
Abstract
The porous carbon-coated LiFePO4 (LFP) nanocrystals synthesized by in situ plasma-assisted pyrolysis are reported. The particle size of LFP nanoparticles is well controlled through the coating of polyaniline (PANI) on FePO4. The effect of PANI content in FePO4/PANI on the morphology and electrochemical performance of LiFePO4 particles is extensively investigated. Results show that the optimized amount of PANI in FePO4/PANI is 10.16% and the corresponding carbon content in activated porous carbon-coated LiFePO4 (LFP/AC-P4) is 9.27%. The primary particle size of LFP/AC-P4 is 20~50 nm which are wrapped and connected homogeneously and loosely by activated porous carbon. The LFP/AC-P4 composite delivers a capacity of 166.9 mAh g−1 at 0.2 C, which is much higher than carbon-encapsulated LiFePO4 nanocomposite (LFP/C) synthesized without the assistance of plasma pyrolysis (163.5 mAh g−1). Even at high rate of 5 C, a specific capacity of 128.4 mAh g−1 is achievable with no obvious capacity fading after 250 cycles.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lithium ion batteries (LIBs) have been widely used in portable electronic devices in our daily life, such as handphones, digital cameras, laptops, etc., since they were first introduced in the 1990s. However, more efforts are still needed to improve the power and energy density of LIBs for their further applications in electric vehicles and hybrid electric vehicles. The cathode material is the key factor to determine the performance of LIBs. Since the pioneering work reported by Goodenough et al. in 1997, orthorhombic olivine LiFePO4 has been considered to be a promising cathode material for LIB due to its high theoretical capacity (170 mAh g−1), long cycle life, low raw material cost, high safety, and environmental friendliness [1]. Unfortunately, the intrinsic poor electronic conductivity (< 10−9 S cm−1) and low ion diffusion coefficient (10−14 to 10−16 cm2 s−1) of LiFePO4 greatly affect its high rate capability, which limits the commercial potential use in certain fields [2,3,4]. Great efforts have been made to overcome these drawbacks. One effective way is coating the surface of LiFePO4 particles with conductive material, which would effectively facilitate the electronic conductivity by allowing electrons to move over the particle surface. Many kinds of carbon precursors have been used for coating of LiFePO4 particles, such as sucrose, glucose, polyethylene glycol, or electrochemically active polymers [5,6,7]. Recently, carbon materials with different dimensions were also adopted to modify the LiFePO4 particles, such as 1D carbon nanotubes or carbon fibers [8,9,10], 2D graphene-based materials [11,12,13,14], and 3D graphene skeleton [15,16,17]. Based on the results of the above studies, a critical rule can be observed. The structure of the coated or modified carbon can affect the performance of the resultant cathode material. Moreover, compared with amorphous carbon, graphitic carbon is more desirable to enhance the electronic conductivity, which can influence the final performance of the LIBs. The other way is minimizing the particle size of LiFePO4 to shorten the Li-ion diffusion path length and increase the ion diffusion coefficient [2, 3]. LiFePO4 particles with controllable sizes and shapes can be fabricated via various synthetic techniques, including hydrothermal processes, solvothermal processes, sol-gel methods, or solid-state reactions [18,19,20]. However, coating with carbon is still required for most of these methods after the formation of crystals, which is a partially coating and complicated process. Moreover, carbon-modified (such as graphene or carbon nanotube) LiFePO4 particles can be developed through an in situ approach, involving the nucleation and growth of FePO4 or LiFePO4 nanoparticles on carbon-based materials and final annealing processes [11, 21, 22]. However, the particle size and morphology cannot be precisely controlled during this procedure, which would greatly influence the performance of prepared cathode materials. Therefore, homogeneous coating of carbon layer on LiFePO4 particles with controlled size can improve both electronic conductivity and ion diffusion coefficient, both of which are critical to strengthen the performance of cathode material.
Here, LiFePO4 nanocrystal was fabricated via an in situ polymerization restriction method. It is the first time to discover the relationship between the amount of introduced aniline and the size of wrapped particle. Then porous carbon-coated LiFePO4 nanocrystals can be fabricated following an in situ plasma-assisted pyrolysis process. Interestingly, more desired graphitic structure as well as the porous structure can be formed during the in situ plasma-assisted pyrolysis procedure compared with the normal calcination process without the assistance of plasma, which is critical to improve the performance of resultant cathode material. At the same time, the effect of the PANI content in FePO4/PANI on the morphology and electrochemical performance of LiFePO4 particles is extensively investigated in the following sections.
Experimental procedure
Preparation of LiFePO4 nanocomposite coated with porous carbon nanosheets by plasma-assisted pyrolysis
The prepared FePO4/PANI precursors (Detailed preparation process is described in the Supporting information) with different amounts of PANI coating (the content of coated PANI were 0, 3.11, 5.21, and 10.16%, respectively, synthesized from the FePO4/PANI (x) (x = 0, 5, 10, and 15)), and equimolar of CH3COOLi were mixed with a certain proportion of sucrose (32 wt.% vs. LiFePO4), which were further grinded uniformly and compressed into a sheet. The fabricated sheet was then sintered at 400 °C for 2 h by plasma discharge under N2 (5% H2) gas protection. After the sample is cooled, it was grinded and pressed again. The regained sheet was then sintered at 600 °C for another 4 h by plasma discharge under N2 (5% H2) gas protection to obtain the LFP@AC nanocomposites, which should have a core-shell structure. In this work, the LFP@AC nanocomposites coated with different amounts of porous carbon nanosheets, which were synthesized from the FePO4/PANI (x) (x = 0, 5, 10, and 15)) as precursor during the plasma pyrolysis process, were synthesized. The carbon content for prepared LFP@AC nanocomposites was 4.93, 6.84, 8.28, and 9.27%, which was signed as LFP@AC-P1, LFP@AC-P2, LFP@AC-P3, and LFP@AC-P4, respectively. The in situ plasma-assisted pyrolysis procedure was completed in a plasma pyrolysis reactor as illustrated in Fig. 1a.

