
Abstract
Polymer blend electrolytes based on poly(ethylene oxide) (PEO) and poly(vinylidene fluoride-hexafluoropropylene) (PVdF-HFP) were prepared by using different lithium salts LiX (X = ClO4, BF4, CF3SO3, and N [CF3SO2]2) using solution casting technique. To confirm the structure and complexation of the electrolyte films, the prepared electrolytes were subjected to X-ray diffraction (XRD) and Fourier transform infrared spectroscopy (FTIR) analysis. Alternating current (AC) impedance analysis was performed for all the electrolyte samples at various temperatures from 303 to 343 K. The result suggests that among the various lithium salts, LiN[CF3SO2]2-based electrolytes exhibited the highest ionic conductivity at 8.20 × 10−4 S/cm. The linear variation of the ionic conductivity of the polymer electrolytes with increasing temperature suggests the Arrhenius-type thermally activated process. Activation energies were found to decrease when doping with lithium imide salt. The dielectric behavior has been analyzed using dielectric permittivity (ε*), electric modulus (M*), and dissipation factor (tanδ) of the samples. Cyclic voltammetry has been performed for the electrolyte films to study their cyclability and reversibility. Thermogravimetric and differential thermal analysis (TG/DTA) was used to ascertain the thermal stability of the electrolytes, and the porous nature of the electrolytes was identified using scanning electron microscopy via ion hopping conduction. Surface morphology of the sample having maximum conductivity was studied by an atomic force microscope (AFM).
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lithium-ion battery quickly becomes dominant in the market of small electronic devices due to their advantages with respect to other types of batteries, such as being compact, light weight, high average discharge rate (∼3.7 V), and showing the highest energy density among all current systems. Comparing the energy densities of different secondary systems, the lithium ones currently present a specific energy density of 200 Wh/kg, which is about three times higher than the energy density of NiH battery and five times that of lead acid. Additional important advantages of Li-ion batteries in comparison with other battery systems are being more environmentally friendly and the absence of memory effects [1]. In lithium-ion battery systems, the separator plays a key role with respect to device performance as it prevents short circuits between the electrodes while allowing ionic conduction [2]. A solid polymer electrolyte (SPE) must meet certain requirements such as being an electronic insulator, ionic conductor, mechanically strong, dimensionally stable, readily wetted by the electrolyte, and chemically resistant to electrolyte and impurity degradation, among others [3]. Poly(ethylene oxide) (PEO)-based polymer electrolyte is of current interest for high energy density and high-power lithium-ion batteries due to their easy formation of complex with lithium salts, high mobility of charge carriers, and stable chemical properties [4]. In polymer electrolytes, metal salts are the main source of charge carriers. The salts mostly used are salts of alkali metals; these metal cations can coordinate with the oxygen atoms in polyether, resulting in their good salvation [5]. The salt solubility in polymer directly determines the number of charge carriers and their mobility, which has a significant effect on the ionic conductive behavior. Moreover, the addition of salt has a highly disturbing effect on the arrangement of the polymer chains and the ensuing conductivity. In order to realize a high lithium-ion conduction, (i) a polymer should have compatibility with inorganic salts and their dissociate ions, (ii) a polymer should provide a connected polar domain as the conduction path, and (iii) a polymer should not interact with carrier ions too strongly in order to avoid complete trapping of carrier ions [6]. In order to focus on the above parameters, the most common solvent-free polymer/salt complexes are PEO/salt electrolytes. Polyethylene oxide-based electrolytes are the first generation of solvent-free solid polymer electrolytes. PEO can complex with lithium salts (Li+) to form polymer electrolytes. Its ethylene oxide (EO) units have a high donor number for Li+ and high chain flexibility which are important for promoting ion transport [7]. PEO is a linear polymer of ethylene oxide with a high degree of crystallinity around 70∼80 % due to the regularity of the unit. PEO has a Tm around 65 °C and a Tg around 