
Abstract
In this paper, we prove an efficient and facile synthesis method for preparing PtRu nanoparticles (NPs)/graphene nanosheets (GNS) catalysts for methanol electrooxidation. Our approach employed carboxylated-graphene nanosheets (C-GNS) as the support which prepared by the Friedel-Crafts reaction between succinic anhydride and GNS. The reaction conditions are mild without cumbersome pretreatment of GNS. The morphology and component of PtRu NPs/C-GNS catalysts were characterized by transmission electron microscopy (TEM) and inductively coupled plasma-atom emission spectroscopy (ICP-AES), respectively. The TEM observation reveals that PtRu NPs with an average diameter of ca. 4.0 ± 0.5 nm uniformly distributed on the edges and wrinkles of C-GNS. The further electrochemical characterizations including cyclic voltammograms (CV) and chronoamperometry (CA) methods show that PtRu NPs/C-GNS catalysts have significantly higher electrocatalytic activity and stability toward methanol electrooxidation compared to the PtRu catalysts supported on the unmodified GNS. This provides an easy approach to synthesize GNS-based electrode materials for high-performance energy conversion devices in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Over past decades, with the decrease in the supply of fossil fuels and the ever-increasing environmental issues from these fuels, fuel cells are receiving much attention [1,2,3]. Fuel cells are considered as one of the most important power sources because of their low emissions, high energy efficiency, and ease of handling of liquid fuel. Due to easy transportation and storage, small organic molecules such as methanol and ethanol have been widely studied as potential candidates for use in fuel cells. As is well known, nanosized platinum and platinum alloys are the most important electrocatalysts in these fuel cells. However, the high cost of Pt and the gradual decrease in their catalytic activity caused by carbonaceous intermediate products such as COads being chemisorbed on these catalysts are the main barriers to commercialize direct methanol fuel cells (DMFCs) [4,5,6,7,8]. To alleviate the CO poisoning of the Pt, one of the strategies is to employ bimetallic catalysts that combine Pt with other metals such as Ru [4, 5, 9], Fe [10], Co [11], Cu [12], and so on. In particular, PtRu alloy is one of the superior binary Pt-based electrocatalysts for DMFCs. Ru is highly oxophilic, and therefore can form oxygenated species at lower potentials than Pt, which will facilitate the removal of CO species adsorbed on the Pt and then release the occupied reaction active sites [1, 2, 5].
On the other hand, it is necessary to further maximize the electrocatalytic performance of the PtRu catalysts and minimize the usage of Pt. So, it is important to prepare uniform dispersion of PtRu nanoparticles (NPs) with small particle size and narrow size distribution because of its large surface-to-volume ratio and facile access to each catalyst particle. However, because of large metal-metal cohesion, PtRu NPs tend to agglomerate. This leads to a decrease in electrochemically active surface area (ESA), which is unfavorable to enhance the catalytic activity. To overcome this problem, it is necessary to develop suitable catalysts supporting materials that possess a high surface area, excellent electronic conductivity, and strong affinity toward catalysts particles to obtain high dispersion and stability of catalysts NPs. Besides, the support also contributes in altering the electronic properties and geometry of the catalysts particles. Graphene is a single-atom-thick sheet of hexagonally arrayed sp2-bonded carbon atoms. It has been intensively studied and received much attention for its applications in a wide variety of areas, such as fuel cells, lithium ions batteries, dye-sensitized solar cells, and chemo/biosensors. Especially, owing to its unique properties such as large specific surface area, high electronic conductivity, great mechanical strength, and outstanding chemical and electrochemical stability, graphene is widely used as the support material for metal NPs catalysts [10, 13,14,15,16,17,18,19,20,21,22,23,24,25,26].
Usually, metal catalysts with support are often prepared in solution-phase. In order to uniformly anchor and grow the metal NPs, the supports must have good dispersion without aggregation for high available surface areas. Unfortunately, graphene nanosheets (GNS) as support materials have been hindered by their agglomeration and poor dispersibility in solvents [17, 22, 26]. What is more, due to its inert nature of the graphitized surface, GNS have insufficient binding sites for anchoring the metal ions or NPs. Consequently, metal catalysts on GNS usually have large particle size and poor dispersion.
To disperse metal catalysts and control particle size, the most efficient strategy is surface functionalization of GNS to introduce more binding sites. One of the most common methods is using graphene oxide (GO) as the support material. There are abundant oxygen-containing functional groups on GO surface which can act as the binding sites. However, the poor conductivity and electrochemical stability of GO result in a loss of the ESA and reduced the durability of the supported metal catalysts during fuel cells works. Recently, functionalization of GNS such as π-π stacking, modification with ionic liquid [19], graft poly(styrenesulfonic acid-g-pyrrole) [26] or poly(pyrogallol) [22], nitrogen-doped [15], etc. have been developed to support uniform metal catalysts. Although great progress has been made, the great challenges still remain. Especially, it is highly desirable to develop rational functionalization method that can attach high dispersion of metal catalysts with small particle size on GNS for high electrocatalytic activity and stability.
