
Abstract
(Ni0.8Mn0.1Co0.1)(OH)2 and Co(OH)2 secondly treated by LiNi0.8Mn0.1Co0.1O2 have been prepared via co-precipitation and high-temperature solid-state reaction. The residual lithium contents, XRD Rietveld refinement, XPS, TG-DSC, and electrochemical measurements are carried out. After secondly treating process, residual lithium contents decrease drastically, and occupancy of Ni in 3a site is much lower and Li/Ni disorder decreases. The discharge capacity is 193.1, 189.7, and 182 mAh g−1 at 0.1 C rate, respectively, for LiNi0.8Mn0.1Co0.1O2-AP, -NT, and -CT electrodes between 3.0 and 4.2 V in pouch cell. The capacity retention has been greatly improved during gradual capacity fading of cycling at 1 C rate. The noticeably improved thermal stability of the samples after being treated can also be observed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lithium-ion batteries for electric vehicles (EVs) and hybrid electric vehicles (HEVs) require cathode materials with higher energy and power, besides of well thermal stability, excellent cycle life, inexpensive, and other performance as a low initial irreversible capacity, low kinetic, and transport resistances [1,2,3,4]. As one of the most promising cathode materials, Ni-based layered oxides of LiNi1-x-yCoxMnyO2 (0 ≤ x + y ≤ 1) have been widely investigated and commercially used for their higher specific energy by enhancing the nickel content, such as LiNi1/3Co1/3Mn1/3O2, LiNi0.5Co0.2Mn0.3O2, LiNi0.6Co0.2Mn0.2O2, LiNi0.7Co0.15Mn0.15O2, especially LiNi0.8Co0.1Mn0.1O2, and so on [5,6,7,8]. However, thermal stability of the LiNi1-x-yCoxMnyO2 materials also turns to be poor with the enhancement of the nickel content, for the augment generation of Ni2+ instead of Ni3+ and migration of Ni2+ into the Li layer interfered with the Li-ion pathway during Li intercalation/deintercalation, which results in poor electrochemical performance [9,10,11]. One of the other important factors with poor thermal stability for the Ni-enriched materials LiNi1-x-yCoxMnyO2 is the residual content of lithium on the surface, including the concentration of lithium carbonate (Li2CO3 or CO3 2−) and lithium hydroxide (LiOH·H2O or OH−). The CO2 exposure time and the electrochemical performance closely relate to CO3 2− content; so, it is necessary to limit the carbonation reaction or lithium content on the surface [12,13,14]. Other factors as structural stability and constituent homogeneity may also cause great influence to the comprehensive properties.
Much effort has been devoted to overcome these problems. Among which, metal substitution of Al and/or Mg could reduce Li+/Ni2+ cation mixing, resulted in improved electrochemical cycling behavior, structural stability, and thermal stability compared to pristine LiNi0.8Mn0.1Co0.1O2 [15]. Ti-substituted LiNi0.8Mn0.1Co0.1O2 alleviated cracking from secondary particles to primary ones and exhibited better multifaceted performances with a relatively high capacity, good rate capability, and acceptable cycling stability [16]. Full concentration gradient (FCG) Ni-enriched materials exhibited better cycling performances and thermal stability at the expense of reversible capacity, whereas the rate capability and low-temperature performance were significantly deteriorated by increasing outer layer thickness [17, 18]. Oxygen non-stoichiometry and Li-excess amount also played a key role to Li+/Ni2+ disorder, structural instability, cycling stability, and rate capability [19, 20]. Electrolyte plus additives of vinylene carbonate (VC)-based blend improved capacity retention compared to cells with PES211 with reducing parasitic reactions and improved safety when cycled to the same upper cutoff potentials for LiNi0.8Mn0.1Co0.1O2 [21]. On the other side, homogeneous coating was widely used for modification to the lithium-ion cathode materials [22,23,24,25,26]. LiNi0.5Mn0.2Co0.3O2 encapsulated in a continuous lithium lanthanum titanium oxide thin layer could effectively suppress side reactions between the charged cathode active material and the electrolyte solution, resulted in substantial reduction of the gas generation, good capacity retention, and rate performance [24]. SiO2-modified layer LiNi0.8Mn0.1Co0.1O2 reduced the polarization gap and the charge-transfer resistance, presented better cycling performance as well as the rate capability even at elevated cut-off voltage and high temperature (60 °C) [25]. However, coating mediums are generally insulated for lithium-ion conduction and increase lithium-ion length, unfavorable for interfacial charge transfer of the electrode. Therefore, lithium-ion conductive materials have been investigated as surface coating agents to improve the cycling performance and thermal stability of cathode materials. The cycling performance of LiNi0.8Mn0.1Co0.1O2 has been effectively enhanced by lithium-ion conductive material Li2TiO3 thin film coating, for which suppressed the direct contact between the active materials and the electrolytes, enhanced the lithium-ion diffusion between the electrode/electrolyte interface, and prevented the pulverization of the active materials during repeated charging/discharging [26].