60 °C. The oxygen atom in the ether unit of PEO has a strong ability to coordinate effectively with the alkali metal cations, which makes it a good solvating medium for polymer electrolytes. The acceptable level of ionic conductivity of PEO electrolytes can only be achieved at temperatures above Tm, but the destruction of crystal at a high temperature leads to the liquid state of electrolytes which cannot function as a separator by itself. The approach to modify PEO is by forming a block copolymer, comb-branch copolymer, or cross-linked networks [8]. Blend-based electrolytes consist of blends of PEO and different classes of polymers. These polymers have, as a common characteristic, a high glass transition temperature. This allows structural stability at the molecular level, although the presence of such supports does not seem to have an incisive effect on the PEO crystallization degree. The polymers must have some well-defined requisites: (i) compatibility between them and the salt, (ii) interactions with the cation not too strong in order to avoid ion trapping, and (iii) both components should preferably contain polar domains [9]. The blend of the PEO and P(VdF-HFP) can hinder the crystalline of the PEO and achieve a good combination of high ionic conductivity. Hence, the poly(vinylidene fluoride-hexafluoropropylene) (PVdF-HFP) has drawn the attention due to its appealing properties that it comprises both amorphous and crystalline phases of the polymer that helps for higher ionic conduction. It has a high anodic stability due to strong electron withdrawing functional group and has a high dielectric constant of ɛ = 8.4 that helps for greater ionization of lithium salt. The unique polymer structure of carbon, hydrogen, and fluorine atoms found in poly(vinylidene fluoride) resins produces polymers that have extremely high thermal and chemical stability and yet retain their thermoplastic nature allowing ease of processing for fabrication [10]. One of the main advantages of fluorinated polymer is their ability to be tailored in different geometries including very thin cells [11]. The choice of the lithium salt is very important for PEO-based electrolytes. Some simple lithium salts, such as lithium chloride (LiCl), do not provide high ionic conductivity. Generally, the bulkier the anion of the lithium salt, the higher is the ionic conductivity. Therefore, an anion with a well delocalized negative charge and low basicity is preferred for improving the ionic conductivity and plays a dominant role in the salt-in-polymer electrolyte (PEO-LiX) [12]. To improve the ionic conductivity, an attempt has been made to identify the effects of different anions, namely ClO4 ¯, CF3SO3 ¯, BF4 ¯, and N(CF3SO2)2 ¯. Even though the commercial lithium salts LiX(X = ClO4, BF4, CF3SO3) are successful in portable lithium batteries, they still have some limitations. The electrolyte composed of LiClO4 presents high conductivity and good thermal stability, but chlorine in LiClO4 is in its highest valence state and gets strongly oxidized. LiClO4 can react strongly with organic solvent under extreme conditions (such as at elevated temperatures or high charging/discharging current density) [13]. LiBF4 had a low anodic oxidation potential and its electrochemical window was only about 3.5 V vs Li/Li+, which mean that it could not be used in a high-voltage battery. Thus, the search for an alternative salt for lithium batteries is necessary to maintain comprehensive and excellent performance. The most appropriate salts for polymer electrolytes are expected be those composed of a polarizing cation and a large anion of delocalized charge such as lithium super acid salts LiCF3SO3 ¯ and LiN(CF3SO2)2 ¯. Especially the imide family salts with large and flexible anions in which PEO polymer electrolyte with lithium bis(trifluoromethylsulfonyl)imide [LiN (SO2CF3)2] exhibited a high conductivity at room temperature. These salts exhibits low lattice energies in the solid state due to delocalization of the negative charge on the anion, which in turn results from the highly electron withdrawing character of trifluoromethyl substituents. It was found that these lithium imides slow down the recrystallization kinetics of polymer electrolytes [14]. In the present work, an attempt is made to investigate this new polymer blend electrolyte based on PEO (6.25)-PVdF-HFP (18.75) and the plasticizer PC (67) with different anions. The prepared electrolytes were subjected to various characterization procedures to optimize the anion that achieves the highest conductivity.
