
Abstract
This study describes the results of the characterization of solid polymer electrolytes using chitosan matrix plasticized with glycerol and doped with cerium and lithium triflate binary salt composition. The electrolytes were prepared by solvent casting method and characterized by thermal analysis (thermogravimetric analysis—TGA and differential scanning calorimetry—DSC), impedance measurements, X-ray diffraction (XRD), scanning electron microscopy (SEM), atomic force microscopy (AFM), and electron paramagnetic resonance (EPR). Samples are thermally stable up to 128–153 °C, and most of them are amorphous. In some cases, the appearance of crystalline peaks is due to the diffraction of salt domains, which makes the samples less conductive. The room temperature conductivity maximum of 10−6 S cm−1 at 30 °C was obtained for the samples with the same total salt mass of 0.15 g (ChitCeTrif0.05LiTrif0.10 and ChitCeTrif0.10LiTrif0.05). Finally, the EPR analysis suggests that the local coordination environment of the paramagnetic Ce3+ is not the same in different samples. In summary, beside the modest conductivity values of these samples, they are still adequate for some electrochemical applications.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Solid polymer electrolytes (SPEs) appear as substitutes for liquid electrolytes because they present very little problems of leakage or pressure-related distortions. Moreover, they are materials with good electrode-electrolyte contact and good mechanical and adhesive properties, and they can be prepared easily and in different forms [1]. Due to their interesting properties such as lightweight, flexibility, thin film-forming ability, and no leakage they find several applications as for example in batteries, fuel cells, and other electrochemical devices among others [2]. The most SPEs extensively investigated are PEO (poly(ethylene oxide))-based systems containing lithium salts [3]. More recently, electrolytes based on natural polymers have been alternatively proposed due to their biodegradability, low production cost, good physical and chemical properties, and good performance as solid polymer electrolytes [4]. Most of these new systems are based on polysaccharides such as cellulose and its derivatives [5], starch [6], agar [7], gelatin [8], or chitosan [9].
The SPEs of the present work were prepared by solvent casting technique, and they consist of a polymer doped with binary salt composition and a plasticizer. Chitosan (Chit) was chosen as polymer host because it is a natural, non-toxic, and biodegradable polymer and it can be used as polymer matrix for ionic conduction [10]. Chit is a chitin derivative obtained by its deacetylation, and it is soluble in dilute aqueous solution of a weak acid [11]. In its structure, the presence of lone pair electrons on the nitrogen and oxygen atoms of the amine and hydroxyl functional groups, respectively, enables the chelation of a proton donor provided by salt [12].
Over past years, the authors’ research have been focused on different approaches with the aim to achieve higher room conductivity values of the SPE samples. This is done by blending polymers [13], crosslinking [14], inserting fillers [15], or plasticizing [16] processes, for example. Among all these methods, plasticization seems to be the simplest and cost-effective way to improve the ionic conductivity of SPEs [17]. The role of the plasticizer is not only to decrease the crystallinity of polymer electrolytes but also to enhance ion dissociation and reduce Tg, which leads to the increase in number of mobile ions; the polymer electrolyte becomes flexible at a reduced temperature resulting in higher ionic conductivity [18].
In this work, a novel polymeric electrolyte system was developed. It contains mobile trivalent rare earth cations, but it is known that the ionic mobilities of multivalent cations are generally low in polymeric matrices probably due to the strong electrostatic force between the multivalent cations and the polar groups of the polymer [19]. However, it was already shown in other studies on mixed salt systems that they display better ionic conductivity than the single salt system [20]. Therefore, in this paper a binary salt system composed of two inorganic salts: cerium and lithium triflates was incorporated in a chitosan matrix with the aim to increase the conductivity values of the samples. Lithium and cerium salts are chosen because their cations are easily coordinated and solvated, resulting in ionic conductivity character of the samples [21]. Additionally, there was added a plasticizer because small organic and compatible molecule as glycerol improve the ionic conductivity values too [22].
Beside the ionic conductivity values evaluation of these new polymer electrolyte systems, the purpose of this work was to verify what kind of changes occur when binary salt system of lithium and cerium triflates is added to the electrolyte formula. These changes were evaluated in terms of thermal stability, ionic conductivity, diffraction patterns, surface morphologies, and electron paramagnetic resonance (EPR). EPR is a sensitive spectroscopic technique for investigating paramagnetic species, such as transition metal ions and lanthanide ions, in a variety of solid-state systems [23,24,25]. In the present work, pulsed EPR technique was used to obtain the absorption EPR line shape of Ce3+ in membranes of Chit:Ce(CF3SO3)3. Hyperfine and super-hyperfine interactions between the electron spin and remote magnetic nuclei present in the surroundings of the paramagnetic Ce3+ ions were investigated by electron spin echo envelope modulation (ESEEM) and hyperfine sublevel correlation experiment (HYSCORE) techniques [26, 27].
Materials and methods
Solid polymer electrolyte preparation
The solid polymer electrolytes were synthesized according to the method described previously [28]. A synthesis scheme, pictured in Fig. 1, shows that 0.20 g of chitosan (Sigma-Aldrich, medium Mw) was dispersed in 10 mL of 1% acetic acid solution (Sigma-Aldrich) and stirred overnight under magnetic stirring for complete dissolution of polymer. Next, different quantities of cerium (III) trifluoromethanesulfonate (CeTrif, HuiChem, 98%) and lithium trifluoromethanesulfonate (LiTrif, Sigma-Aldrich, 98%) were added to the solution and left under stirring for more few minutes. Finally, the plasticizer (0.25 g), in this case glycerol (Himedia, 99.5%), was added and the solution was again left under magnetic stirring. When the solutions were homogeneous, they were poured on plastic Petri plates and cooled at room temperature for a week until film formation, and then, they were subjected to a drying procedure in an oven whose temperature was raised from 25 to 60 °C for 2 days, while the oven was periodically evacuated and purged with dry argon. Samples were represented by the notation ChitCeTrif n LiTrif m , where n and m are the mass of cerium and lithium triflate, respectively. The electrolyte membranes, shown in Fig. 1, were transparent; their thicknesses were determined with a micrometer (Mitutoyo, Japan) and ranged between 0.045 and 0.180 ± 0.001 mm. Finally, the samples were stored in a glovebox under argon atmosphere and removed only before each characterization analysis.

