical oxidation of methanol on the poly-p-PD/Ni(II)/GE surface
Effect of scan rate
It is important to know the type of electron movement of species between the electrolyte and the poly-p-PD/Ni(II)/GE. This can be determined by studying the effect of scan rate (30–90 mV/s) of 0.5 M methanol in 0.1 M of NaOH (pH = 12.0) on the poly-p-PD/Ni(II)/GE surface as can be seen in Fig. 8. The anodic peak current of methanol clearly increases as the scan rate increases. Besides, the potential of the peak is slightly shifted to more positive values, indicating the electrochemical reaction oxidation of methanol under a kinetic limitation [11]. Inset Fig. 8A, on the other hand, indicates the linear dependency of the peak current on the square root of scan rates in the 50 to 90 mV/s range, Ip = 0.313 υ0.5 + 4.239 with R2 = 0.998. This means that the electrooxidation of methanol at the Poly-P-PD/Ni(II)/GE in alkaline media is a diffusion-controlled process [33].

CVs of 0.5 M methanol in 0.1 M of NaOH (pH = 12.0) on the poly-p-PD/Ni(II)/GE surface at various scan rates (from (a) to (g): 30, 40, 50, 60, 70, 80 and 90 mV/s). A Variation of Ip versus square root of scan rates. B Variation of Ep versus lnυ
The total number of electrons for methanol oxidation reaction at the poly-p-PD/Ni(II)/GE can be calculated using the slope of this linear relationship and the compensation in the following equation [34]:
where Ip is the peak current, n is the number of electrons transferred, α is the transfer coefficient, A is the electrode surface area (cm2), D is the diffusion coefficient (5.06 × 10−4 cm2/s as measured by chronoamperometry (see below), υ is the scan rate (mV/s) and C is the bulk methanol concentration (0.5 M). Assuming that nα = 1[10], the number of electrons can be obtained by knowing the value of the transfer coefficient (α). The slope of EP against lnυ (Fig. 8B) can be used for this purpose using the following equation [35]:
where R represents the gas constant (J/mol K), T is the absolute temperature (K), n is the number of electrons transfer and F is the Faraday constant (C/mol). Therefore, the α value can be determined to be 0.183 and consequently n ≈ 1 [36, 37].
Chronoamperometry study
One of the important techniques of electrochemistry is chronoamperometry, which is employed to determine the value of the diffusion coefficient. Figure 9A illustrates the chronoamperograms of different methanol concentrations in 0.1 M of NaOH obtained by setting the working electrode potential at –0.5 V (first step) and 1.4 V (second step) for the poly-p-PD/Ni(II)/GE. With a dramatic increase of methanol concentration, the current densities clearly increase [38]. When the transient current was plotted against the inverse of the square root of time, as seen in Fig. 9B, a linear relationship was obtained. The value of the diffusion coefficient (D) of methanol under diffusion control can be calculated using the average slope of I vs. t−0.5 in the Cottrell equation [39]:

A Chronoamperograms of poly-p-PD/Ni(II)/GE in 0.1 M of NaOH for different concentrations of methanol (from (a) to (d): 0.3, 0.4, 0.5, and 0.8 M), where first and second potential steps were − 0.5 and + 1.4 V vs. SCE, respectively. B The dependency of current on t−0.5 at the same concentrations of methanol
where n is the number of electrons (n = 1), I is the current dominated by methanol diffusion, F is the Faraday constant (C/mol), A is the working electrode’s surface area (cm2), C is the bulk methanol concentration (M) and t is the time (s). The mean value of D was calculated to be 1.59 × 10−4 cm2/s. Table 1 compares the values of diffusion coefficient reported in the literature and the value obtained in this paper. It is possible to deduce that a high diffusion coefficient indicates the high electro-catalytic methanol oxidation at the suggested electrode.
Stability of the poly-p-PD/Ni(II)/GE
The long-term stability of the created electrode, which was simple and low-cost, was tested for 15 days. Figure 10 illustrates the CVs of the poly-p-PD/Ni(II)/GE, at a scan rate of 50 mV/s, in 0.1 M NaOH (pH = 12.0) containing 0.5 M methanol. There is no discernible difference between the original CV and the one measured after 15 days. In fact, the presence of the p-PD polymer layer may prevent the electrode active surface from becoming poisoned during storage.