Materials and methods
The polymer poly(ethylene oxide) (PEO) of an average molecular weight Mw∼8000, poly(vinylidene fluoride-co-hexafluropropylene) (PVdF-co-HFP) of an average molecular weight Mw∼110,000, propylene carbonate (PC), and lithium salts LiClO4, LiBF4, LiCF3SO3, and [LiN (CF3SO2)2] were procured from Sigma-Aldrich chemicals limited, USA. The obtained PEO, PVdF-co-HFP, and lithium salts were dried at 55 °C for 4 h to remove moisture. PEO and PVdF-HFP with different lithium salts and PC-based electrolytes were prepared by solvent casting technique with acetone as a solvent. The polymers PEO and PVdF-HFP were dissolved at 40 °C and room temperature, respectively. After that, the polymer-salt complex was stirred well for 24 h with the help of a magnetic stirrer and degassed to remove air bubbles and poured on a well-cleaned petri dish. The solvent acetone was allowed to evaporate slowly at room temperature for 48 h. The free-standing films were obtained. The films are harvested and stored in highly evacuated desiccators to avoid the moisture absorptions. Flexible thin films with a thickness of about 0.22 mm were obtained. The prepared films were subjected to a.c. impedance analysis, in order to calculate the ionic conductivity. This was carried out with the help of stainless steel blocking electrodes using a computer-controlled microauto lab type III potentiostat/galvanostat in the frequency range of 100 Hz-300 kHz over the temperature range of 303–343 K. The amorphicity of the polymer electrolytes has been investigated by X-ray diffraction (XRD) analysis with the help of X’pert PRO PANanalytical X-ray diffractometer. The complex formation between the polymer and the salt has been confirmed by Fourier transform infrared spectroscopy (FTIR) spectra using SPECTRA RXI PerkinElmer spectrophotometer in the range of 400–4000 cm−1. To investigate the cyclability and reversibility of the electrolyte films, cyclic voltammetry and linear sweep voltammetry studies have been performed using computer-controlled microauto lab type III potentiostat/galvanostat. Thermal stability of the polymer electrolyte was carried out by thermogravimetric and differential thermal analysis (TG/DTA) by using PYRIS DIAMOND from room temperature to 500 °C with the scan rate of 10 °C min−1. The surface morphology of the electrolyte film was examined by Hitachi S3000H scanning electron microscope. Roughness parameter was observed by atomic force microscope (AFM) with the help of the atomic force microscope (A100SGS).
Results and discussion
X-ray diffraction analysis
XRD patterns of pure PEO, PVdF-HFP, LiClO4, LiBF4, LiCF3SO3, and LiN [CF3SO2]2 and prepared polymer electrolytes are shown in Fig. 1. Two broad peaks are found at 2θ = 19.2° and 23.15° which confirm the semicrystalline nature of PEO. The peaks at 17.5°, 18.5°, 20.2°, and 39° reveal the crystalline peaks of PVdF. This confirms the partial crystallization of PVdF units in the copolymer and gives a semicrystalline PVdF-HFP. The d values of the pure lithium salts have been compared well with JCPDS data and the hexagonal (LiClO4), monoclinic (LiCF3SO3), and orthorhombic (LiN [CF3SO2]2) [15]; orthorhombic (LiBF4) structure of lithium salts have also been confirmed [16]. The salt LiClO4 shows high intense peaks at angles 2θ = 20.9°, 22.92°, 26.56°, 32.75°, and 35.4° which reveals the crystalline nature of the ionic salt. Crystalline peaks of lithium triflate (LiCF3SO3) salt are located at 2θ = 16.5°, 19.75°, 20.4°, 22.55°, 25.3°, and 33.15°. LiBF4 salt peaks are observed at the diffraction angle of 14°, 21°, 23°, 26°, 28°, 32°, 39°, 44°, and 55°. The diffraction pattern of LiN[CF3SO2]2 shows intense peaks at 2θ = 13.6°, 15.9°, 18.9°, and 21.4° which reveals the crystalline nature of the salt.