Meanwhile, secondly treating process with lithium-ion conductive material of metal hydroxide precusor has seldom adopted for LiNi0.8Mn0.1Co0.1O2 modification with cycling and thermal stability. In this study, LiNi0.8Mn0.1Co0.1O2 cathode materials were prepared from reaction between co-precipitated synthesized (Ni0.8Mn0.1Co0.1)(OH)2 precusor and LiOH·H2O. Li-excess was applied to sufficient reacting for crystallization and fluxing agent. Then, two different lithium-ion conductive materials with (Ni0.8Mn0.1Co0.1)(OH)2 and Co(OH)2 were used mainly focused on improvement of cycling and thermal stability through decreasing of the residual content of lithium compounds and surface modification. The reason for the selection is that they basically keep the same structural characteristic as LiNi0.8Mn0.1Co0.1O2; they can also react with residual lithium, and their resultants exhibit identical properties such as high capacity, excellent structural, or cycling stability. X-ray diffraction (XRD) and scanning electron microscopy (SEM) were used to investigate the structural characteristic and surface topography. Half cells (coin cells) and full cells (pouch cells) were fabricated for evaluating initial efficiency, rate capability, and cycling stability. Thermogravimetric and differential scanning calorimetry analysis (TG-DSC) were used for thermal stability analysis.
Experimental
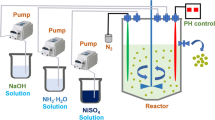
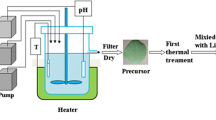
The metal hydroxide precusor (Ni0.8Mn0.1Co0.1)(OH)2 was prepared via co-precipitation. An aqueous solution of NiSO4·6H2O, MnSO4·5H2O, and CoSO4·7H2O was pumped into a continuously stirred tank reactor under a N2 atmosphere. At the same time, a NaOH solution (aq.) and a desired amount of NH3·H2O solution (aq.) as a chelating agent were also separately fed into the reactor. The spherical (Ni0.8Mn0.1Co0.1)(OH)2 particles were filtered, washed, and dried in air at 120 °C to remove adsorbed water. Finally, the LiNi0.8Mn0.1Co0.1O2 powders were prepared by a conventional solid-state reaction using (Ni0.8Mn0.1Co0.1)(OH)2 and lithium hydroxide (LiOH·H2O). The ingredients were mixed for 120 min by ball-milling in a polyurethane lining pot with alumina balls (2:1 to mixed samples in wt%), and then, the sample was separated from the balls by a 300 mesh sieve. The mixture was calcinated at 500 °C for 5 h and 850 °C for 15 h then allowed to cool naturally. The processes of heating up, constant temperature, and cooling down were under flowing oxygen. Besides, an excess of lithium 1.04 in mole ratio was used to compensate for lithium loss during the calcination. The as-prepared powder was named sample LiNi0.8Mn0.1Co0.1O2-AP. (Ni0.8Mn0.1Co0.1)(OH)2 and Co(OH)2 with primary particles of nanometer scale and secondly particles of 2–3 μm approximately were used for treating medium separately with the constant amount of 1.5 wt%. And then, LiNi0.8Mn0.1Co0.1O2-AP and (Ni0.8Mn0.1Co0.1)(OH)2 or Co(OH)2 were mixed for another 120 min by ball-milling, separated by sieve, and calcinated at 850 °C for 8 h. The other processes were kept the same as LiNi0.8Mn0.1Co0.1O2-AP synthesis. LiNi0.8Mn0.1Co0.1O2-NT and LiNi0.8Mn0.1Co0.1O2-CT were named for (Ni0.8Mn0.1Co0.1)(OH)2 and Co(OH)2 treated sample, respectively.
The concentrations of CO3 2− and OH− for the samples were measured by titrimetric analysis. The weighed samples (m, 30 g) which being 4 h baked were firstly dissolved by deionized water in 250-ml constant volume bottle under argon atmosphere. After fully stirring and filtrating, some filtrate (Vt, 25 ml) was fetched out for ultimate titration. Two drops of phenolphthalein ethanol indicator (1 g L−1) were added, and standard hydrochloric acid solution (CHCl, 0.05 mol L−1) was used for titrating. When the color of filtrate changes from red to colorless, the volume was recorded with V1. Then, two drops of methyl red ethanol indicator (2 g L−1) were added for further titrating. After the color of filtrate changes from yellow to orange, the filtrate was heated until the color returns to yellow for evaporation of CO2. Afterwards, the standard hydrochloric acid solution was continually used for titrating while the color of filtrate changes from yellow to orange, the volume was recorded with V2. On other sides, the V0 was stood for volume of standard hydrochloric acid solution used in blank experiment. Finally, the content of Li2CO3 and LiOH was calculated by formula (1) and (2), then translated the concentrations of CO3 2− and OH− accordingly.