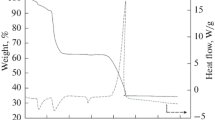
a In situ plasma-assisted pyrolysis reactor. b TGA and c DSC images of FePO4/PANI precursor with different coating contents of PANI
For comparison, a nanocomposite LFP/C using FePO4/PANI as precursor (the content of coated PANI was 10.16%) was also prepared under the same preparation parameters without the assistance of plasma. The carbon content of resultant nanocomposite was 8.37%, marked as LFP/C.
Physical characterization
Hitachi S4800 field emission scanning electron microscopy was used to observe the morphology of the samples. Transmission electron microscopy (TEM) observations were carried out on a JEOL, JEM-2100F (Japan) electron microscope. The phase analysis and crystal structure characterization of the material were carried out by a Bruker D8 Advance X-ray powder diffractometer, which used CuKα radiation (λ = 1.5418 Å) operated at voltage 40 kV and current 250 mA at a scan rate (2θ) of 2° min−1. The carbon content of the synthetic products was determined by CS800 high-frequency infrared carbon and sulfur analyzer. Raman spectra were acquired on a Raman spectrometer (Renishaw, RM-1000 Invia) with a 2.41-eV excitation energy. Fourier transform infrared spectrometer (FTIR) spectra were collected by a Bruker IFS-66V/S spectrometer.
Electrochemical characterization
Electrochemical performances of LFP@AC and LFP@C samples were evaluated using CR2032-type coin cells. Laminated electrode was prepared as follows: the active material with acetylene black and polyvinylidene fluoride (PVDF) in a weight ratio of 85:10:5 were mixed and dissolved in N-methyl-2-pyrrolidinone (NMP) to form a slurry with the assistance of ultrasound. Then the above-prepared slurry was coated onto the Al foil homogeneously. The load quality of fabricated electrode was about 2 mg cm−2, which was further dried at 120 °C for 24 h under a vacuum atmosphere. The button cell was assembled in an argon-filled glove box with the above electrode as cathode, lithium metal as anode, polypropylene film as separator, and 1 mol L−1 LiPF6 in EC/DEC (1:1, volume ratio) solution as electrolyte. At room temperature, the galvanostatical charge-discharge cycling test was carried out in the LAND battery test system. The charge-discharge current rates were 0.2, 0.5, 1, 2, 5, 10, and 20 C, respectively, over a potential range of 2.5–4.2 V (vs. Li/Li+). The electrochemical impedance spectra and cyclic voltammograms (CVs) were recorded on Zahner IM6e electrochemical workstation. The electrochemical impedance test was performed over a frequency range of 1 × 105~0.01 Hz with an applied amplitude of 5 mV. The CV measurement was carried out between 2.5 and 4.5 V.
Results and discussion
Control the particle size of PANI-wrapped FePO4
A different amount of aniline monomer was introduced during the synthesis process of polyaniline (PANI)-coated FePO4 (FePO4/PANI) precursor as mentioned in the experimental part. And the obtained FePO4/PANI precursor was denoted as FePO4/PANI (x) (x = 0, 5, 10, 15, 30, and 47), where x represents the mass of added aniline monomer. The TGA/DSC images of FePO4/PANI precursors with different coating contents of PANI are shown in Fig. 1b, c. The weight of sample FePO4/PANI (0) (Fig. 1b), synthesized without the addition of aniline monomer, decreased by 16.73 wt.% from room temperature to 800 °C, which is mainly due to the loss of adsorbed and crystal water of FePO4 [7]. Based on the result of Fig. 1b, it can be concluded that the weight loss of precursors FePO4/PANI (5), FePO4/PANI (10), FePO4/PANI (15), FePO4/PANI (30), and FePO4/PANI (47) were 19.84, 21.94, 26.89, 27.05, and 27.02%, respectively. Combined with the weight loss of precursor FePO4/PANI (0), it can be summarized that the weight percentage of coated PANI in precursors FePO4/PANI (5), FePO4/PANI (10), FePO4/PANI (15), FePO4/PANI (30), and FePO4/PANI (47) were 3.11, 5.21, 10.16, 10.29, and 10.32%, respectively. It is easy to note that when the quality of introduced aniline reaches 15 g, the content of coated PANI in precursors FePO4/PANI (30) and FePO4/PANI (47) were similar as that in FePO4/PANI (15), which did not increase with the further increase of added aniline monomer as in samples FePO4/PANI (5) and FePO4/PANI (10). Because, the oxidization polymerization of aniline was initiated by the Fe3+ ions on the outer surface of precipitated FePO4 particles [13]. Therefore, once the FePO4 particles are completely coated with a layer of PANI, the polymerization of aniline will be prevented. It can be concluded that the coating content of PANI in FePO4/PANI composite has a maximum value.
Serious agglomeration and uneven particle size distribution can be found in Fig. 2a, which shows the SEM image of prepared FePO4 nanoparticles without coated PANI. On the contrary, when PANI is coated on the surface of FePO4 nanoparticles, the particle size of FePO4 becomes uniform as shown in Fig. 2b–f. Moreover, the particle size decreased with the increase of introduced aniline monomer from 5 to 15 g, and each agglomerated cluster contains a large number of FePO4 nanoparticles interconnected by PANI. However, when the amount of aniline monomer reaches 15 g, the particle size of resultant FePO4 nanoparticles are no longer further reduced (Fig. 2d–f). This is because the polymerization of aniline is blocked after the FePO4 particles are completely coated with a layer of PANI [7, 13].

SEM images of FePO4/PANI composites with different coating contents of PANI: a FePO4/PANI (0), b FePO4/PANI (5), c FePO4/PANI (10), d FePO4/PANI (15), e FePO4/PANI (30), and f FePO4/PANI (47)
Figure S1 shows the EDX result of FePO4/PANI nanocomposites with different amounts of coated PANI. Only O, P, and Fe elements can be found for FePO4/PANI (0) in Fig. S1a, indicating the absence of PANI. Different from FePO4/PANI (0), element N can be found for samples FePO4/PANI (5), FePO4/PANI (10), FePO4/PANI (15), FePO4/PANI (30), and FePO4/PANI (47) as shown in Fig. S1b–f, which could further prove the success coating of PANI on the surface of FePO4 nanoparticles. It can be summarized from Fig. S1b–f that the N content of corresponded samples increased with the increase of the added aniline monomer. However, when the amount of aniline monomer reached 15 g, the N content of obtained FePO4/PANI nanoparticle remained constant. This discovery could further confirm our previous speculation that the polymerization of aniline will be stopped after the synthesized FePO4 nanoparticles are completely coated with a layer of PANI.