XRD patterns of pure PEO, pure PVdF-HFP, pure LiClO4, pure LiBF4, pure LiCF3SO3, pure LiN[CF3(SO2)2], and the prepared electrolytes Pr1, Pr2, Pr3, and Pr4
The characteristic peaks of lithium salts in the prepared electrolytes were absent, which confirms the complete dissolution of the salt in the complex matrix, implying that the salt does not have any separate phase in the electrolytes. The addition of a plasticizer into the blend complex enhances the amorphous region, thus permitting the free flow of ions from one site to another; hence, the overall conductivity of the electrolyte has been significantly improved. The increase in the broadness of the peak reveals the amorphous nature of the complexed system. These results can be interpreted in terms of Hodge et al. [17] criterion which has established a correlation between the height of the peak and the degree of crystallinity. The increased broadness and decreased intensity has been found to be more for LiN [CF3SO2]2 doped PEO-PVdF-HFP polymer electrolyte systems compared to other prepared electrolytes. The order of increasing amorphous nature of the polymer electrolyte is LiN [CF3SO2]2 ¯ > BF4 ¯ > ClO4 ¯ > CF3SO3 ¯. In comparison, all the prepared electrolytes show an amorphous nature indicating that the lithium salt used in the polymer matrix is dissolved completely because all the characteristic peaks corresponding to the lithium salts are diminished in the complexes which LiN[CF3SO2]2 exhibits more amorphicity.
FTIR analysis
In order to understand the interactions between Li+ ions and the polymer hosts in the polymer electrolytes, the IR spectra of pure materials and polymer-salt-plasticizer samples are presented. On addition of salt into the polymer host, the cation of the salt is expected to coordinate with the polar groups in the host polymer matrix resulting in the complexation. This type of interaction will influence the local structure of the polymer backbone, and certain infrared active modes of vibration will be affected. In this context, the infrared spectroscopic studies will give the evidence of the complexation. Figure 2 shows the IR spectra of pure PEO, PVdF-HFP, LiClO4, LiBF4, LiCF3SO3, LiN [CF3SO2]2, and PEO/PVdF-HFP complexes with various lithium salts. FTIR transmission spectra of the complexes were recorded at room temperature in the region 4000–400 cm−1.

FTIR spectra of pure PEO, pure PVdF-HFP, pure LiClO4, pure LiBF4, pure LiCF3SO3, pure LiN[CF3(SO2)2], and the prepared electrolytes Pr1, Pr2, Pr3, and Pr4
A large broad band at 2962 cm−1 of PEO is shifted to (2967, 2969, 2968, 2956 cm−1). Two narrow bands of lower intensity at 2735 and 2695 cm−1 are inherent bands of asymmetric (CH) stretching vibrations of PEO is shifted to (2738, 2739, 2742, 2740 cm−1) and (2694, 2698,2701, 2703 cm−1), respectively. The bands at 2238, 2163, and 1967 cm−1 which are characteristic peaks of PEO [18], get shifted to (2240, 2242, 2242, and 2243 cm−1), (2172, 2175, 2179, 2180 cm−1) and (1975, 1972, 1969, 1966 cm−1), respectively. The band at 1483 cm−1 represents the C–H bending of CH2 in PEO shifted at (1485, 1484, 1488, 1472 cm−1). A very small intensity band at 1799 cm−1 corresponds to the ether oxygen group of PEO. The bands around 1359 and 1343 cm−1 are CH2 wagging and CH2 bending, which are characteristic peaks of PEO, get