Diagram of chitosan-based electrolyte synthesis and physical appearance of the samples
Characterization techniques
The thermal stability was studied using TGA-Q500 instrument in 25 to 900 °C temperature range. For differential scanning calorimetry (DSC) measurements with Mettler DSC 821e, the samples were placed in 40-μL aluminum cans, and the analyses were carried out under a flowing argon atmosphere between − 60 and 200 °C with a heating rate of 5 °C min−1.
The total ionic conductivity was determined by introducing a chitosan electrolyte disk between two 10-mm diameter ion-blocking gold electrodes (Goodfellow, > 99.95%) to form a symmetrical cell electrode/chitosan electrolyte/electrode assembly that was placed in a Büchi TO51 tube-oven. A K-type thermocouple was localized close to electrolyte disk to measure the sample temperature. Before each analysis, the samples were heated to 60 °C for 2 hours to improve contact between the electrodes and the electrolyte. The impedance measurements were carried out using an Autolab PGSTAT-12 (Eco Chemie) at frequencies between 65 kHz and 500 mHz and over the room temperature to about 100 °C.
X-ray diffraction measurements were performed in a PANalytical X’Pert Pro diffractometer equipped with an X’Celerator detector at room temperature and over a scattering angle (2θ) range from 3 to 60°. The samples were placed on a Si wafer to minimize diffuse scattering from the substrate and exposed to monochromated CuKα radiation with λ = 1.541 Å.
The surface morphology of the films was evaluated using SEM micrographs obtained in a LEO 440 instrument. All film samples were pre-coated with a conductive layer of sputtered gold, and the micrographs were taken at 10 kV and at different magnifications.
AFM analysis was done with a Nanosurf easyScan 2 AFM System in the non-contact mode and using silicon AFM probes, a force constant of 48 Nm-1, and a resonance frequency of 190 kHz.
X-band continuous wave (CW-EPR) and pulsed EPR experiments were carried out on a Bruker Elexsys E580 spectrometer operating at 9.478 GHz. CW-EPR measurements were performed at 50 K and the pulsed experiments at 6 K. Electron spin echo envelope modulation (ESEEM) decay was recorded 4500 G using a three-pulse sequence, (π/2)–τ–(π/2)–T–(π/2)–(echo), with (π/2) pulse width of 12 ns. The ESEEM spectrum was obtained from the Fourier transform of the spin echo decay. The echo-detected field-sweep absorption spectra, EDFS, were recorded using a three-pulse sequence with a (π/2) pulse width of 12 ns. The integrated echo intensities were measured as a function of the magnetic field strength over the range of 100–12,100 G. Hyperfine sublevel correlation experiments (HYSCORE) was performed at 4500 G using the pulse sequence (π/2)–τ–(π/2)–t 1–(π)–t 2–(π/2)–τ–(echo), with (π/2) pulse width of 12 ns and τ = 120 ns. Both time intervals t1 and t2 were incremented in 20 ns to generate a 128 × 128 matrix which, after apodization, was Fourier-transformed to generate the two-dimensional HYSCORE spectrum. Numerical simulation was performed using the function saffron from the EasySpin package [29].
Results and discussion
Thermal characterization (TGA and DSC)
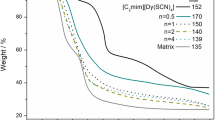
TGA is an effective tool for identification on the thermal stability, and the thermograms of the samples are presented in Fig. 2(a–c). The obtained results reveal that for all the electrolytes the degradation process occurs in three stages. For pure chitosan membrane, the degradation occurs in two stages, and it was thermally stable up to 265 °C [30]. In an early study with cerium triflate, the sample of chitosan matrix also presented two degradation stages, but when salt is added the decomposition occurs in three stages that started in the interval of 128 and 143 °C [31]. In a study of chitosan-NH4NO3-EC (ethylene carbonate) membrane, the authors obtained two-stage decomposition [32]. The first one corresponds to desorption process, which occurs at 50–100 °C and has a weight loss of about 15%. This first stage is followed by a total decomposition that occurs in between 150 and 200 °C with 60% of the weight loss. The total decomposition began at 174 °C, which is a relatively lower value when compared with most pure chitosan membranes where the total decomposition occurs at 200 °C, and this difference is probably due to the presence of EC plasticizer. In the case of this study, the addition of lithium salt to cerium electrolytes does not cause significant changes in the thermogram aspect, and the degradation process of these samples also occurs in three stages. The curves of the samples with and without lithium triflate in Fig. 2 (b1) are practically overlapped, and the main difference is the remaining residue that is higher in lithium-free sample. In relation to the degradation temperature, the addition of lithium makes the samples more stable in almost all cases as shown by the degradation temperature graphs present in Fig. 2 (a2), (b2), and (c2). In samples containing two salts, the degradation started in the interval of 136–153 °C, which is a relatively higher temperature range than that obtained in samples containing only cerium triflate. In both samples, with and without lithium, the decomposition occurs below 200 °C, which is probably due to the presence of glycerol as plasticizer. This first predominant stage of thermal degradation was caused probably by depolymerization of chitosan chains. The second stage occurred between 200 and 300 °C, and the degradation process ended at about 500 °C; however, the weight loss continued slowly as the temperature was increased up to 900 °C. The fact that degradation occurs in three stages may be due to the different interactions of the salts and the plasticizer with polysaccharide chains [31]. The remaining residues were in the range of 24–30% for the samples without and 11–18% with lithium triflate in the electrolyte composition.

a–c TGA and degradation temperatures of ChitCeTrif n LiTrif m electrolytes
The DSC results of ChitCeTrif n LiTrif m -based electrolytes are shown in Fig. 3. The DSC measurements were performed in the − 60 to 200 °C temperature interval, and no glass transition temperature was detected within this range, so the samples semi-crystalline nature was confirmed. This behavior is very similar to that obtained with other natural polymer ionically conducting membranes [33, 34]. Although samples have predominantly amorphous character, they presented some peaks that appear at about 175 °C in most of the samples containing cerium triflate, which may be due to the presence of the crystalline domains. These peaks can be related to undissolved salt or a complex formation between the matrix and the salt. Some samples also presented a weak and large endothermic peak, which can be attributed to acetal crosslinking break. As found for other samples, this crosslinking break is due to the catalytic properties of acetic acid residues in the samples [35].