Performance optimization of activated porous carbon-coated LiFePO4 by in situ plasma-assisted pyrolysis
The above-obtained PANI-wrapped FePO4 nanoparticles, coated with different amounts of PANI (0, 3.11, 5.21, and 10.16%), were mixed with equimolar CH3COOLi and a certain amount of sucrose, which acted as the carbon precursor (32 wt.% vs. LiFePO4). Then activated porous carbon-coated LiFePO4 (LFP@AC) nanocomposites were fabricated through in situ plasma-assisted pyrolysis process. Due to the plasma-assisted pyrolysis reaction, the coated activated porous carbon was expected to be further graphitized. The carbon content of synthetic products donated as LFP@AC-P1, LFP@AC-P2, LFP@AC-P3, and LFP@AC-P4 were 4.93, 6.84, 8.28, and 9.27%, respectively, which were detected by CS800 high-frequency infrared carbon and sulfur analyzer. For comparison, FePO4/PANI with the coated PANI content of 10.16% was chosen as a contrast, which was further coated with carbon through high-temperature pyrolysis reaction but without the assistance of plasma. The total carbon content of resultant carbon-encapsulated LiFePO4 nanocomposite was 8.37%, which was recorded as LFP/C.
Figure S2 shows the EDX images of LFP/AC nanocomposites coated with porous carbon slices and LFP/C nanocomposite fabricated without the assistance of plasma pyrolysis. As can be seen from the EDX images, elements C, O, P, and Fe exist in all synthesized samples, including LFP/AC and LFP/C nanocomposites. And the atomic ratios of P and Fe for all resultant samples are close to 1:1, similar with that in LiFePO4, suggesting the successful synthesis of carbon-coated LiFePO4 nanocomposites. Figure 3a–d shows the SEM images of activated carbon-coated LiFePO4 nanoparticles prepared by in situ plasma-assisted pyrolysis process. As can be seen from Fig. 3a–d, it can be concluded that the particle sizes for resultant LFP/AC nanocomposites became smaller following the increase of coated carbon content. Results show that the optimized amount of carbon in LFP/AC-P4 is 9.27%, and the corresponding particle size of LiFePO4 is 20~50 nm, which are wrapped and connected homogeneously and loosely by activated carbon. As mentioned above, when the surface of the FePO4 particles are completely covered by PANI, the polymerization will terminate, because the exposed Fe3+ ions is the initiators for polymerization. So it can be concluded that too little PANI cannot completely coat the resultant FePO4 particles; however, the amount of PANI coating has an upper limit. Both the coated PANI on the FePO4 particles surface and later introduced sucrose provided the carbon source for the plasma-assisted pyrolysis process. Therefore, samples LFP/AC-P3 and LFP/AC-P4 have the relatively higher carbon content (8.28, and 9.27%, respectively) due to the higher PANI coating content. As is known, carbon sheets will be formed during the high-temperature calcinated process with the pyrolysis of coated PANI and sucrose. The resulting carbon sheets could form graphite and amorphous carbon by ordered or disordered alignment [23, 24]. Combined with the in situ plasma treatment, which can effectively etch the carbon sheets on the surface of the coated carbon to produce porous structures and edges without destroying the core of the fabricated particles, the porous carbon structure can be formed [25,26,27]. Therefore, the obvious porous structure can be found in the coated activated carbon, especially for samples LFP/AC-P3 and LFP/AC-P4, which possessed relatively higher content of coated carbon. On the contrary, the porous carbon structure cannot be searched for sample LFP/C. Therefore, it can be assumed that the plasma-assisted pyrolysis process is a necessary condition for the generation of porous carbon structure.

SEM images of activated porous carbon-coated LiFePO4 nanocomposites: a LFP/AC-P1, b LFP/AC-P2, c LFP/AC-P3, and d LFP/AC-P4. e Carbon-encapsulated LiFePO4 nanocomposite (LFP/C) synthesized without the assistance of plasma pyrolysis
From the transmission electron microscope (TEM) images of LFP/AC-P3 (Fig. 4), it can be seen that the obtained LFP/AC-P3 nanocomposite material was composed of LiFePO4 particles and coated carbon materials. It is worth noting that the synthesized LiFePO4 particles were homogeneously coated with introduced carbon, which has a clear porous structure, and acting as a network to connect the prepared LiFePO4 particles together. Therefore, it can be expected that the homogeneous coated porous carbon network would facilitate the fast transport of electrons and to control the volume deformation of LiFePO4 nanoparticles during charging and discharging processes. However, the desirable porous carbon network has not been found in sample LFP/C (Fig. 4a). As shown in Fig. 4e, the lattice fringe of LFP/AC-3 with a space of 3.69 Å corresponding to the (011) plane of orthorhombic LFP crystal can be found [19, 28]. And combined with the SAED pattern in Fig. 4d, it can be deduced that the porous carbon-coated LiFePO4 particles are polycrystalline with small particles [29].