shifted at (1356, 1355, 1361, 1351 cm−1), (1340, 1339, 1340, 1333 cm−1). The relatively small band at 1236 cm−1 is assigned to CH2 symmetric twisting of PEO and these peaks are found at (1237, 1238, 1235,1230 cm−1). The characteristic vibrational band at 1100 cm−1 was assigned to C–O–C (symmetric and asymmetric) stretching of PEO and are found at (1105, 1099, 1091, 1093 cm−1). The two bands near 947 and 845 cm−1 are assigned to CH2 rocking vibrations of methylene groups and are related to the helical structure of PEO [19]. The most frequently performed observations to confirm the complexation of the polymer host with lithium salts are variation in intensity of the bands, shifting of the bands with respective to the various salts. The small ether oxygen band at 1799 cm−1 is found to smoothen gradually with the addition of various salts in PEO content, which suggests that PEO is disrupted by lithium salts. The 1200–1100 cm−1 region is the location of C–O–C stretching, which decreases its intensity with LiX (where X = ClO4, BF4, CF3SO3, and N[CF3SO2]2) content due to the interaction between the Li+ cations and ether oxygen atoms in PEO. The characteristic peak intensity corresponding to C = O (1651 cm−1) is decreased with the addition of different lithium salts. This indicates the strong interaction of Li+ ions with the carbonyl group [20]. PVdF-HFP contains free electron pairs at the fluorine (F) atoms of CF2 and CF3 groups. The vibrational peaks at 502 and 416 cm−1 are assigned to the bending and wagging vibrations of –CF2, respectively, and get shifted to (506, 504, 503, 512 cm−1) and (411, 422, 418, 414 cm−1). The crystalline phase of the PVdF-HFP polymer is identified by the vibrational bands at 985, 763, and 608 cm−1. Where 763 and 608 cm−1 get shifted to (778, 762, 771, 779 cm−1) and (615, 613, 614, 617 cm−1). The peaks at 1173 and 1390 cm−1 are assigned to the symmetrical stretching of –CF2 and –CH2 groups, respectively [21]. The peak at 881 cm−1 is assigned to the vinylidene group of the polymer and is shifted to (879, 883, 875, 881 cm−1).The peaks at 1483 and 1400 cm−1 are assigned to the CH3 asymmetric bending and C–O stretching vibrations for the plasticizer PC. The band at 625 cm−1 corresponds to the ClO4 − anion [22] which is present in the complex pr1. The vibration mode of free BF4 − anions and that corresponding to the ion pairs of BF4 − appearing at 765 cm−1 [23] in the complex pr2 is found at (753, 761, 773, 779 cm−1). In the region of 1350–1120 cm−1, a strong absorption peak is found at 1184 cm−1 which is attributed to vibrations involving the CF3 group in pure LiCF3SO3 and is shifted to 1183 cm−1 in the complex Pr3 [24]. The vibrational band at 768 cm−1 has been attributed to the triflate anion (CF3SO3)¯ which has shifted to 772 cm−1.The vibrational peak at 1355 cm−1 corresponds to the υ asym (SO2)− plane stretching vibration of SO2 in the imide anion [25] and is shifted in the complex Pr4. Shifting of the band assignments belonging to the anions in the IR region are mainly due to the ion coordination. Both the anion and the cation coordinate with the carbonyl oxygen and carbonyl carbon groups present in the polymer. Some peaks are disappeared and some new peaks are found in the prepared electrolytes. This confirms the complexation between PEO, PVdF-HFP, and lithium salts.