a–c DSC curves of ChitCeTrif n LiTrif m electrolytes
Impedance analysis
Figure 4(a–c) shows the typical impedance plots of ChitCeTrif0.05LiTrif m prepared with 0.05 g of cerium triflate and different quantities of lithium triflate and measured at various temperatures. The impedance plots show a semicircle at high frequencies and a spike at low frequencies. In this figure, one can also observe a disappearance of a depressed semicircle at high frequency with the increase in temperature, which reveals the absence of capacitive nature and just diffusion process [36]. At low frequencies, information about the electrode/electrolyte interface is obtained, and if the interaction is ideal vertical spikes are observed [37]. In the present and other studies, the spike is inclined at an angle less than 90° and may be due to the roughness or non-homogeneous of the electrode/electrolyte interface [38].

Complex impedance plots of ChitCeTrif0.05LiTrif m , measured at different temperatures
The ionic conductivity values can be obtained using Eq. (1).
where l is the thickness of sample, R b is the bulk resistance, and A is the contact area between the electrolyte and the electrode. The R b values were calculated from the intercept of the semicircle or the intercept of the spike with the real axis. The bulk resistance values plotted in Fig. 4 decrease with the increase of the temperature indicating an increase of the ion mobility and the thermal movement of polymer chains [39]. The R b values are higher for lower conductivity values, but an increase in salt concentration increases the number of mobile charge carriers, which leads to the decrease in R b values. However, the salt excess leads to the recombination of ions, a decrease in the amorphousness and, consequently, an increase in R b values and a decrease in ionic conductivity.
The evolution of the ionic conductivity as a function of temperature is shown in Fig. 5. The ionic conductivity depends on factors such as salt concentration and temperature [40]. The higher conductivity value obtained in this work shows that the ionic conductivity can be increased by using a mixture of salts. The best ionic conductivity values were obtained for the samples with the same total amount of salt of 0.15 g. Thus, ChitCeTrif0.05LiTrif0.10 and ChitCeTrif0.10LiTrif0.05 are the samples with higher, but still modest, conductivity values namely 2.63 × 10−6 and 2.28 × 10−6 S cm−1 at 30 °C. At higher temperatures, thermal movement of polymer chain segments and dissociation of salts would be improved, which results in an increase of the conductivity, and values of 9.63 × 10−5 and 5.84 × 10−5 S cm−1 were obtained at 90 °C for the samples referred above. The difference in conductivity values may also be attributed to the different ratio of CeTrif-LiTrif used in the present work. In the case of the ChitCeTrif0.05LiTrif0.10, the incorporation of lithium triflate enhances the room temperature ionic conductivity by one order of magnitude, which may be attributed to the contribution of Li+ ions. The effect of the addition of LiTrif in SPEs was also evaluated in another study, where the authors obtained a greater increase of three to four orders of magnitude in conductivity values at room temperature [41]. The ionic conductivity increases with the increase in total salts amount to reach a maximum, and this is probably due to the increase in the number of mobile charge carriers. However, for higher salt levels a decrease in the conductivity values is certainly related to undissolved or recrystallized salt. On the other hand, at higher salt levels the ions are able to recombine and form neutral ion pairs, leading to a decrease in the number density of mobile ions and a decrease in the ionic conductivity [42].

a–c Conductivity plots of ChitCeTrif n LiTrif m -based electrolytes as a function of inverse of temperature
From the temperature dependence of ionic conductivity, it is possible to deduce that practically all samples displayed almost linear (r 2 ~ 1) relationship of log σ vs 1000/T. This indicates that the ionic conductivity is thermally activated, and the plots obey the Arrhenius equation (2).
where σ 0 is a pre-exponential factor, E a is the activation energy for ion transport, R is the gas constant (8.314 J mol−1 K−1), and T is the temperature. The E a value was calculated using the slope of the fittings of obtained data from Arrhenius equation (2). For the electrolyte ChitCeTrif0.10LiTrif0.05 the E a value is 49.45 kJ mol−1, which is a very similar value to that obtained in a study using other natural polymer as host [43].
XRD analyses
The XRD analysis is a useful tool to determine the structure and crystallization of the polymer matrices [43]. XRD analysis has been performed in order to investigate the effect of the mixed salts in the system, and the respective diffraction patterns of the samples ChitCeTrif n LiTrif m are shown in Fig. 6(a–c). Figure 6 reveals that all the samples presented a peak at 2θ = 10°, which may be attributed to the cerium triflate salt as previously demonstrated in another study [31]. On the other hand, the incorporation of lithium triflate makes some samples more crystalline as in the case of ChitCeTrif0.05LiTrif0.25 and ChitCeTrif0.10LiTrif0.04. The new peaks that appear in XRD data of these samples are certainly related to the diffraction of salt domains. In a study of U(600)nLiCF3SO3 xerogels, some samples also present new peaks with lithium addition, namely at approximately 34°, which are also related with Li+ domains [44]. The XRD obtained results corroborate with conductivity results since the samples with the presence of crystalline domains, namely ChitCeTrif0.05LiTrif0.25, ChitCeTrif0.25LiTrif0.00, and ChitCeTrif0.10LiTrif0.04 are also those with less conductivity. On the other side, the samples with similar XRD patterns, ChitCeTrif0.10LiTrif0.00 and ChitCeTrif0.10LiTrif0.05, also present very similar conductivity values.

a–c XRD of ChitCeTrif n LiTrif m -based electrolytes
SEM analyses
Figure 7 shows the SEM micrographs of the polymer electrolytes films containing only one salt (cerium triflate) and a mixture of two salts (cerium and lithium triflate).

a–c SEM micrographs of ChitCeTrif n LiTrif m -based electrolytes
As shown in Fig. 7 (a1), (b1), and (c1), for the samples without lithium triflate, the surface morphologies are very similar to those obtained with other natural polymers [45, 46]. Also, in this case, the electrolytes present a uniform surface morphology without any phase separation.
The samples containing a binary system of two salts have less homogeneous surfaces, which is clear in the remaining images of Fig. 7.