TEM images of a LFP/C and b, c LFP/AC-P3; d SAED pattern of LFP/AC-P3; e HRTEM image of LFP/AC-P3
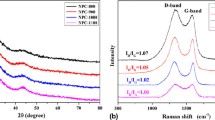
Figure 5a shows the XRD patterns of LFP/AC and LFP/C. All samples show the same peaks, which can match the pattern of orthorhombic olivine phase well (JCPDS No. 83-2092) [20, 30]. The lattice parameter for a, b, and c is 1.0329, 0.601, and 0.4694 nm, respectively, and the cell volume is 0.291 nm3, all of which is consistent with the literature report [1]. Moreover, the impurity peaks located at 2θ = 43.8° and 51° representing Li3Fe2(PO4)3 are not found in the XRD spectra, indicating that the synthesized LFP/AC and LFP/C have very high purity. At the same time, the diffraction peaks of crystalline or amorphous carbon were not observed in the XRD spectra, which is due to low carbon content in LFP/AC and LFP/C as well as the relatively low sensitivity of X-ray diffraction to graphitic materials. In this work, the precursor FePO4/PANI was produced through the in situ polymerization of PANI coating method. Based on the above investigation result, it can be concluded that this preparation method can make the precursor to nanoscale mixing and achieve better uniformity, which can effectively avoid the formation of Fe3O4, FeO, Li3PO4, and other unfavorable impurity [19, 31]. This is very critical for the preparation of LFP/AC and LFP/C nanocomposites with high purity. Therefore, it is necessary to strictly control the feeding order, the feeding speed, and the amount of introduced PANI. At the same time, the introduced carbon precursor (sucrose) cannot only inhibit the formation of Fe3+ but also prevent the crystallization of larger particle size, which is helpful to control the particle size of LiFePO4 under 50 nm. The small particle size can greatly improve the ion diffusion rate of cathode materials. Figure 5b shows the FTIR spectra of LFP/AC and LFP/C. It can be seen from the FTIR spectra that with the increase of coated PANI in the precursor FePO4/PANI, two obvious peaks located at 1626 and 1552 cm−1 can be found for corresponding LFP/AC nanocomposites, which corresponded to sp2 hybrid carbon layer structure. However, this sp2 hybrid carbon layer structure did not show up in LFP/C, which was synthesized without the assistance of plasma pyrolysis. Therefore, it can be assumed that the plasma-assisted pyrolysis could strengthen the graphitization degree of coated carbon in fabricated LFP/AC nanocomposites. This assumption can be further confirmed by the Raman spectra in Fig. 5c. The carbon materials formed from the decomposition of PANI and sucrose are composed of sp2 and sp3 hybrid atomic bonds, which represent ordered graphite phases and disordered amorphous phases, respectively. And the conductivity of ordered graphite phase is greatly higher than that of disordered amorphous phases. This principle was introduced into the study of carbon coating by Doeff et al. [32]. It is considered that the peak height or peak area ratio of the D peak and the G peak in the Raman spectrum can represent the proportion of graphitization of the carbon structure [33, 34]. Therefore, larger value of peak height or peak area ratio of the D peak and the G peak indicates higher degree of graphitization, which should led to higher electrical conductivity of target carbon material. The peak height ratios of the D peak and the G peak for LFP/AC are obviously higher than that of LFP/C based on the Raman spectra in Fig. 5c, suggesting higher degree of graphitization of coated carbon in samples LFP/AC [34, 35]. Therefore, it can be concluded that the conductivities of prepared LFP/AC nanocomposites are higher than that of LFP/C nanocomposite. And the improved conductivity could significantly improve the rate performance of electrode materials.

a XRD patterns, b FTIR spectra, and c Raman spectra of LFP/AC and LFP/C
In order to examine the electrochemical performance of LFP/AC and LFP/C, the coin cells were assembled and their charge and discharge performance was tested at room temperature. Figure 6a, b and Fig. S3 show the initial charge/discharge curves of resultant LFP/AC and LFP/C nanocomposite at the cutoff voltage of 2.5–4.2 V vs. Li/Li+ at different rates in 1 M LiPF6/EMC-DEC (1:1 in v/v) at 25 °C. All the samples exhibit a flat voltage plateau at around 3.45 V, corresponding to the Li+ insertion and extraction process between Fe3+ and Fe2+ [36]. With the increase of wrapped PANI, the corresponding LFP/AC nanocomposites (LFP/AC-2, LFP/AC-3, LFP/AC-4) still exhibit an obvious flat voltage plateau, suggesting a higher capacitance than that of LFP/AC-1 under high charging-discharging rate. Moreover, the voltage difference (∆V) between the flat charge and discharge plateaus was related to the polarization of the assembled coin cell system, which was caused by slow lithium diffusion in the electrode material and increases in the resistance of the electrolyte following the increase of the charging-discharging rate [7]. It is obvious to see that the ∆V of synthesized LFP/AC nanocomposites decreased with the increase of wrapped PANI, indicating that the kinetics of the electrode material was improved [17]. Because the wrapped LiFePO4 particle size decreased with the increase of introduced PANI (as mentioned in the SEM analysis), which could accelerate the lithium diffusion in the electrode material. Moreover, the lower electrochemical polarization suggested an enhanced conductivity, which should be due to the well-connecting carbon network deduced from the homogeneous coated carbon with the increase of wrapped PANI. As illustrated in Fig. 6c, the rate capabilities of the all-prepared samples were assessed by charging-discharging process at rates varying from 0.2 to 20 C in continuous cycling at room temperature. As can be seen from Fig. 6c, the electrochemical property of the LFP/AC-P1 prepared by the FePO4 precursor without PANI coating is the worst. This is because the particle size of synthesized LiFePO4 grew bigger without the control of wrapping PANI, which would slow down the lithium diffusion inside the LiFePO4 particles. Moreover, no coating of PANI on prepared FePO4 particles led to the absence of homogeneous carbon coating on the resultant LiFePO4 particles, which is critical to improving the electronic conductivity of the insulate LiFePO4 particles. The electrochemical performance of LFP/AC nanocomposites (LFP/AC-P2, LFP/AC-P3, LFP/AC-P4) is improved with the increase of the introduced PANI, wrapping on the surface of the precursor FePO4, and LFP/AC-P4 shows the best performance. The same trend can be found for the average value of ten times’ discharge at corresponding rates in Fig. 6d. The LFP/AC-P4 composite delivers a capacity of 166.9 mAh g−1 at 0.2 C. Most impressively, the LFP/AC-P4 exhibits an outstanding rate capability with reversible capacities of 166.8, 163.4, 159.4, 143.3, 114.3, and 57.4 mAh g−1 observed when charged/discharged at the current rates of 0.5, 1, 2, 5, 10, and 20 C, as shown Fig. 6c. Similar with the preparation of LFP/AC-P4, LFP/C was prepared without the assistance of the in situ plasma-assisted pyrolysis. It shows in Fig. 6c that the sample LFP/AC-4 exhibits higher capacitances under the same charge/discharge rates than those of the sample LFP/C (163.5 mAh g−1 at 0.2 C). LFP/C exhibits the reversible capacities of 156.4, 152.4, 146.3, 134.0, 112.7, and 40.2 mAh g−1 when charged/discharged at the current rates of 0.5, 1, 2, 5, 10, and 20 C, as shown Fig. 6c. Figure 7a shows that the LFP/AC-4 exhibits even better rate capacity (below 10 C) and cycling stability than most other carbon-integrated LFP cathodes reported previously (More detailed comparisons are given in Table S1) [37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55].