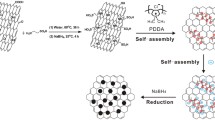
Herein, we report a facilely covalent functionalization of GNS strategy to uniformly disperse PtRu catalysts because of wide use as anode catalysts in DMFCs. Our approach is graft carboxyl-terminated functional groups onto GNS surface by the Friedel-Crafts reaction between GNS and succinic anhydride (SA) (Scheme 1). These carboxyl groups improve the hydrophilicity of the C-GNS. Thus C-GNS can offer more accessible specific surface area for supporting catalysts. More importantly, the carboxyl groups on the surface of GNS could provide enough active sites for anchoring the catalysts precursors (PtCl62− and Ru3+). So, PtRu NPs can firmly and uniformly fix on the surface of carboxylated-graphene nanosheets (C-GNS) and resist aggregate after reduction. The TEM characterization indicated the as prepared PtRu catalysts distributed well on the C-GNS surface. The further electrochemical experiments showed that the PtRu NPs/C-GNS catalysts have enhanced activity, more stability, and anti-poison ability for methanol oxidation than PtRu NPs supported on GNS without modification. These metal NPs/GNS nanohybrids were expected to work as the new anode electrocatalysts for DMFCs application.

Schematic diagram of the C-GNS and preparation of PtRu NPs/C-GNS catalysts.
Experimental
Materials
Graphite powder (diameter 1–10 μm, 99.9% purity) was purchased from Nanjing XFNANO Materials Tech Co., Ltd., China. H2PtCl6·6H2O, RuCl3, and succinic anhydride were purchased from Alfa Aesar. Other chemicals were of analytical grade and used as received.
Synthesis of PtRu NPs/C-GNS catalysts
GO was prepared from pristine graphite powder according to Hummers’ method with some modifications. Briefly, 5 g of graphite powder and 3.75 g of NaNO3 were added to 230 mL of concentrated H2SO4 (98%) with stirring in an ice-water bath. Then, 15 g of KMnO4 was slowly added over about 1 h, so that the temperature of the mixture was maintained less than 10 °C. After the mixture was stirred at 35 °C for 5 days, 500 mL of 5 wt.% H2SO4 aqueous solutions were added over about 1 h under magnetic stirring and the temperature was kept at 98 °C for 2 h. Then, the temperature was reduced to 60 °C, 15 mL of 30 wt.% H2O2 solutions were added, and the mixture was stirred for 2 h at 30 °C. The collected precipitates were repeatedly washed with 3 wt.% H2SO4 + 0.5 wt.% H2O2 solutions, 5 wt.% HCl aqueous solution, and distilled water. Finally, the products were dried in a vacuum oven for 3 days to obtain GO. To obtain GNS, chemical reduction of the suspension of GO was carried out with hydrazine monohydrate (0.1 mL, 0.3 g GO) for 24 h at 80 °C. The synthesized samples was washed with distilled water and dried under vacuum oven for 24 h at 40 °C.
The preparation procedure for the C-GNS was as follows: GNS (200 mg), succinic anhydride (1.5 g) were refluxed with AlCl3 (2 g) in dried N-methyl-2-pyrrolidone at approximately 110 °C under the dry nitrogen atmosphere for 6 h. Then, the reaction mixture was stirred at 150 °C for 48 h. After the reaction, the mixture was decomposed with double-distilled water followed by 0.5 M HCl aqueous solutions. The obtained samples were then washed with double-distilled water five times and following adjusted the pH to 9 with 1.0 M KOH aqueous solutions. Finally, the filtered solid was dried under vacuum for 12 h at 40 °C to obtain C-GNS.
A microwave-assisted reduction process in ethylene glycol solution was used for deposition of PtRu NPs on the C-GNS. The details were as follows: 40 mg of C-GNS was mixed with 876 μL H2PtCl6 (38.6 mM) and 700 μL RuCl3 (48.2 mM) in ethylene glycol solution. After ultrasonication for 40 min, the pH value of the solution was adjusted to 9 with 1.0 M KOH aqueous solution. The mixture was placed in a microwave oven and heated by microwave irradiation for 30 min at 120 °C. The product was allowed to cool to room temperature. Then, it was centrifuged and washed three times with distilled water. The obtained sample was dried in vacuum oven at 55 °C for 24 h and denoted as PtRu NPs/C-GNS catalysts. For comparison, PtRu NPs supported on the unmodified GNS, labeled as PtRu NPs/GNS catalysts, was prepared under the same procedure as described above.