CVs of 0.5 M methanol in 0.1 M of NaOH (pH = 12.0) at the poly-p-PD/Ni(II)/GE with a scan rate 50 mV/s
In an alkaline solution, Wasmus and Vielstich [8] investigated the methanol oxidation reaction (MOR) at the Pt/C and Pt–Ru/C. The peak current of methanol at the poly-p-PD/Ni(II)/GE is larger than the obtained values concerning the Pt/C and Pt–Ru/C. PtRu/C, PtRu/MWCNTs and PtRu/C60MWCNTs40 have been reported for MOR by Quan, DL, and Le [44]. The peak potential for these studied electrodes is about 200 mV smaller than the results obtained with poly-p-PD/Ni(II)/GE. When compared to the PtRu/C and SnO2-modified PtRu/C catalysts prepared by Wang et al. [45], the peak potential is about 100 mV smaller than the results obtained, indicating that the proposed electrode has a greater MOR activity than carbon-supported Pt or Pt/Ru.
Conclusion
In this paper, a polymer film of p-PD and Ni2+ on a GE surface was obtained through electropolymerization. SEM, EDS, TGA and CV with the redox probe (ferricyanide) were used to analyse the improved sensor, which had a poly-p-PD layer and Ni ions successfully coated into the GE surface. The proposed poly-p-PD/Ni(II)/GE shows high catalytic activity towards the methanol oxidation under a diffusion-controlled process. The transformation of Ni2+/Ni3+ cations produces quasi-reversible redox couple peaks observed at + 0.25 and + 0.117 V. The surface coverage (Γ) of the electrochemical activity of the adsorbed redox cations was calculated to be 4.1 × 10−6 mmol/cm2 at the surface of the poly-p-PD/Ni(II)/GE. The important parameters such as electro-active surface area, transfer coefficient, number of electrons transferred and diffusion coefficient have been calculated to be 0.562 cm2, 0.183, 1 and 1.59 × 10−4 cm2/s, respectively. These results suggest that the poly-p-PD/Ni(II) modified graphite electrode could be applied as a direct methanol fuel cell catalyst. The findings indicated that the novel electrode was made with a unique composition that is low-cost, simple, sensitive, selective, repeatable, stable and rapid towards methanol oxidation.
References
Rostami T, Jafarian M, Miandari S, Mahjani MG, Gobal F (2015) Synergistic effect of cobalt and copper on a nickel-based modified graphite electrode during methanol electro-oxidation in NaOH solution. Chin J Catalysis 36(11):1867–1874. https://doi.org/10.1016/S1872-2067(15)60959-7
Mahapatra SS, Datta J (2011) Characterization of Pt-Pd/C electrocatalyst for methanol oxidation in alkaline medium. Int J Electrochem 2011:16 pages. https://doi.org/10.4061/2011/563495.
Abdel Hameed RM, El-Khatib KM (2010) Ni–P and Ni–Cu–P modified carbon catalysts for methanol electro-oxidation in KOH solution. Int J Hydrogen Energy 35(6):2517–2529. https://doi.org/10.1016/j.ijhydene.2009.12.145
Vigier F, Coutanceau C, Perrard A, Belgsir EM, Lamy C (2004) Development of anode catalysts for a direct ethanol fuel cell. J Appl Electrochem 34:439–446
Zhou W, Song S, Li W, Zhou Z, Sun G, Xin Q, Douvartzides S, Tsiakaras P (2005) Direct ethanol fuel cells based on PtSn anodes: the effect of Sn content on the fuel cell performance. J Power Sources 140(1):50–58. https://doi.org/10.1016/j.jpowsour.2004.08.003
Kim HC, Hong JW (2021) Highly Porous Au–Pt bimetallic urchin-like nanocrystals for efficient electrochemical methanol oxidation. Nanomater 11:112. https://doi.org/10.3390/nano11010112
Prathap A, Udayan M, Sawant SN (2021) Facile one-pot synthesis of CuO nanosheet for application as electrocatalyst for methanol oxidation. J Phys Chem Solid 150:109883.