Powder XRD (Rint-1000, Rigaku, Japan) using Cu-Kα radiation was used to identify the crystalline phase of the synthesized materials. XRD data were obtained (2θ = 10°–90°) with a step size of 0.02°. More than 15 diffraction peaks were obtained for all the samples. The lattice parameters were calculated by the Rietveld refinement method using the FULLPROF program via the WinPLOTR interface, which were developed by the group of Rodriguez-Carvajal (Laboratoire Leon Brillouin). The composition in terms of transition metal contents in the materials was determined by the inductively coupled plasma (ICP, Thermo Electron Corporation). The pH values were tested by pH tester. The particle size and morphology were measured by SEM (SU8010) with an accelerating voltage of 15 kV and transmission electron microscopy (TEM, Philips CM200 microscope). The X-ray photoelectron spectroscopy (XPS, Perkin-Elmer, PHI 5600) measurements were performed to get surface information of the materials.
Thermogravimetry-Differential Scanning calorimeter (TG-DSC) experiments were conducted on the powders using a 200 PC (NETZSCH, Germany) instrument. The curves were recorded between room temperature and 500 °C at a scan rate of 5 °C min−1. An empty stainless steel capsule was used as a reference pan. The weight of each sample was measured before and after the experiment. The weight was constant in all cases, indicating that there were no leaks during the experiments.
The CR2016 coin cell tests were performed. For positive electrode fabrication, the prepared materials were mixed with 5 wt% of carbon black and 5 wt% of polyvinylidene fluoride (PVDF) in N-methyl pyrrolidinone (NMP) solvent until the slurries were obtained. The blended slurries were pasted onto an aluminum current collector, and the electrode was dried at 120 °C for 12 h in the air. Then, the electrode pieces were cut to 16 mm in diameter. The test cell consisted of the positive electrode and the lithium foil negative electrode separated by a porous polypropylene film, and 1 mol L−1 LiPF6 in EC and DMC (1:1 in volume) as the electrolyte. The assembly of the cells was carried out in a dry Ar-filled gloved box. The test was carried out using an automatic galvanostatic charge-discharge unit NEWWARE battery cycler between 3.0 and 4.3 V, 3.0 and 4.4 V with the current density of 20 mA g−1 (~0.1C), 40, 100, and 200 mA g−1 versus Li/Li+ electrodes at room temperature.
The 063048-size LiNi0.8Mn0.1Co0.1O2/graphite pouch cells (1200 mAh) were manufactured by Hunan Research Institute of Nonferrous Metals (Changsha City, Hunan Province, China) and tested mainly. The positive electrode consisted of 97.5% LiNi0.8Mn0.1Co0.1O2 powders, 1.0% conductive agent, and 1.5% of binder. The negative electrode of these cells was 97% artificial graphite FSN-4 which commercially purchased from Shanghai Shanshan Technology Company (Shanghai City, China), 1.5% conductive agent, and 1.5% of binder. Before being filled with electrolyte, the cells were cut just below the heat seal and dried at 80 °C under vacuum for 12 h to remove residual water. Then, the cells were transferred to an argon-filled glove box where they were filled with electrolyte and vacuum-sealed. Subsequently, they were placed in a temperature box to allow for the completion of wetting. Then, the cells were charged at 0.01 C to 3.5 V for aging and transferred into the glove box to release any gas generated and vacuum sealed again. On next step, they were charged at 0.01 C to 4.2 V for formation. The test was carried out using an automatic galvanostatic charge-discharge unit NEWWARE battery cycler between 3.0 and 4.2 V with the current density of 18 mA g−1 (~0.1 C), 36, 90, 180, and 360 mA g−1 versus graphite electrodes at room temperature. Cycling properties were rested between 3.0 and 4.2 V at 1 C rate.
Results and discussion
As shown in Table 1, the measured compositions of the samples carried out by ICP-MS test are close to the target compositions, on which (Ni0.8Mn0.1Co0.1)(OH)2 treated sample LiNi0.8Mn0.1Co0.1O2-NT exhibits slightly higher Ni, Mn, Co contents than that of LiNi0.8Mn0.1Co0.1O2-AP and Co(OH)2 treated sample LiNi0.8Mn0.1Co0.1O2-CT presents the highest Co content. Therefore, the nominal compositions are used to describe the materials throughout this paper for simplicity.
Table 2 shows the variation of pH values and concentrations of CO3 2− and OH− for different samples. As we can see, after secondly treating process by lithium-ion conductive material (Ni0.8Mn0.1Co0.1)(OH)2 or Co(OH)2, residual lithium contents of resulted samples LiNi0.8Mn0.1Co0.1O2-NT and -CT decrease drastically especially for concentrations of CO3 2−. The reason is that residual CO3 2− and OH− in LiNi0.8Mn0.1Co0.1O2-AP reacted with (Ni0.8Mn0.1Co0.1)(OH)2 or Co(OH)2, along with their decomposition. Residual lithium compounds have great influence on gas generation, sensitivity to moisture, and interfere with diffusion of Li+ thus do bad damage to electrochemical performances for Ni-based cathode materials [14, 24]. Thereby, improvement of thermal stability, process stability, storage performances, and cycling properties for lithium batteries has been anticipated.