Materials characterization
The surface chemical compositions of the C-GNS were analyzed by Fourier transform infrared spectrometry (FTIR) (Nicolet, 6700). The Raman spectrum (inVia Reflex, Renishaw, England) was also used to study the integrity and structure of the C-GNS and the prepared catalysts. The structure and composition of the PtRu NPs/C-GNS and PtRu NPs/GNS catalysts were determined by powder X-ray diffraction (XRD, Bruker AXS X-ray diffractometer) and inductively coupled plasma-atom emission spectroscopy (ICP-AES, Spectro Ciros), respectively. The morphology of the catalysts was investigated by transmission electron microscopy (TEM, JEOL-3010) and scanning electron microscope (SEM, Quanta450&IE250X-Max50).
For electrochemical investigation, a glassy carbon (GC, 5 mm diameter) electrode was polished with the slurry of 0.5 and 0.03 μm alumina successively and washed ultrasonically in double-distilled water prior to use. The catalysts ink was prepared by dispersing 5 mg of catalysts in 5 mL of water by sonication. When a dark homogeneous dispersion was formed, 40 μL of the ink was dropped onto the GC electrode using micro-syringe. The total loading mass of catalysts on glassy carbon electrode is 203.82 μgcm−2. After dried in air, the electrode was coated with 10 μL of 0.05 wt.% Nafion ethanol solution to fix the catalysts powder. All electrochemical measurements were performed on a CHI660D electrochemical workstation (Chenhua Instrument Company of Shanghai, China). A conventional three-electrode glass cell was used with a platinum wire as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode. The electrochemical surface area (ESA) and the electrochemical performance of the electrocatalysts were evaluated by cyclic voltammetry. CO stripping voltammograms were obtained on PtRu NPs/C-GNS and PtRu NPs/GNS catalysts at a scan rate of 50 mV s−1 in a 0.5 M H2SO4 solution. The electrocatalysts on the working electrode were pre-adsorbed of CO at − 0.16 V for 10 min. Then, the dissolved CO was removed by bubbling N2 into solution for 20 min, and the stripping voltammograms were collected at a scan rate of 50 mV s−1. All the potentials reported herein were in respect to SCE. Double-distilled water was used throughout.
Results and discussion
Figure 1 shows the FTIR patterns of GNS, SA, C-GNS, PtRu NPs/C-GNS, and PtRu NPs/GNS catalysts recorded in 4000–500 cm−1. The characteristic features of GNS are the absorption band that corresponded to C–O vibration at 1062 cm−1 and C–C skeleton vibration of carbon ring in graphene at 1593 cm−1. Similarly, the above two characteristic absorption peaks of GNS appear in the PtRu NPs/GNS catalysts, except for a slight shift in the peak position. The characteristic peaks of SA that occurred at 2975 cm−1 can be ascribed to the saturated C-H stretching vibration. The peaks at 1805 and 1784 cm−1 are the asymmetric and symmetric vibration of C=O, respectively. For the FTIR spectra of C-GNS, a broad peak occurred at 3435 cm−1, which assigns to O-H stretching vibration. One at 2916 cm−1 assigns to asymmetric and symmetric C-H vibrations; and peak at 1605 cm−1 assigns to the stretching vibration of C=O in carboxyl group. The characteristic peaks of SA at 1850 and 1784 cm−1 were not observed in C-GNS, which suggested that the SA rings are completely opened. Obviously, the PtRu NPs/C-GNS catalysts exhibit the same FTIR absorption characteristic peaks with the C-GNS support materials. This result revealed that the carboxyl acid groups were successfully grafted on the surface of GNS. These surface functional groups on the C-GNS and GNS supports can be preserved during the process of catalyst preparation.