Wasmus S, Vielstich W (1993) Methanol oxidation at carbon supported Pt and Pt-Ru electrodes: an on line MS study using technical electrodes. J Appl Electrochem 23:120–124
Berger DJ (1999) Fuel cells and precious-metal catalysts. Science 286(5437):49. https://doi.org/10.1126/science.286.5437.49c
Mousavi Z, Benvidi A, Jahanbani S, Mazloum-Ardakani M, Vafazadeh R, Zare HR (2016) Investigation of electrochemical oxidation of methanol at a carbon paste electrode modified with Ni(II)-BS complex and reduced graphene oxide nano sheets. Electroanal 28(12):2985–2992. https://doi.org/10.1002/elan.201501183
Raoof JB, Ojani R, Hosseini SR (2013) An electrochemical investigation of methanol oxidation on nickel hydroxide nanoparticles. S Afr J Chem 66:47–53
Liu Y, Wei W, Zhai X, Zeng J (2008) A novel carbon nanotube-modified biosensor containing a dsDNA-Ni(II) complex membrane, and its use for electro-catalytic oxidation of methanol in alkaline medium. Microchim Acta 162:245–251. https://doi.org/10.1007/s00604-007-0757-4
Jeon J-S, Yu I-K, Kim W, Choi S-H (2020) Electrocatalytic oxidation of methanol by a polymeric Ni complex-modified electrode prepared by a one-step cold-plasma process. Front Chem 8:595616. https://doi.org/10.3389/fchem.2020.595616
Rageh H, Abdel-sabour M (2017) Pharmaceutical electrochemistry: the electrochemical behaviour of paracetamol at ZnO nanoparticales/1, 2-napthaquinone-4-sulphonic acid glassy carbon modified electrode. Anal Bioanal Electrochem 9(3):351–364
Kausaite A, Ramanaviciene A, Ramanavicius A (2009) Polyaniline synthesis catalysed by glucose oxidase. Polymer 50(8):1846–1851. https://doi.org/10.1016/j.polymer.2009.02.013
Kumar SA, Chen S-M (2007) WITHDRAWN: direct electrochemistry and electrocatalysis of myoglobin on redox-active self-assembling monolayers derived from nitroaniline modified electrode. Biosens Bioelectron 22(12):3042–3050. https://doi.org/10.1016/j.bios.2007.01.003
Nobutoki H, Koezuka H (1997) Theoretical study of hyperpolarizabilities in crystalline m-nitroaniline. J Phys Chem A 101(20):3762–3768. https://doi.org/10.1021/jp962102a
Abou-Krisha MM, Assaf FH, Toghan AA (2007) Electrodeposition of Zn–Ni alloys from sulfate bath. J Solid State Electrochem 11(2):244–252. https://doi.org/10.1007/s10008-006-0099-x
Toghan A, Abo-bakr AM, Rageh HM, Abou-Krisha MM, Abd-Elsabour M (2018) Electro-oxidation and detection limit of catechol as a precursor for pharmaceutical applications at a glassy carbon electrode. J Pharm Appl Chem 4(2):133–7. https://doi.org/10.18576/jpac/040208.
El-Shafei AA (1999) Electrocatalytic oxidation of methanol at a nickel hydroxide/glassy carbon modified electrode in alkaline medium. J Electroanal Chem 471(2):89–95. https://doi.org/10.1016/S0022-0728(99)00235-1
Laghrib F, Boumya W, Lahrich S, Farahi A, El Haimouti A, El Mhammedi MA (2017) Electrochemical evaluation of catalytic effect of silver in reducing 4-nitroaniline: Analytical application. J Electroanal Chem 807:82–87. https://doi.org/10.1016/j.jelechem.2017.11.024
Amare M, Admassie S (2012) Polymer modified glassy carbon electrode for the electrochemical determination of caffeine in coffee. Talanta 93:122–128. https://doi.org/10.1016/j.talanta.2012.01.058
Abdel Rahim MA, Hassan HB, Abdel Hameed RM (2007) Graphite electrodes modified with platinum-nickel nano-particles for methanol oxidation. Fuel Cells 7(4):298–305. https://doi.org/10.1002/fuce.200600046
Zheng L, Jf S (2009) Curcumin multi-wall carbon nanotubes modified glassy carbon electrode and its electrocatalytic activity towards oxidation of hydrazine. Sens Actuators B 135(2):650–655. https://doi.org/10.1016/j.snb.2008.09.035
Garcia-Miranda Ferrari A, Foster CW, Kelly PJ, Brownson DAC, Banks CE (2018) Determination of the electrochemical area of screen-printed electrochemical sensing platforms. Biosensors 8:53–62
Lyons MEG, Keeley GP (2006) The redox behaviour of randomly dispersed single walled carbon nanotubes both in the absence and in the presence of adsorbed glucose oxidase. Sensors 6:1791–1826
Fleischmann M, Korinek K, Pletcher D (1971) The oxidation of organic compounds at a nickel anode in alkaline solution. J Electroanal Chem Interf Electrochem 31(1):39–49. https://doi.org/10.1016/S0022-0728(71)80040-2
Morallon E, Rodes A, Vazquez JL, Perez JM (1995) Voltammetric and in-situ FTIR spectroscopic study of the oxidation of methanol on Pt(hkl) in alkaline media. J Electroanal Chem 391(1–2):149–157. https://doi.org/10.1016/0022-0728(95)03965-J
Risbud MS, Baxter S, Skyllas-Kazacos M (2012) Preparation of nickel modified carbon fibre electrodes and their application for methanol oxidation. Open Fuel Energy Sci J 5:9–20. https://doi.org/10.2174/1876973X01205010009
Javan H, Asghari E, Ashassi-Sorkhabi H, Moradi-Haghighi M (2020) Nickel nanoparticles decorated on carbon quantum dots as a novel non-platinum catalyst for methanol oxidation; a green, low-cost, electrochemically-synthesized electrocatalyst. Chem Eng Sci 217:115534. https://doi.org/10.1016/j.ces.2020.115534
Golabi SM, Nozad A (2003) Electrocatalytic oxidation of methanol at lower potentials on glassy carbon electrode modified by platinum and platinum alloys incorporated in poly(o-aminophenol) film. Electroanal 15(4):278–286. https://doi.org/10.1002/elan.200390035
Tong X, Qin Y, Guo X, Moutanabbir O, Ao X, Pippel E, Zhang L, Knez M (2012) Enhanced catalytic activity for methanol electrooxidation of uniformly dispersed nickel oxide nanoparticles carbon nanotube hybrid materials. small 8(22): 3390–95. DOI: https://doi.org/10.1002/smll.201200839.