The powder XRD and Rietveld refinement patterns of the samples are presented in Fig. 1. Despite the minor differences in their transition metal contents, all the diffraction peaks for the LiNi0.8Mn0.1Co0.1O2-AP, -NT, and -CT samples are similarly well-defined layer structure based on a hexagonal R \( \overline{3} \) m structure and the splitting of (108)/(110), (006/102) peaks are observed. Nevertheless, some minor difference or slight shits for main diffraction peaks can be found between 2θ regions of 18–19.5° and 43.5–45.5°, especially for LiNi0.8Mn0.1Co0.1O2-NT sample, indicating that the crystal structure of samples being treated has also slightly changed accordingly. Rietveld fitting results for the XRD patterns of LiNi0.8Mn0.1Co0.1O2 based on hexagonal LiMO2 structure are shown in Table 3. Lattice parameters of a, c, and Wyckoff position of Z value in O are corresponding to the main diffraction peaks. S. W. Woo et al. investigated that Al3+ doping was well occupied in the transition metal layer while Mg2+ both in the Li and/or Ni layers, resulting in variation of a and c because of the ionic radius discrepancy [15]. R. Du et al. showed that of a and c monotonically expanded as the amount of Ti-doping increased for the radius of Ti4+ (0.605 Å) was larger than that of Ni3+ (0.56 Å), Co3+ (0.545 Å), and Mn4+ (0.53 Å) and smaller than that of Li+ (0.74 Å) [16]. On the other hand, SiO2 or Li2TiO3 coating did not affect on main diffraction peaks shit or change the lattice parameters [25, 26]. In our study, the structural parameters of LiNi0.8Mn0.1Co0.1O2-CT basically keep well with as-prepared sample LiNi0.8Mn0.1Co0.1O2-AP, which means lithium-ion conductive materials Co(OH)2 full or partially reacted with residual content of lithium compounds Li2CO3 and LiOH·H2O in the secondly treating process, resulting in a coating layer of nano scale particle LiCoO2 or Co2O3 probably outside the LiNi0.8Mn0.1Co0.1O2 particles. Differently, evident shit can be observed from 2θ regions of 18–19.5° and 43.5–45.5° for LiNi0.8Mn0.1Co0.1O2-NT. Lattice parameters of a and c have also obviously increased and Wyckoff position of Z value in O decreased correspondingly. The ionic radius of Ni2+ (0.69 Å) is a little greater than that of Ni3+ (0.56 Å). Therefore, we consider that sample LiNi0.8Mn0.1Co0.1O2-NT after secondly treating process may possess more Ni2+ content compared to Ni3+ content in the transition metal layer of crystal structure than that of LiNi0.8Mn0.1Co0.1O2-AP. Li/Ni disorder usually occurs in Ni2+ and Li+ for their analogous ionic radius. Consequently, Li/Ni disorder decreases with an increase in the number of Ni2+ content. More Ni3+ content in the transition metal layer tends to lower bond energy than Co3+ and Mn4+ and possess much high possibility to contain oxygen deficiency [19]. Similar analysis has been carried out by F. Wu et al. about the effect of Ni2+ content on Li/Ni disorder for Ni-rich cathode materials [20]. In our research, it can also be confirmed by the data of occupancy of Ni in 3a site from Rietveld fitting results. Occupancy of Ni in 3a site for LiNi0.8Mn0.1Co0.1O2-NT is much lower than that of LiNi0.8Mn0.1Co0.1O2-AP, while the value of LiNi0.8Mn0.1Co0.1O2-CT is lower than that of LiNi0.8Mn0.1Co0.1O2-AP either. For the two different treating processes, Co(OH)2 treating resulting in surface coating, while Li(Ni0.8Mn0.1Co0.1)(OH)2 treating tends to combine with LiNi0.8Mn0.1Co0.1O2-CT. Anyhow, Li/Ni disorder is decreased for both two samples.