FTIR spectra of GNS, PtRu NPs/GNS, SA, C-GNS and PtRu NPs/C-GNS
Raman spectroscopy is a useful tool for the characterization of carbon materials, especially for analyzing the surface structure and distinguishing sp2 and sp3 hybridized forms of carbon. Figure 2 displays Raman spectroscopy of GNS, PtRu NPs/GNS, C-GNS, and PtRu NPs/C-GNS. The appearance of two prominent peaks at 1368 cm−1 and 1590 cm−1 could be ascribed to the D and G bands of graphene [17], respectively. The D band corresponds to defects in the curved graphene sheet, while the G band is related to the stretching mode of crystal graphite. It can be deduced that there are large number of structural defects in C-GNS since the D band is very intense. On the contrary, the D band is greatly suppressed in the Raman spectrum of GNS, indicating that the GNS prepared by the chemical route exhibits perfect ordered graphite structure. The extent of the defects in graphite materials can be quantified by the intensity ratio of the D to G bands (i.e., ID/IG). It can be obtained from Fig. 2 that the values of the ID/IG ratio are 0.82 and 1.13 for the GNS and C-GNS, respectively. It is noted that C-GNS shows higher ID/IG value than that of GNS. In combination with the FTIR spectra results, it can be deduced that C-GNS have been grafted massive carboxyl groups by the Friedel-Crafts reaction process, which led to the more structural damage of GNS. Moreover, the values of the ID/IG ratio of PtRu NPs/GNS and PtRu NPs/C-GNS catalysts are 0.80 and 1.09, respectively, which are slightly less than that of corresponding support materials. It can be attributed that some of defect sites on the surface of the GNS or C-GNS are covered by the supported PtRu NPs. In addition, comparing PtRu NPs/GNS catalysts, the more reduced ID/IG value of PtRu NPs/C-GNS catalysts than that of their support may be ascribed to the more high dispersion of PtRu NPs that covered more defect sites on the surface of C-GNS.

Raman spectrum of GNS, PtRu NPs/GNS, C-GNS, and PtRu NPs/C-GNS
As mentioned in above, the excellent dispersibility in solvents is one of the necessary conditions for GNS as superior support materials. Hence, a comparison of the dispersibility of GO, GNS, and C-GNS in water was carried out and the corresponding result is shown in Fig. S1 (Supporting information). The suspension of GO, GNS, and C-GNS in pure water with concentrations of 1 mg mL−1 placed for 1 month after ultrasonic treatment. As shown in Fig. S1, there are no signs of phase separation for GO and C-GNS when the above solution sitting undisturbed for 1 month. In comparison, the dispersion of GNS produced black precipitation at the bottom of tube. This demonstrates that the more carboxyl groups improves obviously the dispersion of C-GNS in water, which is helpful for anchoring and growing PtRu catalysts, and C-GNS are the more excellent support materials for noble metal catalysts in fuel cells.
Figure 3 shows the TEM images of the PtRu NPs/C-GNS and PtRu NPs/GNS catalysts. It can be seen from Fig. 3 that the C-GNS is decorated successfully with lots of well-dispersed PtRu NPs. Their size distribution was evaluated statistically through measuring the diameter of 100 PtRu NPs in the selected TEM images. It is noted that the particle size of PtRu NPs distributes mainly between 2.2 and 6.2 nm with an average diameter of ca. 4.0 ± 0.5 nm. Notably, no NPs aggregation is clearly observed on the C-GNS surface. However, for the unmodified GNS, PtRu NPs do not disperse uniformly on the GNS surface and have a broad distribution (5.5–15.5 nm) with an average diameter of ca. 10.0 ± 2.0 nm. The reasons for this finding should be as follows. For the unmodified GNS, there are few defect sites on GNS surface with uneven distribution. When PtRu NPs are deposited on the GNS, PtRu NPs tend to deposit on these localized defect sites, thus leading to poor dispersion and aggregation. However, for the GNS with SA modification, the carboxyl-terminated groups on the surface of GNS produces a uniform distribution of the carboxylic groups that serve as functional groups for the immobilization of catalysts precursors (PtCl62− and Ru3+) on the surface of the GNS through electrostatic interaction and coordination. Therefore, a much more uniform distribution of PtRu NPs is observed on the surface of the C-GNS supports. Moreover, the SEM is also used to investigate the morphology of the catalysts and support materials (Supporting information). Figure S2a and b are the high magnified SEM images of GNS and C-GNS, respectively. Both of them display obviously layered structure of reduced grapheme oxide. The high magnification SEM images of GNS (Fig. S2c, 2e) and C-GNS (Fig. S2d, 2f) indicate that PtRu NPs on the surface of GNS and C-GNS. Comparing the accumulation-mode PtRu NPs on the GNS, the PtRu NPs uniformly dispersed on the C-GNS with small particle size. Figure S2g and h (i and j) show the elemental mapping of Pt (Ru) of PtRu NPs. Both elements are evenly distributed in the whole surface of GNS and C-GNS. The results of TEM and SEM indicate that carboxylic groups play key roles in obtaining PtRu NPs with high dispersion and small particle size supported on the GNS surface. On the other hand, the composition of the prepared catalysts was determined by ICP-AES and the corresponding result is shown in Table S1 (Supporting information). There are 12.63 and 6.39 wt.% of Pt and Ru in PtRu NPs/C-GNS, whereas PtRu NPs/GNS has 12.58 and 6.44 wt.% of Pt and Ru. The results of ICP-AES show that the PtRu NPs/C-GNS and PtRu NPs/GNS catalysts have the same Pt and Ru loadings.