Figure 7a shows the results of the samples with the same cerium amount of 0.05 g and varied quantity of lithium salt of 0.10 and 0.25 g. As already seen above, the addition of 0.10 g of lithium triflate made the samples more conductive (Fig. 5a) probably due to the presence of lithium ions. On the other hand, when a large amount of lithium is added, in this case 0.25 g, the ionic conductivity decreases probably due to undissolved or recrystallized salt, which justifies the clearer lines in SEM images (Fig. 7 (a3)) and crystalline peaks in XRD (Fig. 6a).
Figure 7b presents the SEM micrographs of samples with 0.25 g of cerium triflate and different quantities of lithium triflate. The addition of a small amount of lithium salt (0.05 g) results in a less regular structure and in a more conductive sample. At low magnification, Fig. 7 (b2.1) and (b2.2), a granular structure is observed. In Fig. 7 (b2), it is seen that some crystalline aggregates are adjacent taking shape of a semi-crystalline morphology, which corroborates XRD data (Fig. 6b). In a study of blends with different mass ratios of PMMA/PVdF, the SEM images at different magnifications revealed a granular structure at low magnification [47]. The extreme PMMA/PVdF compositions of 80/20 and 30/70 are more amorphous and crystalline structures. In 60/40 blend ratio, the samples have semi-crystalline morphology because PVdF crystalline aggregates are adjacent.
When lithium triflate is added to chitosan-based samples with fixed 0.10 g of cerium triflate, the resulting SEM images are presented in Fig. 7c. The first increment of lithium (0.04 g) results in a poorly conductive sample, which can be explained by recrystallized or undissolved salt. This is confirmed by the peaks observed in XRD data (Fig. 6c) and the irregular surface presented in SEM micrograph (Fig. 7 (c2)). Also in the case of higher lithium amounts (0.10 g), although the samples show a porous morphology (Fig. 7 (c4)), the conductivity is close to that obtained for 0.04 g of lithium salt and again it may be due to undissolved salt. The increments of 0.05 g of lithium salt in DSC and XRD investigations suggest a lower degree of crystallinity and predominance of the amorphous phase, which results in a higher value of ionic conductivity.
All the prepared samples also appeared translucent and showed very good adhesion properties.
AFM analysis
Aiming to evaluate the homogeneity and changes in the surface topography of the electrolytes the AFM analyses were carried out. Therefore, morphological characteristics of chitosan doped with one salt (cerium triflate) and two salts (cerium and lithium triflate) were examined by AFM, and Fig. 8 displays characteristic images of these samples. The scanning area of 2.00 μm × 2.00 μm and the roughness, expressed in roughness mean square (RMS) of chitosan electrolytes are presented in Fig. 8. In the case of the samples containing 0.05 g of cerium triflate and different quantities of lithium triflate, Fig. 8a, the RMS varies between 7.10 and 124 nm. The more conductive sample (ChitCeTrif0.05LiTrif0.10) presented smaller roughness of 7.10 nm, while the less conductive sample (ChitCeTrif0.05LiTrif0.25) showed higher roughness of 124 nm. From these results, it was stated that the most conductive sample exhibits a lower surface roughness value, and the same remarks were done for samples of chitosan doped with europium triflate [48]. For the samples with 0.25 g of cerium triflate and different quantities of lithium triflate the AFM results are presented in Fig. 8b. Similar to conductivity results also in this case the roughness values are very close each other. The RMS value varies between 8.43 and 10.2 nm, and the most conductive sample (ChitCeTrif0.25LiTrif0.05) exhibits a higher surface roughness of 10.2 nm. This behavior is opposite to those referred in the literature for samples with chitosan doped with cerium triflate [31].

AFM images of ChitCeTrif n LiTrif m -based electrolytes
EPR analysis
Trivalent cerium ion, Ce3+, is a paramagnetic ion with 4f 1 electronic configuration and 2F free-ion ground state. Spin-orbit coupling causes the separation between 2F7/2 and 2F5/2 levels, the latter being the ground state separated by 2200 cm−1 (~ 0.27 eV) from the 2F7/2 excited state. Since the unpaired electron is in the 4f shell and there are two further filled shells (5s and 5p), crystal field effects are smaller than in transition metal ions, where the unpaired electron is in the latest occupied shell. In an axial field, the sixfold J = 5/2 from the 2F5/2 level splits into three degenerate Kramer doublets, MJ = ± 1/2, ± 3/2, and ±5/2. For tetragonal symmetries, the expected g values are g || = 0.86 and g ⊥ = 2.57 for a MJ = ± 1/2 doublet ground state; g || = 2.57 and g ⊥ = 0 for a MJ = ± 3/2 ground state; and g || = 4.29 and g ⊥ = 0 for a MJ = ± 5/2 ground state [49,50,51]. Sometimes, admixture of J levels should be considered to describe experimentally observed g values [52]. As natural cerium has no isotope with non-zero nuclear spin, the EPR spectrum of Ce3+ is composed of a single line without hyperfine structure. Reliable EPR spectra of Ce3+ have been reported almost solely for single crystals and polycrystalline materials [50,51,52,53,54].
No signals were observed in the CW-EPR spectra of the three samples investigated. The absorption EPR line shape of Ce3+ of sample ChitCeTrif0.05LiTrif0.00 was obtained by the pulsed EPR technique EDFS (Fig. 9a). The observed broad signal (~ 104 G) was previously attributed to Ce3+ions in glasses and polycrystalline materials [55, 56]. The principal values of the g tensor, g zz = g || = 0.3 and g xx = g yy = g ⊥ = 1.61, were obtained from the EDFS spectrum. The observed g values are close to those calculated for a MJ = ± 1/2 doublet ground state. The discrepancy can be explained by assuming admixture with the first excited state 2F7/2 [52]. It is interesting to note that the g values of Ce3+ in the chitosan:Ce(CF3SO3)3 membranes (g || = 0.3, g ⊥ = 1.61) are rather different from those reported for Ce3+ in La(CF3SO3)3 crystals (g || = 3.81, g ⊥ = 0.26 for the MJ = ± 5/2 doublet and g || = 0.977, g ⊥ = 2.15 for the MJ = ± 1/2 doublet) [50], suggesting that the local coordination environment of the paramagnetic Ce3+ is not the same in this system.