XRD patterns of LiNi0.8Mn0.1Co0.1O2 -AP (a), -NT (b), and -CT (c)
SEM images of the metal hydroxide precursor and LiNi0.8Mn0.1Co0.1O2 samples at different magnifications are shown in Fig. 2. All the samples show analogous morphology of a quasi-spherical shape and 5–10 μm particle size approximately with the metal hydroxide precursor, which are composed of agglomerates of much smaller primary particles. Therefore, ball-milling and sintering process did not damage the particle size and morphology. From the high-resolution images of samples, we can observe that the surface of particles exhibits some ultra-fine particles most likely residual lithium compounds for LiNi0.8Mn0.1Co0.1O2-AP, smooth without any coating layer for LiNi0.8Mn0.1Co0.1O2-NT, and clear coating layer outside the LiNi0.8Mn0.1Co0.1O2-CT particles, which indicates distinct difference for pre and after treated samples. TEM images of the samples are shown in Fig. 3. A thick layer can be observed tightly outside the LiNi0.8Mn0.1Co0.1O2-AP particle, which is most likely residual lithium compounds or oxygen defects film resulted from surface decomposing reaction [19]. But for sample LiNi0.8Mn0.1Co0.1O2-NT, the surface is smooth, and hardly, anything can be found outside the particles, which reveals that treating process consumed residual lithium and surface side reaction were suppressed. As for sample LiNi0.8Mn0.1Co0.1O2-CT, a clear coating layer is adhered to the surface of particle. Unfortunately, the coating layer seems to be suspended, but not combined tightly with the core particle. It indicates that reaction resultant LiCoO2 or Co3O4 after treating process may suspend outside the particle, even probably mix with the LiNi0.8Mn0.1Co0.1O2 powders.

SEM images of the metal hydroxide precursor (a), LiNi0.8Mn0.1Co0.1O2-AP (b), -NT (c), and -CT (d) at different magnifications

TEM images of LiNi0.8Mn0.1Co0.1O2-AP (a), -NT (b), and -CT (c) particles
XPS was used to analyze the components on the LiNi0.8Mn0.1Co0.1O2 surfaces as shown on Fig. 4. Two types of chemical environments were identified for carbon and oxygen. The XPS wide spectra (a), spectra of Ni (b), Co (c), and Mn (d) present that the of Ni, Co, and Mn are weak for sample LiNi0.8Mn0.1Co0.1O2-AP. On the other sides, the spectra intensities of C (e) and O (f) are distinct for it. The phenomena indicate that LiNi0.8Mn0.1Co0.1O2-AP particles were covered by some other thick things, such as residual lithium of LiOH·H2O and Li2CO3. However, for sample LiNi0.8Mn0.1Co0.1O2-NT and -CT, the spectra patterns for each element almost keep the same. Spectra of O (e) and C (f) further prove the existence of residual lithium compounds. O 1s spectra comprise two peaks around 531.5 and 529 eV, corresponding to the absorbed oxygen coming from the surface of CO3 2− or OH− and the lattice oxygen [27, 28]. For sample LiNi0.8Mn0.1Co0.1O2-AP, only a very weak peak around 529 eV can be seen, revealing that the oxygen are all in absorbed state as CO3 2− or OH−. C 1s spectra around 285 eV peak are used as internal calibrator, while spectra around 289 eV peak are attributed to carbonate compound, generally as Li2CO3 [19, 25]. We can see the obvious shift, narrowed, and weakened spectra around 289 eV peak for sample LiNi0.8Mn0.1Co0.1O2-NT and -CT. Take the hypothesis that all the samples contain the same amount of internal calibrator carbon, the intensities of O 1s and C 1s from Li2CO3 outside the particles are greatly weakened for sample LiNi0.8Mn0.1Co0.1O2-NT and CT. The results are in accordance with SEM, TEM, and concentrations of CO3 2− and OH− measured by titrimetric analysis.

XPS wide spectra of LiNi0.8Mn0.1Co0.1O2 (a) and spectra of Ni (b), Co (c), Mn (d), O (e), and C (f)
Nowadays, the pouch cell has been widely commercially used as one of the main lithium-ion batteries and the cycling performance which is more representative. Therefore, in our study, electrochemical performance characterization mainly focused on pouch cell. Coin cell test only for LiNi0.8Mn0.1Co0.1O2-AP sample was selected for comparison of initial discharge capacity and efficiency between coin cell and pouch cell. Figure 5 and Table 4 present discharge curves and rate capability of LiNi0.8Mn0.1Co0.1O2-AP between 3.0 and 4.3 V, 4.4 V in coin cell. Besides of higher discharge capacity and initial efficiency for testing carried out between 3.0 and 4.4 V, no obvious discrepancy as voltage plateau, or rate capability is observed for different voltage range. Two distinct plateaus around 4.2 and 3.7 V are shown for both samples at 0.1 C rate, in which the former indicates reduction of Co4+/Co3+ and the latter of Ni4+/Ni3+/Ni2+ mainly [25]. With the increase of discharge rate, the plateaus decrease correspondingly for the deterioration of polarization. The initial efficiency and discharge capacity between 3.0 and 4.3 V at 0.1 C rate are 88.7% and 210.1 mAh g−1, while the discharge capacity is also increased to 202.8, 192.1, and 182.4 mAh g−1 at 0.2, 0.5, and 1 C rate compared with other studies described [15, 16, 19, 25, 26]. The account for the predominance may contribute to the difference of metal hydroxide precursor as-prepared and the synthesis conditions, such as reacting temperature and protecting atmosphere.