Sunitha M, Asha S, Ramachandran T (2020) Nickel boride and cobalt boride coated stainless steel gauze for enhanced electrochemical oxidation of methanol. Ionics 26:1875–1884. https://doi.org/10.1007/s11581-019-03410-w
Bard AJ, Faulkner LR (1980) Electrochemical methods, fundamentals and applications. John Wiley and Sons, New York
Wang H, Zhang M, Cheng F, Xu C (2008) Pt supported on Ti for methanol electrooxidation by magnetron sputter method. Int J Electrochem Sci 3:946–952
Anthon GE, Barrett DM (2004) Comparison of three colorimetric reagents in the determination of methanol with alcohol oxidase. Application to the assay of pectin methylesterase. J agr food chem 52(12):3749–53. https://doi.org/10.1021/jf035284w.
Harraz FA, Faisal M, Jalalah M, Almadiy AA, Al-Sayari SA, Al-Assiri MS (2020) Conducting polythiophene/α-Fe2O3 nanocomposite for efficient methanol electrochemical sensor. Appl Surf Sci 508:145226. https://doi.org/10.1016/j.apsusc.2019.145226
Ferdowsi GhS, Seyedsadjadi SA, Ghaffarinejad A (2015) Ni nanoparticle modified graphite electrode for methanol electrocatalytic oxidation in alkaline media. J Nanostruct Chem 5:17–23. https://doi.org/10.1007/s40097-014-0124-z
Bard AJ, Faulkner LR (2001) Electrochemical methods: fundamentals and applications. New York: Wiley and Sons 5:12–14
Jafarian M, Moghhaddam RB, Mahjani MG, Gobal F (2006) Electro-catalytic oxidation of methanol on a Ni–Cu alloy in alkaline medium. J Appl Electrochem 36:913–918. https://doi.org/10.1007/s10800-006-9155-6
Galal A, Atta NF, Hassan HK (2012) Graphene supported-Pt-M (M = Ru or Pd) for electrocatalytic methanol oxidation. Int J Electrochem Sci 7:768–784
Hassaninejad-Darzi SK, Rahimnejad M (2014) Electrocatalytic oxidation of methanol by ZSM-5 nanozeolite modified carbon paste electrode in alkaline medium. J Iran Chem soc 11:1047–1056. https://doi.org/10.1007/s13738-013-0373-7
Wang Y, Chen W, Pan D, Xu Q, Ma J, Zheng J, Li R (2017) Methanol electrooxidation reaction in alkaline medium on glassy carbon electrode modified with ordered mesoporous Ni/Al2O3. Int J Electrochem Sci 12)2:(194–206. https://doi.org/10.20964/2017.03.47.
Quan, DL, Le PH, (2021) Enhanced methanol oxidation activity of PtRu/C100−xMWCNTsx (x = 0–100 wt.%) by controlling the composition of C‐MWCNTs support. Coat 11:571. https://doi.org/10.3390/coatings11050571.
Wang G, Takeguchi T, Zhang Y, Muhamad EN, Sadakane M, Shen Y, Ueda W (2009) Effect of SnO2 deposition sequence in SnO2-modified PtRu/C catalyst preparation on catalytic activity for methanol electro-oxidation. J Electrochem Soc 156(7):B862–B869. https://doi.org/10.1149/1.3133249
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University for funding this work through Research Group no. RG-21-09-79.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Abd-Elsabour, M., Alhamzani, A.G. & Abou-Krisha, M.M. Fabrication of novel nickel-modified electrodes and their application for methanol oxidation in fuel cell. Ionics 28, 1915–1925 (2022). https://doi.org/10.1007/s11581-022-04447-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-022-04447-0