Echo-detected field-sweep absorption spectra, EDFS (a) and electron spin echo envelope modulation, ESEEM (b), decay and spectrum of sample ChitCeTrif0.05LiTrif0.00. ESEEM experiment was performed at 6 K at a magnetic field of 4500 G, showing a feature at ~ 19.2 MHz assigned to 1H and a weak feature at ~ 5 MHz assigned to 13C nuclei
In order to clarify the local environment of Ce3+ in the chitosan:Ce(CF3SO3)3 membranes, we have resorted to the spin-echo-pulsed EPR techniques ESEEM and HYSCORE. Figure 9b shows the ESEEM decay and spectrum of sample ChitCeTrif0.05LiTrif0.00. The envelope decay curve is obtained from the electron spin-echo amplitude recorded as a function of the pulse separation. These decay curves often exhibit a periodic modulation whose frequencies correspond to the Larmor frequencies of nuclear species surrounding the paramagnetic electron spin. Figure 9b shows the spectrum obtained from the Fourier transform of the electron spin echo decay. The identification of the nuclear species is performed by comparing the frequency of the peak in the ESEEM spectrum with the Larmor frequencies of nuclei at the magnetic field of the ESEEM experiment. The intense peak at around 19 MHz in Fig. 9b corresponds to 1H nuclei (nuclear spin I = ½, gyromagnetic ratio γ = 4.2576 kHz/G, natural abundance 100%). The weak peak at around 5 MHz can be assigned to 13C nuclei (I = ½, γ = 1.07 kHz/G, natural abundance 1.1%). It is interesting to note that there are no peaks corresponding to 19F nuclei (I = ½, γ = 4.0055 kHz/G, natural abundance 100%) in the ESEEM spectra of Fig. 9b suggesting that the CF3SO3 − group does not participate in the complexation of the Ce3+ ion.
Information on the magnetic nuclei coupled to the paramagnetic Ce3+ in the chitosan:Ce(CF3SO3)3 membranes was obtained with the two-dimensional pulse technique HYSCORE. The spectrum of sample ChitCeTrif0.05LiTrif0.00 depicted in Fig. 10 exhibits an intense signal close to the (19.16 MHz) point of the diagonal line. This isotropic feature results from the interaction between the Ce3+ electronic spin and the 1H nuclei. No features are observed at the Larmor frequency corresponding to the 19F.

Hyperfine sublevel correlation, HYSCORE, spectrum of sample ChitCeTrif0.05LiTrif0.00 at 6 K with magnetic field strength of 4500 G and τ = 120 ns. Dashed lines mark the position of features due to 1H
This interpretation is supported by the observation of the proton peak in the three-pulse ESEEM spectrum (Fig. 9b). Numerical simulation of the experimental HYSCORE spectrum was performed using a spin Hamiltonian which includes the electronic and nuclear Zeeman interactions and the hyperfine interaction between the Ce3+ unpaired electron spin and the 1H nuclear spin [57, 58]:
where β e is the Bohr magneton, β n is the nuclear magneton and I H is the 1H nuclear spin. The other symbols have their usual meaning. Euler angles (α, β, γ) were included to account for the fact that the g and AH tensors are not diagonal at the same coordinate system and are defined according to the EasySpin package [29, 59].
The best-fit for the experimental spectrum is achieved with the axial g tensor interacting with the 1H nucleus. The measured and the simulated HYSCORE spectra of sample ChitCeTrif0.05LiTrif0.00 are shown in Fig. 11. For the simulations, we wrote the AH tensor as a iso1 + T, where a iso is the isotropic hyperfine coupling and T is a traceless tensor containing only dipolar hyperfine coupling. The values used to generate the best-fit simulation are listed in Table 1. The intense proton peaks found in the HYSCORE spectra belong to protons of chitosan with hyperfine couplings a iso = 0.6 MHz, T ⊥ = 3.4 MHz, which are comparable to those reported for Cu(H2O)6 in MCM-41 (a iso = 1.5 MHz, T ⊥ = 4.8 MHz) [27] and pyridine–copper complex (a iso = 4 MHz, T ⊥ = 2.6 MHz) [58].

HYSCORE spectrum of sample ChitCeTrif0.05LiTrif0.00 (blue contours) and its numerical simulation (green contours). The dashed line marks the position of features due to 1H. Both experimental and numerical spectra were normalized. The parameters used to generate the numerical simulation are displayed in Table 1
Assuming that the anisotropic part of the hyperfine interaction (T ⊥ = 3.4 MHz) can be described by a point-dipolar interaction, the distance (r) between the unpaired electron of Ce3+ ion and the protons of the chitosan macromolecule can be estimated from the following eq. [57, 60, 61]:
where h is the Planck constant, g e = 2.0023 is the free electron g factor and g n = ħγ n/β n = 5.5857 is the 1H nuclear g factor. A Ce−H distance of 2.83 Å (0.28 nm) was found which is consistent with the distance between the cation and macromolecular protons in polymer electrolytes [62].
Conclusion
This paper reports thermal analysis, conductivity, XRD, SEM, AFM, and EPR measurements in electrolytes formed by chitosan, prepared from acetic acid solution, and containing glycerol and binary system of two salts: cerium and lithium triflates. The obtained results were compared to the membranes with only one salt system. All prepared samples presented good flexibility, homogeneity, and transparency. The thermal analysis revealed that the degradation occurs in three stages, and the samples are thermally stable up to 128–153 °C. The best room temperature ionic conductivity, 10−6 S cm−1 (at 30 °C), was obtained for the samples with 0.15 g of total salt amount. Most of the electrolytes are amorphous, and the appearance of crystalline peaks in XRD data are due to the diffraction of salt domains. Because there was no signal in CW-PR spectra, pulsed EPR technique EDFS was used to study the interaction of Ce3+ ion with chitosan network. The observed broad signal (~ 104 G), attributed to Ce3+ions, had values of the g tensor, g zz = g || = 0.3 and g xx = g yy = g ⊥ = 1.61, which were close to those calculated for a MJ = ± 1/2 doublet ground state and explained by assuming admixture with the first excited state 2F7/2. To investigate a local coordination environment of the paramagnetic Ce3+, the spin-echo-pulsed EPR techniques ESEEM and HYSCORE technique were used. The results revealed no peaks corresponding to 19F, suggesting that the CF3SO3 − group does not participate in the complexation of the Ce3+ ion. HYSCORE numerical simulation revealed that the paramagnetic Ce3+ in the chitosan:Ce(CF3SO3)3 membranes is coupled to 1H nuclei of chitosan, and an Ce−H distance is 2.83 Å (0.28 nm).