Ionic conductivity
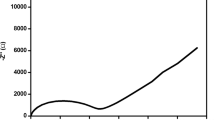
Impedance spectroscopy is employed to establish the conduction mechanism, observing the participation of the polymeric chain, mobility, and carrier generation processes. Using the measured bulk resistance, the conductivity of the polymer electrolyte is calculated using the relation σ = ℓ/R b A, where ℓ is the thickness of the film, A is the surface area of the film, and R b is the bulk resistance of the film. For PEO-based electrolytes, the EO units have a high donor number for Li+ and also high chain flexibility for promoting rapid ion transport. With the process of breaking/forming lithium-oxygen (Li-O) bonds, ion transport occurs by intrachain or interchain hooping in the PEO-based electrolyte accompanied by the gradual replacement of the ligands for the solvation of Li+; the continuous segmental rearrangement results in a long-range displacement of lithium ions [26]. Figure 3 shows the typical impedance plots of PEO-PVdF-HFP-LiX, where (X = ClO4 ¯, BF4 ¯, CF3SO3 ¯, and N [CF3SO2]2 ¯) based polymer electrolyte membranes with different lithium salts. Z′ and Z″ represent the real and imaginary part of the impedance data at room temperature, respectively. In this work, the semicircular portions corresponding to the high-frequency region are absent indicating that the majority of the current carriers in the electrolyte medium are ions and the total conductivity is mainly due to ion conduction [27]. The obtained spur corresponding to the lower frequency region is ascribed to double-layer capacitance in a cell with an ion blocking electrode configuration. The maximum room temperature ionic conductivity value of the order of 8.20 × 10−4 S/cm is obtained for the sample PEO/PVdF-HFP/LiN [CF3SO2]2. Table 1 shows that regardless of the polymer matrix, LiN [CF3SO2]2 has better conductivity when blended with PEO and PVdF-HFP. The blend of the PEO and PVdF-HFP can hinder the crystalline of the PEO and achieve a good combination of high ionic conductivity with LiN[CF3SO2]2 which exhibits great charge delocalization favorable to ionic dissociation in solvating polymers such as PEO. Besides, this present work has significant advantages such as higher ionic conductivity at low activation energy. The temperature-dependent ionic conductivity plots of the electrolytes containing different lithium salts are shown in Fig. 4.

Room temperature complex impedance plot of the prepared samples

Temperature-dependent ionic conductivity plot of the prepared samples
The conductivity values obtained for all the blend polymer electrolyte films in the temperature range 303–353 K are listed in Table 2. It is evident that among the different lithium salts studied, the film containing LiN[CF3SO2]2 offers maximum ionic conductivity. Among the various anion electrolyte systems, the [CF3SO2]2-based electrolyte exhibited the least bulk resistance. This feature may be due to the delocalization of the formal negative charge in the anion, the strong electron withdrawing nature, and the conjugation between the triflic groups and the lone electron pair on the nitrogen increasing the ionic mobility and dissociation of the imide salt. Increasing the temperature also increased the Li+ mobility due to the free volume enhancement of the electrolyte medium, which provides additional pathways and encourages ionic movement. Several factors affect the ionic conductivity of the polymer electrolyte, such as dissociation of ionic salts, cation radii, ionic mobility, types of (cation and anion) charge carriers, and temperature [40]. As the temperature rises, the conductivity growths for all the polymer electrolytes increases (Fig. 4) due to an increase of free volume in the polymer and an increase of ion and segmental mobility that will assist ion transport and virtually compensate for the retarding effect of ion clouds. The temperature dependence of the ionic conductivity is linear, which indicates that ion conduction follows the Arrhenius-type thermally activated process given by the relation, σ = σ o exp (−E a/KT) where σ o is the pre-exponential factor, E a the activation energy, and K the Boltzmann constant. That is, ion transport in a polymer electrolyte is correlated with polymer segmental motion. The ionic conductivity is closely coupled to the segmental motions of the polymer chains and local flexibility, i.e., in the electrolytes, the ions are coordinated to a macromolecule. It can translate within the system only in conjunction with the segmental motion of the polymer chain. It seems useful to consider the behavior of macromolecular species, in which ions are irreversibly bound to a polymer chain. Thus, the cluster formation is shown to occur when a group of interacting polymer segments move from a random distribution throughout the system [19].