TEM images and size distribution of PtRu NPs of PtRu NPs/C-GNS (a, c) and PtRu NPs/GNS (b, d) catalysts
The crystal structure of GNS, C-GNS, PtRu NPs/C-GNS, and PtRu NPs/GNS catalysts are characterized by the XRD measurements (Fig. 4). It can be seen that the peak at 2θ = 26.3° in curves can be attributed to the C (002) plane of GNS. It is noted that both GNS and C-GNS support materials have very similar XRD pattern and exhibit sharp diffraction peaks at 2θ = 26.3°, which can be assigned to the graphite crystallographic planes (002) of graphene. The three peaks at 39.8°, 46.3°, and 67.8° can be assigned to the crystalline diffractions of Pt (111), Pt (200), and Pt (220), respectively, which indicates the face-centered cubic (fcc) Pt structure of PtRu alloys in catalysts [1]. It should be noted that the Pt (111) bands at 39.8° are broader and weaker for PtRu NPs/C-GNS than that for PtRu NPs/GNS, suggesting that the size of PtRu nanoparticles on C-GNS is smaller. Moreover, the average size of PtRu nanoparticles in both catalysts was calculated based on the Pt (220) diffraction peak according to Scherrer’s formula [1]. The average PtRu crystallite sizes are estimated to be 4.5 and 10.4 nm for PtRu NPs/C-GNS and PtRu NPs/GNS catalysts, respectively. These values agree with the TEM results. It is well known that high dispersion and small size of PtRu nanoparticles on C-GNS supports have larger surface areas and advantages in catalytic activity. Thus, PtRu NPs/C-GNS catalyst presumably possessed excellent electrocatalytic performance toward methanol oxidation.

XRD patterns of GNS, C-GNS, PtRu NPs/C-GNS, and PtRu NPs/GNS catalysts
The electrochemically active surface area (ESA) of catalysts can reflect the intrinsic electrocatalytic activity of the catalysts. Here, the CO stripping voltammetry was used to compare the ESA of PtRu NPs/C-GNS and PtRu NPs/GNS catalysts. Figure 5 shows the CO stripping voltammograms and subsequent cyclic voltammograms for PtRu NPs/C-GNS and PtRu NPs/GNS catalysts in 0.5 M H2SO4 solution at a scan rate of 50 mV s−1. It can be observed from the voltammograms that for the two catalysts in the first scan, the hydrogen desorption peaks are largely suppressed in the lower potential region due to the saturation of Pt surface with COad species. It is noted that the CVs showed the appearance of two peaks for CO stripping on PtRu NPs/C-GNS catalysts, whereas a single oxidation peak appeared on PtRu NPs/GNS catalysts. No CO oxidation was monitored in the second scan for the two catalysts, which conforms the complete removal of COad species. It can be found that for the PtRu NPs/C-GNS catalysts, the onset potential for CO oxidation is 0.25 V, and the two peaks potential are 0.34 and 0.47 V, respectively. It is well known that Ru is not active for CO adsorption. The possible reason for this fact is that some Pt is not alloyed with Ru and still remains beside the PtRu domain. There are surface Pt atoms with Ru atoms or RuOx in adjacent sites. As the first peak (0.34 V) shows a lower CO stripping potential, it is very likely to be the oxidation by OH species that can be generated in adjacent Ru (RuOx) sites at a lower potential. However, PtRu NPs/GNS catalysts have onset potential (0.29 V) and peak potential (0.45 V) for CO oxidation. Comparing PtRu NPs/GNS catalysts, the negative shift in the onset for CO oxidation on PtRu NPs/C-GNS catalysts might indicate that it is more active for CO oxidation, which can be ascribed to the higher Ru loading in the PtRu NPs/C-GNS catalysts. The ESA value of the two catalysts were determined using the CO oxidation charge after subtracting the background current of the subsequent CV curves with the assumption of 420 μC cm−2 as the oxidation charge for one monolayer of CO on smooth Pt surface. The results show that the ESA values of the PtRu NPs/C-GNS and PtRu NPs/GNS catalysts are 64.1 m2 g−1 and 27.8 m2 g−1, respectively. The ESA of PtRu NPs/C-GNS catalysts is significantly higher comparing that of PtRu NPs/GNS because of the nature of higher dispersion and smaller size of PtRu NPs on C-GNS surface. This also demonstrates that the PtRu NPs/C-GNS catalysts are electrochemically more accessible, which is very important for electrooxidation methanol reactions.