References
Ramadan R, Kamal H, Hashem HM, Abdel-Hady K (2014) Gelatin-based solid electrolyte releasing Li+ for smart window applications. Sol Energ Mater Sol Cells 127:147–156. https://doi.org/10.1016/j.solmat.2014.04.016
Karthikeyan S, Sikkanthar S, Selvasekarapandian S, Arunkumar D, Nithya H, Kawamura J (2016) Structural, electrical and electrochemical properties of polyacrylonitrile-ammonium hexaflurophosphate polymer electrolyte system. J Polym Res 23:1–10
Tarascon J-M, Armand M (2001) Issues and challenges facing rechargeable lithium batteries. Nature 414(6861):359–367. https://doi.org/10.1038/35104644
Leones R, Sentanin F, Rodrigues LC, Marrucho IM, Esperança JMSS, Pawlicka A, Silva MM (2012) Investigation of polymer electrolytes based on agar and ionic liquids. Express Polym Lett 6(12):1007–1016. https://doi.org/10.3144/expresspolymlett.2012.106
Regiani AM, Machado GO, LeNest J-F, Gandini A, Pawlicka A (2001) Cellulose derivatives as solid electrolyte matrixes. Macromol Symp 175(1):45–53. https://doi.org/10.1002/1521-3900(200110)175:1<45::AID-MASY45>3.0.CO;2-M
Marcondes RFMS, D'Agostini PS, Ferreira J, Girotto EM, Pawlicka A, Dragunski DC (2010) Amylopectin-rich starch plasticized with glycerol for polymer electrolyte application. Solid State Ionics 181(13-14):586–591. https://doi.org/10.1016/j.ssi.2010.03.016
Raphael E, Avellaneda CO, Manzolli B, Pawlicka A (2010) Agar-based films for application as polymer electrolytes. Electrochim Acta 55(4):1455–1459. https://doi.org/10.1016/j.electacta.2009.06.010
Vieira DF, Avellaneda CO, Pawlicka A (2007) Conductivity study of a gelatin-based polymer electrolyte. Electrochim Acta 53(4):1404–1408. https://doi.org/10.1016/j.electacta.2007.04.034
Khiar ASA, Puteh R, Arof AK (2006) Conductivity studies of a chitosan-based polymer electrolyte. Physica B 373(1):23–27. https://doi.org/10.1016/j.physb.2005.10.104
Navaratnam S, Ramesh K, Ramesh S, Sanusi A, Basirum WJ, Arof AK (2015) Transport mechanism studies of chitosan electrolyte system. Electrochim Acta 175:68–73. https://doi.org/10.1016/j.electacta.2015.01.087
Choudhury NA, Northrop PWC, Crothers AC, Jain S, Subramanian VR (2012) Chitosan hydrogel-based electrode binder and electrolyte membrane for EDLCs: experimental studies and model validation. J Appl Electrochem 42(11):935–943. https://doi.org/10.1007/s10800-012-0469-2
Majid SR, Arof AK (2007) Electrical behavior of proton-conducting chitosan-phosphoric acid-based electrolytes. Physica B 390(1-2):209–215. https://doi.org/10.1016/j.physb.2006.08.038
Shukur MF, Ithnin R, Kadir MFZ (2014) Protonic transport analysis of starch-chitosan blend based electrolytes and application in electrochemical device. Mol Cryst Liq Cryst 603(1):52–65. https://doi.org/10.1080/15421406.2014.966259
Ghosh S, Rasmusson J, Inganäs O (1998) Supramolecular self-assembly for enhanced conductivity in conjugated polymer blends: ionic crosslinking in blends of poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) and poly(vinylpyrrolidone). Adv Mater 10(14):1097–1099. https://doi.org/10.1002/(SICI)1521-4095(199810)10:14<1097::AID-ADMA1097>3.0.CO;2-M
Majid SR, Idris NH, Hassan MF, Winie T, Khiar ASA, Arof AK (2005) Transport studies on filler-doped chitosan based polymer electrolyte. Ionics 11(5-6):451–455. https://doi.org/10.1007/BF02430265
Shukur MF, Ithnin R, Kadir MFZ (2014) Electrical characterization of corn starch-LiOAc electrolytes and application in electrochemical double layer capacitor. Electrochim Acta 136:204–216. https://doi.org/10.1016/j.electacta.2014.05.075
Ng LS, Mohamad AA (2006) Protonic battery based on a plasticized chitosan-NH4NO3 solid polymer electrolyte. J Power Sources 163(1):382–385. https://doi.org/10.1016/j.jpowsour.2006.09.042
Mishra K, Pundir SS, Rai DK (2017) Effect of polysorbate plasticizer on the structural and ion conduction properties of PEO-NH4PF6 solid polymer electrolyte. Ionics 23(1):105–112. https://doi.org/10.1007/s11581-016-1790-2
Morita M, Araki F, Kashiwamura K, Yoshimoto N, Ishikawa M (2000) Ionic structure and conductance behavior of plasticized polymeric electrolytes containing multivalent cations. Electrochim Acta 45(8-9):1335–1340. https://doi.org/10.1016/S0013-4686(99)00341-2
Ramesh S, Arof AK (2000) Electrical conductivity studies of polyvinyl chloride-based electrolytes with double salt system. Solid State Ionics 136-137(1-2):1197–1200. https://doi.org/10.1016/S0167-2738(00)00598-1
Ramesh S, Wong KC (2009) Conductivity, dielectric behaviour and thermal stability studies of lithium ion dissociation in poly(methyl methacrylate)-based gel polymer electrolytes. Ionics 15(2):249–254. https://doi.org/10.1007/s11581-008-0268-2
Pawlicka A, Mattos RI, Tambelli CE, Silva IDA, Magon CJ, Donoso JP (2013) Magnetic resonance study of chitosan bio-membranes with proton conductivity properties. J Membr Sci 429:190–196. https://doi.org/10.1016/j.memsci.2012.11.048
Pilbrow JR (1990) Transition ion electron paramagnetic resonance. Clarendon Press, Oxford
Griscom DL (1980) Electron spin resonance in glasses. J Non-Cryst Solids 40(1-3):211–272. https://doi.org/10.1016/0022-3093(80)90105-2