The initial charge/discharge curves of a LFP/AC-P4 and b LFP/C at the cutoff voltage of 2.5–4.2 V vs. Li/Li+ at different rates in 1 M LiPF6/EMC-DEC (1:1 in v/v) at 25 °C. c Rate capabilities and cycle performance of the LFP/AC and LFP/C. d Discharge capacity at different rates of LFP/AC and LFP/C nanocomposites. The data present in figure d are the average value of ten times’ discharge at corresponding rates. e Cycling performance of LFP/AC and LFP/C under the charge/discharge rate of 5 C. f The cyclic voltammograms (CV) curves of LFP/AC and LFP/C at a scan rate of 0.1 mV s−1

a Comparison of the rate capability of the LFP/AC-P4 (this work) with those of the previously reported LFP/C composites synthesized by different methods with different LFP crystal morphology and composite structure. b EIS specra of LFP/AC and LFP/C in the frequency range between 200 kHz and 10 mHz. c The relationship between ZRe and ω−1/2 at low frequency. The inset of (b) shows an equivalent circuit
This may be due to two main reasons. First, the in situ plasma-assisted pyrolysis can strengthen the graphitization of coated carbon in LFP/AC-P4, which should improve the conductivity of prepared LFP/AC-P4. And the improved conductivity could significantly improve the rate performance of electrode materials. Second, LFP/AC-P4 shows obvious porous structure caused by the plasma-assisted pyrolysis process, which would facilitate the penetration of electrolyte in electrode material to decrease the resistance of the electrolyte [56, 57]. However, the porous carbon structure cannot be found for sample LFP/C. Figure 6e shows the cyclic performance of LFP/AC and LFP/C under the charge/discharge rate of 5 C at the cutoff voltage of 2.5–4.2 V vs. Li/Li+. From the cycle performance test, it can be found that the charge/discharge stability of LFP/AC-P4 is the best. Even at high rate of 5 C, a specific capacity of 128.4 mAh g−1 is achievable, with no obvious capacity fading after 250 cycles. Because the higher amount of wrapped PANI on the precursor FePO4 would produce the smaller controlled particle size, which will facilitate the diffusion of Li+. Furthermore, the higher amount of introduced PANI is helpful for the homogeneous carbon coating on the final produced LiFePO4 particles, which acts as a carbon network to speed up the electronic conductivity by allowing electrons to move over the particle surface. Figure 6f shows the CV curves of LFP/AC and LFP/C measured at a scanning rate of 0.1 mV s−1 in the potential window range of 2.5 to 4.5 V vs. Li+/Li. Cyclic voltammetry (CV) is a useful technique for investigating the phase transformation and ionic diffusion process during electrode reactions. All the CV curves exhibit a pair of symmetrical anodic/cathodic peaks, which corresponds to the oxidation/reduction transformation between Fe2+ and Fe3+. However, the potential difference between two peaks is relatively larger compared with the potential difference of reversible redox peak for the ideal one electron (0.059 V), suggesting the occurrence of voltage hysteresis [34]. The reason should be due to the low conductivity of LiFePO4, which causes the slightly smaller area of the reduction peak compared with the oxidation peak. Because the peak area of the CV curve reflects the quantity of electricity exchanged by the oxidation and reduction reaction of the electrode active material, therefore the areas of the oxidation and the reduction peaks indicate the polarization state of the electrode and the utilization ratio of electrode active material [14, 22, 58]. Compared with the other three curves, LFP/AC-P4 and LFP/AC-P3 show sharper redox peaks, bigger peak areas, and smaller voltage hysteresis than LFP/AC-P1, LFP/AC-P2, and LFP/C, implying higher capacity, better electrode kinetics, and smaller polarization degree. This result is consistent with the conclusions obtained from the initial charge/discharge curves and the cycling performance curves.
Figure 7b displays the electrochemical impedance spectroscopy (EIS) spectra, which are used to further study the reaction kinetics of electrode materials. A depressed semicircle in the high-frequency region and a sloping line in the low-frequency region can be found in all resultant Nyquist plots. The semicircle in the high-frequency region interprets the charge transfer resistance on the electrolyte/oxide electrode interface. And the sloping lines in the low-frequency region represent the typical Warburg impedance, which is attributed to the diffusion of Li+ in the cathode material [59, 60]. The impedance spectra can be reasonably fitted to the inserted equivalent circuit in Fig. 7b. In this circuit, Rs represents the ohmic resistance and CSEI and RSEI represent the relax capacitance according to the high-frequency region of the semicircle in the Nyquist plots and the SEI film resistance. Cdl and Rct refer to the double-layer capacitance according to the medium-frequency region of the semicircle in the Nyquist plots and the charge transfer resistance. Moreover, W indicates the Warburg resistance. According to the fitting results, the charge transfer impedances (Rct) decreased with the increased of wrapped PANI as shown in Table 1. Moreover, with the same amount of wrapped PANI, the in situ plasma-assisted pyrolysis is helpful to depress the value of Rct. The lower Rct is beneficial to overcome the kinetic limitation during charge/discharge process, which could improve the embedded depth of lithium in electrode material and prevent the distortion of the crystal structure in the LiFePO4 particles [61].