Discharge curves of LiNi0.8Mn0.1Co0.1O2-AP between 3.0 and 4.3 V, 4.4 V at different rates in coin cell rate
Figure 6 and Table 5 show discharge curves and rate capability of different LiNi0.8Mn0.1Co0.1O2 electrodes between 3.0 and 4.2 V in pouch cell. Compared to data of coin cell, the capacity and voltage plateau at various discharging rate for pouch cell are slightly lower down accordingly, owing to lower efficiency of artificial graphite anode material compared with Li negative electrode for coin cell. The discharge capacity is 193.1, 189.9, 182.9, 177.5, and 166 mAh g−1 at 0.1, 0.2, 0.5, 1, and 2 C rate, respectively, for LiNi0.8Mn0.1Co0.1O2-AP electrodes. Two distinct plateaus are down to around 4.1 and 3.6 V for both samples at 0.1 C rate. After secondly treating process, the discharge capacity decreases in certain degree to 189.7 and 182.0 mAh g−1 at 0.1 C for LiNi0.8Mn0.1Co0.1O2-NT and -CT electrodes. Meanwhile, rate capability slightly decreases for LiNi0.8Mn0.1Co0.1O2-NT electrodes and basically holds the line for LiNi0.8Mn0.1Co0.1O2-CT. The minor variation is attributed to reaction resultant LiNi0.8Mn0.1Co0.1O2 or metal oxide for LiNi0.8Mn0.1Co0.1O2-NT, LiCoO2, or Co3O4 for LiNi0.8Mn0.1Co0.1O2-CT after treating process.

Discharge curves of LiNi0.8Mn0.1Co0.1O2-AP (a), -NT (b), and -CT (c) at different rates in pouch cells
Figure 7 and Table 6 show the variation of discharge capacity and capacity retention vs cycle number of the artificial graphite/LiNi0.8Mn0.1Co0.1O2 pouch cells cycled at 1 C (180 mA g−1) between 3.0 and 4.2 V. Although LiNi0.8Mn0.1Co0.1O2-AP electrodes deliver the highest initial discharge capacity of 178.2 mAh g−1, it shows the poorest capacity retention of 85.5% after 200 cycles and 69.8% after 400 cycles during gradual capacity fading of cycling. For the LiNi0.8Mn0.1Co0.1O2-NT electrodes, the capacity retention is 90.8% after 200 cycles and 83.3% after 400 cycles delivered a relative low initial discharge capacity of 172.3 mAh g−1. For the LiNi0.8Mn0.1Co0.1O2-CT electrodes, the capacity retention is 90.4% after 200 cycles and 81.4% after 400 cycles delivered a much lower initial discharge capacity of 168.1 mAh g−1. The improved cycling stability is attributed to greatly decreasing of residual lithium compounds outside the particle layer confirmed by titrimetric analysis, SEM, TEM, and XPS characterization. Studies of relationship between residual lithium compounds and electrochemical performance improvement were referred to either [13, 19]. Furthermore, the LiNi0.8Mn0.1Co0.1O2-NT electrodes reveal relatively better capacity retention than LiNi0.8Mn0.1Co0.1O2-CT, especially after 200 cycles. It can be attributed to the increment of Ni2+ contents in the crystal structure proved by XRD and Rietveld refinement results. The phenomena of Ni2+ content effect on Li/Ni disorder and electrochemical properties are in accordance with similar investigation [20]. The capacity retention beyond 80% after 500 cycles is accepted for commercial application in full cell test conditions. Meanwhile, other studies improved cycling stability through surface coating and so on [15, 16, 19, 25, 26]. The coin cell tests revealed the best capacity retention of 90% after 200 cycles. Therefore, our study presents relative better electrochemical properties such as cycling stability improvement.

Cycling performance of LiNi0.8Mn0.1Co0.1O2 at 1 C in pouch cells
Thermal stability and safety of positive electrode at highly delithiated states are important concerns in determining the suitability of lithium batteries for practical applications. Figure 8 shows the TG-DSC profiles of the chemically delithiated LiNi0.8Mn0.1Co0.1O2 materials (-AP, -NT, -CT) in the presence of electrolyte charged to 4.3 V at a scan rate of 5 °C min−1. The profiles presented can be divided into three steps as of room temperature to around 250 °C (1), 250 °C to about 350 °C (2), and 350 °C to 500 °C (3). Step 1 with weight loss 2% and endothermic behavior corresponds to evaporation of adsorbed water and other surface adhesives [15]. Step 2 of major exothermic reaction represents the structural change of the delithiated positive electrode, oxygen liberation, and volatile combustion of the residual electrolyte [15, 18], which accompanied by massive weight loss of 9.3, 7.8, and 8.3% for sample LiNi0.8Mn0.1Co0.1O2-AP, -NT, and -CT. The treated sample LiNi0.8Mn0.1Co0.1O2-NT and -CT exhibits somewhat lower onset temperature than that of LiNi0.8Mn0.1Co0.1O2 -AP, indicating that the starting temperature of the oxygen evolution from the lattice is deferred. However, the exothermic reaction peak 267 °C of the sample LiNi0.8Mn0.1Co0.1O2-AP shifts to a higher temperature from 277 °C for LiNi0.8Mn0.1Co0.1O2-CT to 281 °C for LiNi0.8Mn0.1Co0.1O2-NT. On step 3 of the heating temperature up to 350 °C, the weight loss for sample LiNi0.8Mn0.1Co0.1O2-NT and -CT keeps unchanged. On the contrary, the weight loss for sample LiNi0.8Mn0.1Co0.1O2-AP is continued all the time, which may be attributed to the decomposition reaction of the positive electrode and remaining electrolyte. It is evident that the LiNi0.8Mn0.1Co0.1O2 treated by (Ni0.8Mn0.1Co0.1)(OH)2 and Co(OH)2 affects the thermal behaviors in terms of onset temperature as well as the exothermic peak or heat generation. The results clearly indicate the noticeably improved thermal stability of the samples after being treated, due to consumed residual lithium and increased coating layer outside the particles. The TG-DSC results are keeping well with other studies. Furthermore, the exothermic reaction peak of the sample is higher than that of previous research of below 250 °C [15, 18], revealing better thermal stability.