Van Doorslaer S, Schweiger A (2000) One- and two-dimensional pulse electron paramagnetic resonance spectroscopy: concepts and applications. Naturwissenschaften 87(6):245–255. https://doi.org/10.1007/s001140050714
Höfer P, Grupp A, Nebenfiir H, Mehring M (1986) Hyperfine sublevel correlation (hyscore) spectroscopy: a 2D ESR investigation of the squaric acid radical. J Chem Phys 132:279–282
Pöppl A, Kevan L (1996) A practical strategy for determination of proton hyperfine interaction parameters in paramagnetic transition metal ion complexes by two-dimensional HYSCORE electron spin resonance spectroscopy in disordered systems. J Phys Chem 100(9):3387–3394. https://doi.org/10.1021/jp9525999
Alves R, Donoso JP, Magon CJ, Silva IDA, Pawlicka A, Silva MM (2016) Solid polymer electrolytes based on chitosan and europium triflate. J Non-Cryst Solids 432:307–312. https://doi.org/10.1016/j.jnoncrysol.2015.10.024
Stoll S, Britt RD (2009) General and efficient simulation of pulse EPR spectra. Phys Chem Chem Phys 11(31):6614–6625. https://doi.org/10.1039/b907277b
Wan Y, Creber KAM, Peppley B, Tam Bui V (2006) Chitosan-based solid electrolyte composite membranes: I. preparation and characterization. J Membr Sci 280(1-2):666–674. https://doi.org/10.1016/j.memsci.2006.02.024
Alves R, Sentanin F, Sabadini RC, Pawlicka A, Silva MM (2016) Influence of cerium triflate and glycerol on electrochemical performance of chitosan electrolytes for electrochromic devices. Electrochim Acta 217:108–116. https://doi.org/10.1016/j.electacta.2016.09.019
Ng LS, Mohamad AA (2008) Effect of temperature on the performance of proton batteries based on chitosan-NH4NO3-EC membrane. J Membr Sci 325(2):653–657. https://doi.org/10.1016/j.memsci.2008.08.029
Alves R, Silva MM (2014) The influence of glycerol and formaldehyde in gelatin-based polymer electrolytes. Mol Cryst Liq Cryst 591(1):64–73. https://doi.org/10.1080/15421406.2013.822739
Leones R, Rodrigues LC, Fernandes M, Ferreira RAS, Cesarino I, Pawlicka A, Carlos LD, de Zea Bermudez V, Silva MM (2013) Electro-optical properties of the DNA-Eu3+ bio-membranes. J Electroanal Chem 708:116–123. https://doi.org/10.1016/j.jelechem.2013.08.031
Lima E, Raphael E, Rodrigues LC, Ferreira RAS, Carlos LD, Silva MM, Pawlicka A (2012) Photo luminescent polymer electrolyte based on agar and containing europium picrate for electrochemical devices. Mater Sci Eng B-Solid 177(6):488–493. https://doi.org/10.1016/j.mseb.2012.02.004
Avellaneda CO, Vieira DF, Al-Kahlout A, Leite ER, Pawlicka A, Aegerter MA (2007) Solid-state electrochromic devices with Nb2O5:Mo thin film and gelatin-based electrolyte. Electrochim Acta 53(4):1648–1654. https://doi.org/10.1016/j.electacta.2007.05.065
Stephan AM, Thirunakaran R, Renganathan NG, Sundaram V, Pitchumani S, Muniyandi N, Gangadharan R, Ramamoorthy P (1999) A study on polymer blend electrolyte based on PVC/PMMA with lithium salt. J Power Sources 81-82:752–758. https://doi.org/10.1016/S0378-7753(99)00148-2
Wu GM, Lin SJ, Yang CC (2006) Preparation and characterization of PVA/PAA membranes for solid polymer electrolytes. J Membr Sci 275(1-2):127–133. https://doi.org/10.1016/j.memsci.2005.09.012
Avellaneda CO, Vieira DF, Al-Kahlout A, Heusing S, Leite ER, Pawlicka A, Aegerter MA (2008) All solid-state electrochromic devices with gelatin-based electrolyte. Sol Energ Mater Sol Cells 92(2):228–233. https://doi.org/10.1016/j.solmat.2007.02.025
Alves RD, Rodrigues LC, Andrade JR, Fernandes M, Pinto JV, Pereira L, Pawlicka A, Martins R, Fortunato E, de Zea Bermudez V, Silva MM (2013) GelatinnZn(CF3SO3)2 polymer electrolytes for electrochromic devices. Electroanalysis 25(6):1483–1490. https://doi.org/10.1002/elan.201300038
Kumar A, Sharma R, Das MK, Gajbhiye P, Kar KK (2016) Impacts of ceramic filler and the crystallite size of polymer matrix on the ionic transport properties of lithium triflate/poly (vinylidene fluoride-co-hexafluoropropene) based polymer electrolytes. Electrochim Acta 215:1–11. https://doi.org/10.1016/j.electacta.2016.08.087
Shukur MF, Ithnin R, Kadir MFZ (2014) Electrical properties of proton conducting solid biopolymer electrolytes based on starch-chitosan blend. Ionics 20(7):977–999. https://doi.org/10.1007/s11581-013-1033-8
Rajendran S, Sivakumar M, Subadevi R (2004) Investigations on the effect of various plasticizers in PVA-PMMA solid polymer blend electrolytes. Mater Lett 58(5):641–649. https://doi.org/10.1016/S0167-577X(03)00585-8
Correia SMG, de Zea Bermudez V, Silva MM, Barros S, Ferreira RAS, Carlos LD, Passos de Almeida AP, Smith MJ (2002) Morphological and conductivity studies of di-ureasil xerogels containing lithium triflate. Electrochim Acta 47(15):2421–2428. https://doi.org/10.1016/S0013-4686(02)00100-7
Alves RD, Rodrigues LC, Andrade JR, Pawlicka A, Pereira L, Martins R, Fortunato E, Silva MM (2013) Study and characterization of a novel polymer electrolyte based on agar doped with magnesium triflate. Mol Cryst Liq Cryst 570(1):1–11. https://doi.org/10.1080/15421406.2012.703041