Based on the inclined line in the Warburg region, the Li+ diffusion coefficient (D (cm2 s−1)) can be calculated using the following equation:
where R is the gas constant, T is the absolute temperature, A is the surface area of the cathode electrode, n is the number of electrons per molecule during the electrochemical reaction, F is the Faraday constant, C is the Li+ concentration (7.69 × 10−3 mol cm−3), and σw is the Warburg impedance coefficient of resultant LiFePO4 nanocomposites [7, 12]. Figure 7c shows the relationship between ZRe and square root of frequency (ω−1/2) in the low-frequency region. From Eq. 2, the Warburg factor σw can be obtained by linearly fitting the plot between ZRe and ω−1/2 as shown in Fig. 7c, and the result is listed in Table 1. As listed in Table 1, the LFP/AC-P4 nanocomposite demonstrates the highest Li+ diffusion coefficient and lowest impedance, which are attributed to the shortened ion diffusion route from the smaller wrapped LFP nanoparticles, the decreased electrolyte resistance caused by the constructed porous structure and the improved electronic conductivity by the assistance of in situ plasma process. Both of the reduced charge transfer resistance and increased Li+ diffusion coefficient of LFP/AC-P4 nanocomposite greatly favor its high capacitance, excellent rate capability, as well as superior cyclic stability.
Conclusion
In summary, the porous carbon-coated LiFePO4 nanocrystals were successfully synthesized by in situ plasma-assisted pyrolysis. The homogeneous coating of PANI on precursor FePO4 is helpful for controlling the size of final produced LFP particles. And the smaller particle size indicates the shorter ion diffusion route in the carbon-wrapped LFP nanoparticles. Moreover, the coated PANI can also act as a carbon network after pyrolysis process to speed up the electronic conductivity by allowing electrons to move over the particle surface. With the adoption of in situ plasma-assisted pyrolysis process, porous structure could be introduced into the carbon slice-coated LFP particles and the graphitization degree of coated carbon could be strengthened, which can decrease the electrolyte resistance and improve the electronic conductivity, respectively. Therefore, compared with LFP/AC-P1 prepared without the introduction of PANI and LFP/C fabricated without the assistance of plasma, LFP/AC-P4 exhibits reduced charge transfer resistance and increased Li+ diffusion coefficient, both of which favor its high capacitance, excellent rate capability, as well as superior cyclic stability. It delivers a capacity of 166.9 mAh g−1 at 0.2 C and exhibits an outstanding rate capability with reversible capacities of 166.8, 163.4, 159.4, 143.3, 114.3, and 57.4 mAh g−1 observed when charged/discharged at the current rates of 0.5, 1, 2, 5, 10, and 20 C. Even at high rate of 5 C, a specific capacity of 128.4 mAh g−1 is achievable, with no obvious capacity fading after 250 cycles.
References
Padhi AK, Nanjundaswamy KS, Goodenough JB (1997) Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J Electrochem Soc 144(4):1188–1194
Liang Y, Wen K, Mao Y, Liu Z, Zhu G, Yang F, He W (2015) Shape and size control of LiFePO4 for high-performance lithium-ion batteries. ChemElectroChem 2(9):1227–1237
Li Y, Meyer S, Lim J, Lee SC, Gent WE, Marchesini S, Krishnan H, Tyliszczak T, Shapiro D, Kilcoyne AL, Chueh WC (2015) Effects of particle size, electronic connectivity, and incoherent nanoscale domains on the sequence of lithiation in LiFePO4 porous electrodes. Adv Mater 27(42):6591–6597
Wang L, He X, Sun W, Wang J, Li Y, Fan S (2012) Crystal orientation tuning of LiFePO4 nanoplates for high rate lithium battery cathode materials. Nano Lett 12(11):5632–5636
Zhu J, Yoo K, El-halees I, Kisailus D (2014) Solution deposition of thin carbon coatings on LiFePO4. ACS Appl Mater Interfaces 6(23):21550–21557
Zhao D, Feng Y-l, Wang Y-g, Xia Y-y (2013) Electrochemical performance comparison of LiFePO4 supported by various carbon materials. Electrochim Acta 88:632–638
Jiang Z, Jiang Z-j (2012) Effects of carbon content on the electrochemical performance of LiFePO4/C core/shell nanocomposites fabricated using FePO4/polyaniline as an iron source. J Alloys Compd 537:308–317
Wang G, Ma Z, Shao G, Kong L, Gao W (2015) Synthesis of LiFePO4@carbon nanotube core–shell nanowires with a high-energy efficient method for superior lithium ion battery cathodes. J Power Sources 291:209–214
Lee B-S, Son S-B, Park K-M, Lee G, Oh KH, Lee S-H, Yu W-R (2012) Effect of pores in hollow carbon nanofibers on their negative electrode properties for a lithium rechargeable battery. ACS Appl Mater Interfaces 4(12):6702–6710
Ji H, Zhang L, Pettes MT, Li H, Chen S, Shi L, Piner R, Ruoff RS (2012) Ultrathin graphite foam: a three-dimensional conductive network for battery electrodes. Nano Lett 12(5):2446–2451
Wi S, Kim J, Park K, Lee S, Kang J, Kim KH, Nam S, Kim C, Park B (2016) Evaluation of graphene-wrapped LiFePO4 as novel cathode materials for Li-ion batteries. RSC Adv 6(107):105081–105086
Zhou Y, Lu J, Deng C, Zhu H, Chen GZ, Zhang S, Tian X (2016) Nitrogen-doped graphene guided formation of monodisperse microspheres of LiFePO4 nanoplates as the positive electrode material of lithium-ion batteries. J Mater Chem A 4(31):12065–12072