CO stripping voltammograms of PtRu NPs/C-GNS (a) and PtRu NPs/GNS (b) catalysts 0.5 M H2SO4 aqueous solution at a scan rate of 50 mV s−1. The dotted line voltammograms refer to the second cycle (after removal of the absorb CO)
The electrocatalytic activity of the PtRu NPs/C-GNS and PtRu NPs/GNS as potential electrocatalysts for DMFCs was characterized by cyclic voltammograms (CVs) and chronoamperometry (CA) measurements in acidic media. Figure 6 shows CVs of PtRu NPs/C-GNS and PtRu NPs/GNS catalysts measured in nitrogen-saturated 0.5 M H2SO4 + 1.0 M CH3OH aqueous solution. As shown in Fig. 6, the voltammogram profiles of both two catalysts exhibit the well-known features of methanol oxidation on PtRu electrocatalysts. As a kinetically controlled reaction, the activity of methanol oxidation on PtRu catalysts can be represented by the magnitude of the anodic peak. The higher anodic current on PtRu NPs/C-GNS catalysts means their higher electrocatalytic activity, which is in agreement with the observation of a larger ESA. It is noted that the forward peak current of methanol oxidation on the PtRu NPs/C-GNS catalysts is 657.3 mA mg−1, being 1.8 times higher than that on the PtRu NPs/GNS catalysts (361.1 mA mg−1), respectively. Moreover, on the other hand, although the two catalysts have the same forward peak potential of methanol oxidation (0.65 V), the onset potential on the PtRu NPs/C-GNS catalysts (0.20 V) shifts more than 75 mV in negative direction comparing that of PtRu NPs/GNS catalysts (0.275 V). These show a noticeable feature that PtRu NPs/C-GNS catalysts exhibit better electrocatalytic performance for methanol oxidation than PtRu NPs/GNS catalysts, which can be ascribed to some superior features of PtRu NPs/C-GNS catalysts over the PtRu NPs/GNS sample: smaller size, better dispersion, and higher ESA of PtRu NPs on the GNS surface. A comparison to the recently reported electrocatalysts [27,28,29] has been summarized in Table 1. Except Pt/TiO2/r-GO catalysts [29], the PtRu NPs/C-GNS catalysts provide the higher electrocatalytic activity toward methanol oxidation with respect to known oxidation peak potential and current density.

Cyclic voltammograms of PtRu NPs/C-GNS (a) and PtRu NPs/GNS (b) catalysts in nitrogen-saturated 0.5 M H2SO4 + 1.0 M CH3OH aqueous solution at a scan rate of 50 mV s−1
Figure 7 shows the amperometric i-t curves for PtRu NPs/C-GNS and PtRu NPs/GNS catalysts at a potential of 0.50 V in a nitrogen-saturated 0.5 M H2SO4 + 1.0 M CH3OH aqueous solution for 60 min. Before each experiment was performed, the electrolyte was deaerated with N2 for 15 min. These curves reflect the activity and stability of the two catalysts in catalyzing the methanol oxidation reaction. The decay current density decreased rapidly in the initial period for all the catalysts because of the formation of the intermediate species (such as COads) during the methanol oxidation reaction. It is noted that during the whole time, the current density of methanol oxidation on PtRu NPs/C-GNS is higher and the current density decay is much slower than that of PtRu NPs/GNS catalysts, though the current decay with time was observed for the two catalysts modified electrodes. The PtRu NPs/C-GNS catalysts retain a current density of 66.8 mA mg−1 at 3600 s, which is much higher than that of PtRu NPs/GNS catalysts (19.74 mA mg−1), indicating that PtRu NPs/C-GNS catalysts is more suitable for long-term operation. Five repetitive measurements on PtRu NPs/C-GNS catalysts gave a standard deviation of 4.2%, showing good reproducibility of the catalysts. Furthermore, the amperometric i-t curves for 24 h were carried out and the morphology of the electrocatalysts after the amperometric tests was investigated and the results are shown in Fig. S3 and S4, respectively. It is noted that the PtRu NPs/C-GNS catalysts exhibited higher long-term stability since their current density of 58.3 mA mg−1 at 24 h. However, the PtRu NPs/GNS catalysts only retain a current density of 18.1 mAmg−1 at 24 h. On the other hand, PtRu NPs in the PtRu NPs/GNS catalyst were seriously aggregated after amperometric tests. The PtRu aggregation (68 (length) × 40 (width) nm) appears in PtRu NPs/GNS catalyst after potential cycling. However, for the PtRu NPs/C-GNS electrocatalyst, the morphology does not change obviously except the slight increase of the particle size (the diameter is increased from 4.0 ± 0.5 to 7.0 ± 1.5 nm). The TEM results confirm the excellent long-term operation stability of the PtRu NPs/C-GNS electrocatalyst mainly because the PtRu NPs in the catalyst can keep their size and morphology. The increased stability also indicates that PtRu nanoparticles fixed strongly on C-GNS via carboxyl groups as interlinker. These results in this study demonstrate that the C-GNS prepared by the Friedel-Crafts reaction between succinic anhydride and GNS can be used as efficient and effective catalysts supports, especially for the development of PtRu electrocatalysts with high loading for the methanol electrooxidation in DMFCs.