Ribeiro G, Conde M, Carvalho J, Rodrigues LC, Alves R, Ferreira RAS, Pawlicka A, Carlos LD, Silva MM (2012) Natural membranes for application in biomedical devices. Mol Cryst Liq Cryst 562(1):147–155. https://doi.org/10.1080/15421406.2012.679172
Nicotera I, Coppola L, Castriota M, Cazzanelli E (2006) Investigation of ionic conduction and mechanical properties of PMMA-PVdF blend-based polymer electrolytes. Solid State Ionics 177(5-6):581–588. https://doi.org/10.1016/j.ssi.2005.12.028
Alves R, de Camargo ASS, Pawlicka A, Silva MM (2016) Luminescent polymer electrolytes based on chitosan and containing europium triflate. J Rare Earth 34(7):661–666. https://doi.org/10.1016/S1002-0721(16)60076-5
Abragam A, Bleaney B (1970) Electron paramagnetic resonance of transition ions. Oxford University Press, London
Petasis DT, Bellesis GH, VanderVen NS, Friedberg SA (1991) Electron paramagnetic resonance of Ce3+ in La(CF3SO3)3·9H2O. J Appl Phys 70(10):5998–6000. https://doi.org/10.1063/1.350073
Pidol L, Guillot-Noel O, Kahn-Harari A, Viana B, Pelenc D, Gourier D (2006) EPR study of Ce3+ ions in lutetium silicate scintillators Lu2Si2O7 and Lu2SiO5O. J Phys Chem Solids 67(4):643–650. https://doi.org/10.1016/j.jpcs.2005.10.175
Yamaga M, Honda M, Shimamura K, Fukuda T, Yosida T (2000) Electron spin-resonance study on Ce3+ in BaLiF3. J Phys-Condens Matter 12(27):5917–5928. https://doi.org/10.1088/0953-8984/12/27/310
Barrie JD, Momoda LA, Dunn B, Gourier D, Aka G, Vivien D (1990) ESR and optical spectroscopy of Ce3+: β″-alumina. J Solid State Chem 86(1):94–100. https://doi.org/10.1016/0022-4596(90)90118-H
Popescu FF, Bercu V, Barascu JN, Martinelli M, Massa CA, Pardi LA, Stefan M, Nistor SV, Nikl M (2010) Study of the Kramers rare earth ions ground multiplet with a large orbital contribution by multifrequency EPR spectroscopy: Ce3+ in PbWO4 scintillator. Opt Mater 32(5):570–575. https://doi.org/10.1016/j.optmat.2009.11.015
Magon CJ, Donoso JP, Lima JF, Eckert H, Zanotto ED, Lumeau J, Glebova L, Glebov L (2016) Electron paramagnetic resonance (EPR) studies on the photo-thermo ionization process of photo-thermo-refractive glasses. J Non-Cryst Solids 452:320–324. https://doi.org/10.1016/j.jnoncrysol.2016.09.012
Chiesa M, Mattsson K, Taccheo S, Robin T, Lablonde L, Mechin D, Milanese D (2014) Defects induced in Yb3+/Ce3+ co-doped aluminosilicate fiber glass preforms under UV and γ-ray irradiation. J Non-Cryst Solids 403:97–101. https://doi.org/10.1016/j.jnoncrysol.2014.07.011
Pöppl A, Hartmann M, Böhlmann W, Böttcher R (1998) Coordination geometry of the copper-pyridine complex in frozen solution as studied by proton and deuterium two-dimensional hyperfine sublevel correlation electron spin resonance spectroscopy. J Phys Chem A 102(20):3599–3606. https://doi.org/10.1021/jp980406m
Carl PJ, Vaughan DEW, Goldfarb D (2002) Interactions of Cu(II) ions with framework Al in high Si:Al zeolite Y as determined from X- and W-band pulsed EPR/ENDOR spectroscopies. J Phys Chem B 106(21):5428–5437. https://doi.org/10.1021/jp0145659
Chechik V, Carter E, Murphy D (2016) Electron paramagnetic resonance. Oxford University Press, Oxford. https://doi.org/10.1039/9781782629436 (http://easyspin.org/easyspin/documentation/eulerangles.html)
Vezin H, Moissette A, Hureau M, Brémard C (2006) trans-Stilbene incorporation in acidic medium-pore ZSM-5 zeolite: a pulsed EPR study. Chem Phys Chem 7:2474–2477
Maurelli S, Berlier G, Chiesa M, Musso F, Cora F (2014) Structure of the catalytic active sites in vanadium-doped aluminophosphate microporous materials: new evidence from spin density studies. J Phys Chem C 118(34):19879–19888. https://doi.org/10.1021/jp505896x
Donoso JP, Bonagamba TJ, Panepucci HC, Oliveira LN, Gorecki W, Berthier C, Armand M (1993) Nuclear magnetic relaxation study of poly(ethylene oxide)-lithium salt based electrolytes. J Chem Phys 98(12):10026–10036. https://doi.org/10.1063/1.464435
Acknowledgements
We are grateful to the Fundação para a Ciência e Tecnologia and FEDER (Ref. UID/QUI/00686/2013 and UID/QUI/0686/2016) and program POCH/FSE for a grant SFRH/BD/97232/2013 (R. Alves) for financial support of this work. The authors are grateful to Mr. Marcos de Oliveira, Jr. for technical assistance. The financial support of the Brazilian agencies Capes and The Brazilian National Council for Scientific and Technological Development (CNPq) are gratefully acknowledged. Research was partially financed by the CeRTEV, Center for Research, Technology and Education in Vitreous Materials, FAPESP 2013/07793-6. M. M. Silva and A. Pawlicka acknowledge the Brazilian National Council for Scientific and Technological Development (CNPq) for grants provided by this institution (PVE grant 406617/2013-9 and PQ grant 305029/2013-4).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Alves, R., Sabadini, R.C., Silva, I.D.A. et al. Binary Ce(III) and Li(I) triflate salt composition for solid polymer electrolytes. Ionics 24, 2321–2334 (2018). https://doi.org/10.1007/s11581-017-2355-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-017-2355-8