As the amorphous region increases, however, the polymer chain acquires faster internal modes in which bond rotations produce segmental motion to favor inter-intrachain ion hopping, and thus, the degree of conductivity becomes high. Among the various anionic-based electrolytes, the PEO/PVdF-HFP/LiN [CF3SO2]2 electrolyte membrane exhibits the highest ionic conductivity (8.20X10−4 S/cm at room temperature). Because all the low molecular anions are counter ions of strong acids, the difference in conductivity is presumably due to the difference in lattice energies. The maximum ionic conductivity of the sample Pr4 may be due to the two different ion solvating species of the salt LiN [CF3SO2]2, which leads to a long-range columbic force producing more free ions. Moreover, it is well established that imide anions can afford a higher degree of negative charge dispersion than other lithium salts in a given salt concentration [41]. Activation energies were found to decrease when doping with lithium imide salt is found to be low of the order of 0.19 eV. In addition, the imide salt can act as a plasticizer at an elevated temperature, further influencing the degree of dissociation; hence, the conductivity of the sample will be higher than that of the other samples. The conductivity has also been significantly improved due to the addition of a plasticizer because it further improves the dissolution of the lithium salt [42].
Dielectric spectra analysis
The dielectric relaxation behavior of the polymer electrolyte brings about important insights into ionic transport phenomenon. The measured impedance data were used to calculate the real and imaginary parts of the complex permittivity using the relation,
where real ɛ '(ω) and imaginary ɛ ” (ω) components are the storage and loss of energy in each cycle of the applied electric field, respectively. Figure 5 represents the frequency dependence of ɛ '(ω) and ɛ ” (ω) for LiN [CF3SO2]2 at different temperatures. From Fig. 5, it is clear that the values of ɛ '(ω) are very high at a low-frequency region. Such high value of dielectric permittivity at low frequencies has been explained by the presence of space charge effects, which is contributed by the accumulation of charge carriers near the electrodes [43].

Frequency-dependent dielectric analysis of the composition PEO/PVdF-HFP/LiN[CF3(SO2)2] at different temperatures
At higher frequencies, ɛ '(ω) has been found to be relatively constant with frequency. This is because periodic reversal of the field takes place so rapidly that the charge carriers will hardly be able to orient themselves in the field direction resulting in a decrease of dielectric constant. The large value of ɛ ” (ω) is also due to the motion of free charge carrier within the material. The variation of ɛ ” (ω) with log ω clearly indicates the β-relaxation due to some local movement of side group dipoles [44]. Increase in the value of dielectric constant and dielectric loss can be observed at higher temperatures and is attributed to the higher charge carrier density. As temperature increases, the degree of salt dissociation and redissociation of ion aggregates, resulting in the increase in the number of free ions or charge carrier density.
Modulus spectra analysis
Figure 6 depicts the frequency dependence of M'(ω) and M″(ω) for LiN[CF3SO2]2-doped PEO/PVdF-HFP at different temperatures. Both plots show an increase at the high-frequency end, but a well-defined dispersion peaks are not observed. This increasing trend in the plot at higher frequencies may be attributed to the bulk effect. With the raise of temperature, the height of the peak decreases suggesting a plurality of relaxation mechanism [45]. At lower frequencies, it is observed that the value of M″(ω) is in the vicinity of zero, indicating that the contribution of electrode polarization is negligible. The presence of this long tail at the low-frequency region also provides evidences of the large capacitance associated with the electrodes.

Frequency-dependent modulus analysis of the composition PEO/PVdF-HFP/LiN[CF3(SO2)2] at different temperatures
Loss tangent spectra
The dielectric loss tangent (tanδ) can be defined by the equation
Tan δ= ɛ ″ /ɛ '
The relaxation parameters of the polymer electrolytes can be obtained from the study of tan δ as a function of frequency. The variation of tan δ with frequency for the LiN [CF3SO2]2-doped PEO/PVdF-HFP at different temperatures is shown in Fig. 7.

Frequency-dependent loss tangent analysis of the composition PEO/PVdF-HFP/LiN[CF3(SO2)2]2 at different temperatures
It has been observed that the tan δ increases with increasing frequency and reaches a maximum. Then it decreases for a further increase of frequency. For maximum dielectric loss at a particular temperature, the absorption peak is described by the relation ωτ = 1 where τ is the relaxation time and ω is the frequency of the applied signal. The relaxation parameter for PEO/PVdF-HFP/LiN [CF3SO2]2 at various temperatures are tabulated in Table 2. As the temperature is increased, the frequency at which tan (δ max) occurred shifted to higher frequencies. This behavior suggests that the system can be represented by a parallel RC element [46].