Chronoamperometry curves of PtRu NPs/C-GNS (a) and PtRu NPs/GNS (b) catalysts in nitrogen-saturated 0.5 M H2SO4 + 1.0 M CH3OH solutions at 0.5 V
Conclusions
In summary, we have successfully developed a new strategy for the synthesis of noble metal NPs/GNS nanohybrids based on the carboxylated-graphene nanosheets. Thanks to the excellent dispersion of C-GNS in water and its much higher density and homogeneity of surface carboxyl groups, small PtRu NPs with an average diameter of ca. 4.0 ± 0.5 nm were uniformly deposited on C-GNS surface. Comparing to PtRu NPs/GNS catalysts, PtRu NPs/C-GNS catalysts toward methanol electrooxidation exhibit significantly enhanced catalytic current density, lower oxidation peak potential, and longer-term operation stability, due to the smaller particle size and higher dispersion of PtRu NPs on C-GNS, higher ESA. The developed C-GNS should be the promising catalysts support for noble metal NPs in fuel cells.
References
Hsin YL, Hwang KC, Yeh C-T (2007) Poly(vinylpyrrolidone)-modified graphite carbon nanofibers as promising supports for PtRu catalysts in direct methanol fuel cells. J Am Chem Soc 129:9999–10010
Chetty R, Kundu S, Xia W, Bron M, Schuhmann W, Chirila V, Brandl W, Reinecke T, Muhler M (2009) PtRu nanoparticles supported on nitrogen-doped multiwalled carbon nanotubes as catalyst for methanol electrooxidation. Electrochim Acta 54:4208–4215
Wu B, Kuang Y, Zhang X, Chen J (2011) Noble metal nanoparticles/carbon nanotubes nanohybrids: synthesis and applications. Nano Today 6(1):75–90
Xue X, Lu T, Liu C, Xu W, Su Y, Lv Y, Xing W (2005) Novel preparation method of Pt–Ru/C catalyst using imidazolium ionic liquid as solvent. Electrochim Acta 50:3470–3478
Wu B, Hu D, Kuang Y, Liu B, Zhang X, Chen J (2009) Functionalization of carbon nanotubes by an ionic-liquid polymer: dispersion of Pt and PtRu nanoparticles on carbon nanotubes and their electrocatalytic oxidation of methanol. Angew Chem Int Ed 48:4751–4754
Antolini E, Gonzalez ER (2011) Effect of synthesis method and structural characteristics of Pt–Sn fuel cell catalysts on the electro-oxidation of CH3OH and CH3CH2OH in acid medium. Catal Today 160(1):28–38
Xiong B, Zhou Y, Zhao Y, Wang J, Chen X, O’Hayre R, Shao Z (2013) The use of nitrogen-doped graphene supporting Pt nanoparticles as a catalyst for methanol electrocatalytic oxidation. Carbon 52:181–192
Calderón JC, García G, Querejeta A, Alcaide F, Calvillo L, Lázaro MJ, Rodríguez JL, Pastor E (2015) Carbon monoxide and methanol oxidations on carbon nanofibers supported Pt–Ru electrodes at different temperatures. Electrochim Acta 186:359–368
Rahsepar M, Pakshir M, Piao Y, Kim H (2012) Synthesis and electrocatalytic performance of high loading active PtRu multiwalled carbon nanotube catalyst for methanol oxidation. Electrochim Acta 71:246–251
Lee E, Kim S, Jang J-H, Park H-U, Matin MA, Kim Y-T, Kwon Y-U (2015) Effects of particle proximity and composition of Pt–M (M = Mn, Fe, co) nanoparticles on electrocatalysis in methanol oxidation reaction. J Power Sources 294:75–81
Zhang J-M, Sun S-N, Li Y, Zhang X-J, Zhang P-Y, Fan Y-J (2017) A strategy in deep eutectic solvents for carbon nanotube-supported PtCo nanocatalysts with enhanced performance toward methanol electrooxidation. Int J Hydrog Energy 42:26744–26751
Kugai J, Okazaki T, Seino S, Nakagawa T, Yamamoto TA, Tanaka S (2017) Effects of carboxylate stabilizers on the structure and activity of carbon-supported Pt–Cu nanoparticles towards methanol oxidation. Int J Hydrog Energy 42:2984–2995
Ahmed AM, Sayed SY, El-Nagar GA, Morsi WM, El-Deab MS, El-Anadouli BE (2019) Enhanced electrocatalytic oxidation of glucose at graphene nanosheets—metal oxides nanoparticles modified GC electrodes. J Electroanal Chem 835:313–323