Wang Y, Wang Y, Hosono E, Wang K, Zhou H (2008) The design of a LiFePO4/carbon nanocomposite with a Core–Shell structure and its synthesis by an in situ polymerization restriction method. Angew Chem Int Ed 47(39):7461–7465
Liu H, Miao C, Meng Y, Xu Q, Zhang X, Tang Z (2014) Effect of graphene nanosheets content on the morphology and electrochemical performance of LiFePO4 particles in lithium ion batteries. Electrochim Acta 135:311–318
Wu G, Ran R, Zhao B, Sha Y, Su C, Zhou Y, Shao Z (2014) 3D amorphous carbon and graphene co-modified LiFePO4 composite derived from polyol process as electrode for high power lithium-ion batteries. J Energy Chem 23(3):363–375
Yang J, Wang J, Wang D, Li X, Geng D, Liang G, Gauthier M, Li R, Sun X (2012) 3D porous LiFePO4/graphene hybrid cathodes with enhanced performance for Li-ion batteries. J Power Sources 208:340–344
Wang B, Al Abdulla W, Wang D, Zhao G (2015) Three-dimensional porous LiFePO4 cathode material modified with nitrogen-doped graphene aerogel for high-power lithium ion batteries. Energy Environ Sci 8(3):869–875
Tao S, Huang W-F, Wu G-X, Zhu X-B, Wang X-B, Zhang M, Wang S-H, Chu W-S, Song L, Wu Z-Y (2014) Performance enhancement of Lithium-ion battery with LiFePO4@C/RGO hybrid electrode. Electrochim Acta 144:406–411
Li J, Zhang L, Zhang L, Hao W, Wang H, Qu Q, Zheng H (2014) In-situ growth of graphene decorations for high-performance LiFePO4 cathode through solid-state reaction. J Power Sources 249:311–319
Mun J, Ha H-W, Choi W (2014) Nano LiFePO4 in reduced graphene oxide framework for efficient high-rate lithium storage. J Power Sources 251:386–392
Zhu P, Yang Z, Zeng P, Zhong J, Yu J, Cai J (2015) Homogeneous precipitation synthesis and electrochemical performance of LiFePO4/CNTs/C composites as advanced cathode materials for lithium ion batteries. RSC Adv 5(130):107293–107298
Liu Y, Gu J, Zhang J, Yu F, Wang J, Nie N, Li W (2015) LiFePO4 nanoparticles growth with preferential (010) face modulated by Tween-80. RSC Adv 5(13):9745–9751
Tian X, Jiang Z, Jiang Y, Xu W, Li C, Luo L, Jiang Z-J (2016) Sulfonic acid-functionalized mesoporous carbon/silica as efficient catalyst for dehydration of fructose into 5-hydroxymethylfurfural. RSC Adv 6(103):101526–101534
Tian X, Zhang LL, Bai P, Zhao XS (2011) Sulfonic-acid-functionalized porous benzene phenol polymer and carbon for catalytic esterification of methanol with acetic acid. Catal Today 166(1):53–59
Liu Z, Zhao Z, Wang Y, Dou S, Yan D, Liu D, Xia Z, Wang S (2017) In situ exfoliated, edge-rich, oxygen-functionalized graphene from carbon fibers for oxygen electrocatalysis. Adv Mater 29(18):1606207
Tao L, Wang Q, Dou S, Ma Z, Huo J, Wang S, Dai L (2016) Edge-rich and dopant-free graphene as a highly efficient metal-free electrocatalyst for the oxygen reduction reaction. Chem Commun 52(13):2764–2767
Tian Y, Ye Y, Wang X, Peng S, Wei Z, Zhang X, Liu W (2017) Three-dimensional N-doped, plasma-etched graphene: highly active metal-free catalyst for hydrogen evolution reaction. Appl Catal A Gen 529:127–133
Du Y, Tang Y, Huang FQ, Chang C (2016) Preparation of three-dimensional free-standing nano-LiFePO4/graphene composite for high performance Lithium ion battery. RSC Adv 6:52279–52283
Choi SH, Lee J-K, Kang YC (2015) Three-dimensional porous graphene-metal oxide composite microspheres: preparation and application in Li-ion batteries. Nano Res 8(5):1584–1594
Kretschmer K, Sun B, Xie X, Chen S, Wang G (2016) A free-standing LiFePO4-carbon paper hybrid cathode for flexible lithium-ion batteries. Green Chem 18:2691–2698
Dokko K, Shiraishi K, Kanamura K (2005) Identification of surface impurities on LiFePO4 particles prepared by a hydrothermal process. J Electrochem Soc 152(11):A2199–A2202
Doeff MM, Hu Y, McLarnon F, Kostecki R (2003) Effect of surface carbon structure on the electrochemical performance of LiFePO4. Electrochem Solid-State Lett 6(10):A207–A209
Dhindsa KS, Mandal BP, Bazzi K, Lin MW, Nazri M, Nazri GA, Naik VM, Garg VK, Oliveira AC, Vaishnava P, Naik R, Zhou ZX (2013) Enhanced electrochemical performance of graphene modified LiFePO4 cathode material for lithium ion batteries. Solid State Ionics 253:94–100
Zhu X, Hu J, Wu W, Zeng W, Dai H, Du Y, Liu Z, Li L, Ji H, Zhu Y (2014) LiFePO4/reduced graphene oxide hybrid cathode for lithium ion battery with outstanding rate performance. J Mater Chem A 2(21):7812–7818
Guo X, Fan Q, Yu L, Liang J, Ji W, Peng L, Guo X, Ding W, Chen Y (2013) Sandwich-like LiFePO4/graphene hybrid nanosheets: in situ catalytic graphitization and their high-rate performance for lithium ion batteries. J Mater Chem A 1(38):11534–11538
Wang B, Liu A, Abdulla WA, Wang D, Zhao XS (2015) Desired crystal oriented LiFePO4 nanoplatelets in situ anchored on a graphene cross-linked conductive network for fast lithium storage. Nanoscale 7(19):8819–8828
Li W, Hwang J, Chang W, Setiadi H, Chung KY, Kim J (2016) Ultrathin and uniform carbon-layer-coated hierarchically porous LiFePO4 microspheres and their electrochemical performance. J Supercrit Fluid 116:164–171
Zhang Y, Huang Y, Wang X, Guo Y, Jia D, Tang X (2016) Improved electrochemical performance of lithium iron phosphate in situ coated with hierarchical porous nitrogen-doped graphene-like membrane. J Power Sources 305:122–127
Yang X, Tu J, Lei M, Zuo Z, Wu B, Zhou H (2016) Selection of carbon sources for enhancing 3D conductivity in the secondary structure of LiFePO4/C cathode. Electrochim Acta 193:206–215