TG-DSC profiles of the electrochemically delithiated LiNi0.8Mn0.1Co0.1O2 charged to 4.3 V in coin cells
Based on low residual lithium compounds, high initial efficiency, high discharge capacity, both excellent cycling, and thermal stability, we believe that the LiNi0.8Mn0.1Co0.1O2-NT and LiNi0.8Mn0.1Co0.1O2-CT cathode material hold great promise for commercial use in lithium-ion batteries within EV and HEV systems.
Conclusions
(Ni0.8Mn0.1Co0.1)(OH)2 and Co(OH)2 secondly treated LiNi0.8Mn0.1Co0.1O2 were prepared via co-precipitation and high-temperature solid-state reaction. The concentrations of CO3 2− and OH−, XRD Rietveld refinement, XPS, TG-DSC, and electrochemical measurements with CR2016 coin cell and 063048-size pouch cells were carried out. After secondly treating process, residual lithium contents decrease drastically especially for concentrations of CO3 2−. The obvious shift, narrowed, and weakened XPS spectra around 289 eV peak is observed. Occupancy of Ni in 3a site is much lower and Li/Ni disorder decreases. The initial efficiency and discharge capacity between 3.0 and 4.3 V at 0.1 C rate are 88.7% and 210.1 mAh g−1 for LiNi0.8Mn0.1Co0.1O2-AP in coin cell. The discharge capacity is 193.1, 189.7, and 182 mAh g−1 at 0.1 C rate, respectively, for LiNi0.8Mn0.1Co0.1O2-AP, -NT, and -CT electrodes between 3.0 and 4.2 V in pouch cell. The capacity retention is 69.8, 83.3, and 81.4% after 400 cycles during gradual capacity fading of cycling at 1 C rate. The noticeably improved thermal stability of the samples after being treated can be observed, with the exothermic reaction peak 267 °C of the sample LiNi0.8Mn0.1Co0.1O2 -AP shifted to a higher temperature from 277 °C for LiNi0.8Mn0.1Co0.1O2-CT to 281 °C for LiNi0.8Mn0.1Co0.1O2-NT.
References
Armand M, Arascon JM (2008) Building better batteries. Nature 451:652
Manthiram A, Murugan AV, Sarkar A, Muraliganth T (2008) Nanostructured electrode materials for electrochemical energy storage and conversion. Energy Environ Sci 1:621
Goodenough JB, Kim Y (2010) Challenges for rechargeable li batteries. Chem Mater 22:587
Dunn B, Kamath H, Tarascon JM (2011) Electrical energy storage for the grid: a battery of choices. Science 334:928
Zheng HH, Sun Q, Liu G, Song XY (2012) Correlation between dissolution behavior and electrochemical cycling performance for LiNi1/3Co1/3Mn1/3O2-based cells. J Power Sources 207:134
He ZJ, Wang ZX, Huang ZM, Chen H, Li XH, Guo HJ (2015) A novel architecture designed for lithium rich layered Li[Li0.2Mn0.54Ni0.13Co0.13]O2 oxides for lithium-ion batteries. J Mater Chem A 3(32):16817
He ZJ, Wang ZX, Chen H, Huang ZM, Li XH, Guo HJ, Wang RH (2015) Electrochemical performance of zirconium doped lithium rich layered Li1.2Mn0.54Ni0.13Co0.13O2 oxide with porous hollow structure. J Power Sources 299:334
Sun ST, Du CQ, Qu DY, Zhang XH, Tang ZY (2015) Li2ZrO3-coated LiNi0.6Co0.2Mn0.2O2 for high-performance cathode material in lithium-ion battery. Ionics 21:2091
Dahn JR, Fuller EW, Obrovac M, von Sacken U (1994) Thermal stability of LixCoO2, LixNiO2 and λ-MnO 2 and consequences for the safety of li-ion cells. Solid State Ionics 69:265
Arai H, Okada S, Ohtsuka H, Ichimura M, Yamaki J (1995) Characterization and cathode performance of Li1−xNi1+xO2 prepared with the excess lithium method. Solid State Ionics 80:261
He ZJ, Ping J, Yi ZJ, Peng C, Shen CS, Liu JS (2017) Optimally designed interface of lithium rich layered oxides for lithium ion battery. J Alloys Compd 708:1038