Chang G, Cai Z, Jia H, Zhang Z, Liu X, Liu Z, Zhu R, He Y (2018) High electrocatalytic performance of a graphene-supported PtAu nanoalloy for methanol oxidation. Int J Hydrog Energy 43:12803–12810
Chen D, He Z, S-e P, L-a H, Shao H, Jin Y, Wang J (2019) Pd nanoparticles supported on N and P dual-doped graphene as an excellent composite catalyst for methanol electro-oxidation. J Alloy Compd 785:781–788
Eshghi A, Kheirmand M, Sabzehmeidani MM (2018) Platinum–iron nanoparticles supported on reduced graphene oxide as an improved catalyst for methanol electro oxidation. Int J Hydrog Energy 43:6107–6116
Jiang F, Yao Z, Yue R, Du Y, Xu J, Yang P, Wang C (2012) Electrochemical fabrication of long-term stable Pt-loaded PEDOT/graphene composites for ethanol electrooxidation. Int J Hydrog Energy 37:14085–14093
Kumar S, Mahajan M, Singh R, Mahajan A (2018) Silver nanoparticles anchored reduced graphene oxide for enhanced electrocatalytic activity towards methanol oxidation. Chem Phys Lett 693:23–27
Liu Y, Huang Y, Xie Y, Yang Z, Huang H, Zhou Q (2012) Preparation of highly dispersed CuPt nanoparticles on ionic-liquid-assisted graphene sheets for direct methanol fuel cell. Chem Eng J 197:80–87
Ng JC, Tan CY, Ong BH, Matsuda A, Basirun WJ, Tan WK, Singh R, Yap BK (2019) Novel palladium-guanine-reduced graphene oxide nanocomposite as efficient electrocatalyst for methanol oxidation reaction. Mater Res Bull 112:213–220
Pushkarev AS, Pushkareva IV, Grigoriev SA, Kalinichenko VN, Presniakov MY, Fateev VN (2015) Electrocatalytic layers modified by reduced graphene oxide for PEM fuel cells. Int J Hydrog Energy 40:14492–14497
Shi Q, Mu S (2012) Preparation of Pt/poly(pyrogallol)/graphene electrode and its electrocatalytic activity for methanol oxidation. J Power Sources 203:48–56
Toh SY, Loh KS, Kamarudin SK, Daud WRW (2018) Facile preparation of ultra-low Pt loading graphene-immobilized electrode for methanol oxidation reaction. Int J Hydrog Energy 43:16005–16014
Yang F, Zhang B, Dong S, Wang C, Feng A, Fan X, Li Y (2019) Reduced graphene oxide supported Pd-Cu-Co trimetallic catalyst: synthesis, characterization and methanol electrooxidation properties. J Energy Chem 29:72–78
Zhang X, Zhang J-W, Xiang P-H, Qiao J (2018) Fabrication of graphene-fullerene hybrid by self-assembly and its application as support material for methanol electrocatalytic oxidation reaction. Appl Sur Sci 440:477–483
Zhao Y, Zhan L, Tian J, Nie S, Ning Z (2011) Enhanced electrocatalytic oxidation of methanol on Pd/polypyrrole–graphene in alkaline medium. Electrochim Acta 56(5):1967–1972
Bai P, Tian F, Wang H, Yang T, Bi X, Chai Z, Wang X (2019) Electrocatalytic enhancement of 0D/1D/2D multidimensional PtCo alloy@cobalt benzoate/Graphene composite catalyst for alcohol electro-oxidation. Adv Mater Interfaces 6:1900946. https://doi.org/10.1002/admi.201900946
Qiu X, Yan X, Cen K, Sun D, Xu L, Tang Y (2018) Achieving highly Electrocatalytic performance by constructing holey reduced Graphene oxide hollow nanospheres sandwiched by interior and exterior platinum nanoparticles. ACS Appl Energy Mater 1:2341–2349
Wu S, Liu J, Ye Y, Tian Z, Zhu X, Liang C (2019) Oxygen defects induce strongly coupled Pt/metal oxides/rGO nanocomposites for methanol oxidation reaction. ACS Appl Energy Mater 2:5577–5583
Acknowledgments
This work was financially supported by NSFC (21303134), China Postdoctoral Science Foundation (2013M532017) and Outstanding Youth Science Fund of Xi’an University of Science and Technology (2018YQ2-13).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 2598 kb)
Rights and permissions
About this article

Cite this article
Wu, B., Wu, C., Zhu, J. et al. Facile synthesis of carboxylated-graphene nanosheets supported PtRu catalysts and their electrocatalytic oxidation of methanol. Ionics 26, 4599–4608 (2020). https://doi.org/10.1007/s11581-020-03608-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-020-03608-3