Pratheeksha PM, Mohan EH, Sarada BV, Ramakrishna M, Hembram K, Srinivas PV, Daniel PJ, Rao TN, Anandan S (2016) Development of a novel carbon-coating strategy for producing core-shell structured carbon coated LiFePO4 for an improved Li-ion battery performance. Phys Chem Chem Phys 19(1):175–188
Liu Y, Gu J, Zhang J, Yu F, Dong L, Nie N, Li W (2016) Metal organic frameworks derived porous lithium iron phosphate with continuous nitrogen-doped carbon networks for lithium ion batteries. J Power Sources 304:42–50
Oh J, Lee J, Hwang T, Kim JM, Seoung K, Piao Y (2017) Dual layer coating strategy utilizing N-doped carbon and reduced graphene oxide for high-performance LiFePO4 cathode material. Electrochim Acta 231:85–93
Cao Z, Zhu G, Zhang R, Chen S, Sang M, Jia J, Yang M, Li X, Yang S (2018) Biological phytic acid guided formation of monodisperse large-sized carbon@LiFePO4/graphene composite microspheres for high-performance lithium-ion battery cathodes. Chem Eng J 351:382–390
Ma H, Xiang J, Xia X (2018) Graphene foam supported LiFePO4 nanosheets composite as advanced cathode for lithium ion batteries. Mater Res Bull 101:205–209
Guzmán G, Vazquez-Arenas J, Ramos-Sánchez G, Bautista-Ramírez M, González I (2017) Improved performance of LiFePO4 cathode for Li-ion batteries through percolation studies. Electrochim Acta 247:451–459
Fischer MG, Hua X, Wilts BD, Castillo-Martinez E, Steiner U (2018) Polymer-templated LiFePO4/C nanonetworks as high-performance cathode materials for lithium-ion batteries. ACS Appl Mater Interfaces 10(2):1646–1653
Du G, Zhou Y, Tian X, Wu G, Xi Y, Zhao S (2018) High-performance 3D directional porous LiFePO4/C materials synthesized by freeze casting. Appl Surf Sci 453:493–501
Jing P, Yao L, Xiang M, Wang Y, Wu J, Wang B, Zhang Y, Wu H, Liu H (2018) Facile synthesis of bowl-like LiFePO4/C composite with high rate-performance. J Electron Mater 47(7):3543–3551
Rosaiah P, Zhu J, Hussain OM, Liu Z, Qiu Y (2018) Well-dispersed rod-like LiFePO4 nanoparticles on reduced graphene oxide with excellent electrochemical performance for Li-ion batteries. J Electroanal Chem 811:1–7
Wang Q, Peng D, Chen Y, Xia X, Liu H, He Y, Ma Q (2018) A facile surfactant-assisted self-assembly of LiFePO4/graphene composites with improved rate performance for lithium ion batteries. J Electroanal Chem 818:68–75
Yang J, Li Z, Guang T, Hu M, Cheng R, Wang R, Shi C, Chen J, Hou P, Zhu K, Wang X (2018) Green synthesis of high-performance LiFePO4 nanocrystals in pure water. Green Chem 20(22):5215–5223
Takeuchi T, Tabuchi M, Nakashima A, Nakamura T, Miwa Y, Kageyama H, Tatsumi K (2005) Preparation of dense LiFePO4/C composite positive electrodes using spark-plasma-sintering process. J Power Sources 146(1):575–579
Jiang Z, Zhang B, Shen Q, Jiang Z-J (2019) In-situ plasma assisted formation of graphitic nanosheet supported N-doped carbon-coated antisite defectless LiFePO4 as a high-performance cathode material for lithium-ion batteries. J Alloys Compd 806:864–873
Wu SX, Chiang CL, Wang CC, Chen CY (2018) Functionalization of MWCNTs by plasma treatment and use as conductive additives for LiFePO4 electrode. J Taiwan Inst Chem Eng 89:208–214
Gao C, Zhou J, Liu G, Wang L (2018) Lithium-ions diffusion kinetic in LiFePO4/carbon nanoparticles synthesized by microwave plasma chemical vapor deposition for lithium-ion batteries. Appl Surf Sci 433:35–44
Wang B, Xu B, Liu T, Liu P, Guo C, Wang S, Wang Q, Xiong Z, Wang D, Zhao XS (2014) Mesoporous carbon-coated LiFePO4 nanocrystals co-modified with graphene and Mg2+ doping as superior cathode materials for lithium ion batteries. Nanoscale 6(2):986–995
Ni H, Liu J, Fan L-Z (2013) Carbon-coated LiFePO4–porous carbon composites as cathode materials for lithium ion batteries. Nanoscale 5(5):2164–2168
Zhang Y, Wang W, Li P, Fu Y, Ma X (2012) A simple solvothermal route to synthesize graphene-modified LiFePO4 cathode for high power lithium ion batteries. J Power Sources 210(4):47–53
Sun L, Deng Q, Fang B, Li Y, Deng L, Yang B, Ren X, Zhang P (2016) Carbon-coated LiFePO4 synthesized by a simple solvothermal method. CrystEngComm 18(39):7537–7543
Long Y, Shu Y, Ma X, Ye M (2014) In-situ synthesizing superior high-rate LiFePO4/C nanorods embedded in graphene matrix. Electrochim Acta 117(4):105–112
Su C, Bu X, Xu L, Liu J, Zhang C (2012) A novel LiFePO4/graphene/carbon composite as a performance-improved cathode material for lithium-ion batteries. Electrochim Acta 64(1):190–195
Funding
Authors acknowledge the supports from the Chinese National Natural Science Foundation (No. 11105078), the Guangdong Provincial Natural Science Foundation (No. 2017A030313092), the Science Foundation of Zhejiang Sci-Tech University (No. 18062245-Y), the “Outstanding Talent and Team Plans Program” and “the Fundamental research funds for the central university” of South China University of Technology (No. 2018ZD25).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tian, X., Chen, W., Jiang, Z. et al. Porous carbon-coated LiFePO4 nanocrystals prepared by in situ plasma-assisted pyrolysis as superior cathode materials for lithium ion batteries. Ionics 26, 2715–2726 (2020). https://doi.org/10.1007/s11581-019-03422-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-019-03422-6