Matsumoto K, Kuzuo R, Takeya K, Yamanaka A (1999) Effects of CO2 in air on li deintercalation from LiNi1-x-yCoxAlyO2. J Power Sources 81–82:558
Shizuka K, Kiyohara C, Shima K, Takeda Y (2007) Effect of CO2 on layered Li1+zNi1−x−yCoxMyO2 (M = al, Mn) cathode materials for lithium ion batteries. J Power Sources 166:233
Zhang XY, Jiang WJ, Zhu XP, Maugera A, Liu Q, Julien CM (2011) Aging of LiNi1/3Mn1/3Co1/3O2 cathode material upon exposure to H2O. J Power Sources 196:5102
Wooa SW, Myungb ST, Banga H, Kima DW, Sun YK (2009) Improvement of electrochemical and thermal properties of Li[Ni0.8Co0.1Mn0.1]O2 positive electrode materials by multiple metal (Al, Mg) substitution. Electrochim Acta 54:3851
Du R, Bia YJ, Yang WC, Peng Z, Liu M, Liu Y, Wu BM, Yang BC, Ding F, Wang DY (2015) Improved cyclingstability of LiNi0.8Co0.1Mn0.1O2 via Ti substitution with a cut-off potential of 4.5 V. Ceram Int 41:7133
Liang LW, Hu GR, Cao YB, Du K, Peng ZD (2015) Synthesis and characterization of full concentration-gradient LiNi0.7Co0.1Mn0.2O2 cathode material for lithium-ion batteries. J Alloys Compd 635:92
Lee EJ, Noh HJ, Yoon CS, Sun YK (2015) Effect of outer layer thickness on full concentration gradient layered cathode material for lithium-ion batteries. J Power Sources 273:663
Bi YJ, Yang WC, Du R, Zhou JJ, Liu M, Liu Y, Wang DY (2015) Correlation of oxygen non-stoichiometry to the instabilities and electrochemical performance of LiNi0.8Co0.1Mn0.1O2 utilized in lithium ion battery. J Power Sources 283:211
Wu F, Tian J, Su YF, Wang J, Zhang CZ, Bao LY, He T, Li JH, Chen S (2015) Effect of Ni2+ content on lithium/nickel disorder for Ni-Rich cathode materials. ACS Appl Mater Interfaces 7:7702
Li J, Downie LE, Ma L, Qiu WD, Dahn JR (2015) Study of the failure mechanisms of LiNi0.8Co0.1Mn0.1O2 cathode. J Electrochem Soc 162(7):A1401
Wang ZG, Wang ZX, Guo HJ, Peng WJ, Li XH, Wang JX (2014) Enhanced high-voltage electrochemical performance of LiCoO2 coated with ZrOxFy. Mater Lett 123:93
Lee EH, Cho JH, Kim JM, Park JH, Lee SY (2014) Facile surface modification of high-voltage lithium-ion battery cathode materials with electroconductive zinc antimonate colloidal nanoparticles. RSC Adv 4:15630
Kim YS (2013) Encapsulation of LiNi0.5Co0.2Mn0.3O2 with a thin inorganic electrolyte film to reduce gas evolution in the application of lithium ion batteries. Phys Chem 15:6400
Meng K, Wang ZX, Guo HJ, Li XH, Wang D (2016) Improving the cycling performance of LiNi0.8Co0.1Mn0.1O2 by surface coating with Li2TiO3. Electrochim Acta 211:822
Liang LW, Hu GR, Jiang F, Cao YB (2016) Electrochemical behaviours of SiO2-coated LiNi0.8Co0.1Mn0.1O2 cathode materials by a novel modification method. J Alloys Compd 657:570
Li JG, Wang L, Zhang Q, He XM (2009) Synthesis and characterization of LiNi0.6Mn0.4-xCoxO2 as cathode materials for Li-ion batteries. J Power Sources 189:28
Moses AW, Garcia Flores HG, Kim JG, Langell MA (2007) Surface properties of LiCoO2, LiNiO2 and LiNi1-xCoxO2. Appl Surf Sci 253:4782
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21501015) and the Hunan Provincial Natural Science Foundation of China (No. 2016JJ3007).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Tang, Z., Zheng, H., Qian, F. et al. Improvement of cycling and thermal stability of LiNi0.8Mn0.1Co0.1O2 cathode material by secondly treating process. Ionics 24, 61–71 (2018). https://doi.org/10.1007/s11581-017-2179-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-017-2179-6