Thermal analysis
Thermogravimetric analysis is very essential to study the thermal stability of polymeric systems under application conditions. During the cell reactions, heat is known to get generated in the cell which can melt or degrade the polymer electrolyte within cell and cause internal short circuits. Hence, thermal stability of a polymer electrolyte is vital for a safe and endurable electrochemical cell. TG/DTA has been used to measure the diffusion characteristics and the moisture uptake. Also, it is used to investigate the thermal degradation, phase transitions, and crystallization of polymers.
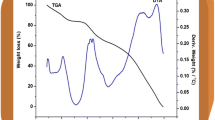
The thermal stability of the prepared samples was studied by TGA and depicted in Fig. 8 (pr1–pr4). The TGA curves indicate that the films containing different lithium salts exhibit weight losses of approximately, 12, 8, 9, and 10 %, respectively, at 100 °C. This weight loss is mainly due to the presence of moisture, which may be acquired during the loading of the sample. It is noted that all the prepared samples show appreciable weight losses of approximately 23, 20, 19, and 10 %, respectively, at 150 °C. The weight loss observed in the temperature range 200–300 °C is primarily due to the degradation of propylene carbonate (PC) because of the boiling point of the PC (242 °C). It may also be due to the blended polymer degradation. Among the different lithium salt-based electrolyte systems, LiN [CF3SO2]2 exhibited the highest thermal stability of approximately 375 °C with weight loss of 7 %. This weight loss is found to be minimum among the other compositions studied which indicates the maximum stability of the imide group-based salt. Presence of two strong electrons withdrawing [CF3SO2]2 − groups of the anion which provides charge delocalization; thus have better interactions with the polymer chains and plasticizers than the other salts which are used in the present investigation. In Fig. 8 the DTA curves of all the samples show first exothermic peaks at 126, 128,136, and 90 and second exothermic peaks at 290, 292, 398, and 399 indicating the decomposition of the electrolytes as evidence by rapid weight loss observed in the TG curve. It is concluded that the addition of imide salt effectively increased the thermal stability of the electrolyte. The nitrogen group present in the imide salt also improved the interaction with the polymer chains; among the different salts, LiN [CF3SO2]2 has the strong interactions since the cleavage of the C–S bond occurs first which could be ascertained by the detected –CF3 group. The –CF3 group of the salt remains unchanged on the surface after heat treatment and withstand up to a high temperature. Furthermore, the interactions between the split products of the LiN [CF3SO2]2 and PC content generate heat and the system reaches the maximum temperature. Hence, the polymer electrolyte based on the LiN [CF3SO2]2 salt showed better thermal stability than the other systems. This result is in close agreement with the literatures [47, 48]. The weight loss observed in the TGA curves is also confirmed from the DTA curves by the presence of endothermic peaks in all the four samples and the DTA results, which were in close agreement with the TGA results.

TG/DTA analysis of the prepared complexes Pr1, Pr2, Pr3, and Pr4
Cyclic voltammetry
Figure 9 shows the electrochemical stability of the blended films containing various salt contents. The cyclic voltammetric studies have been carried out to estimate the electrochemical stability window (ESW) or working voltage limit of the polymer electrolytes. The voltammograms recorded on the cells comprising the electrolytes sandwiched between two symmetrical stainless steel electrode/polymer electrolyte/stainless steel cell coupled with a scan rate of 5 mV/s.

Cyclic voltammograms of the prepared electrolytes: a Pr1, b Pr2, c Pr3, and d Pr4 using stainless steel cell couple as working electrode
A substantial value of about 2.5 V was optimized from an electrochemical stability point of view when imide salt was added to the polymer electrolyte. Such enhanced value of EWS was thought to be sufficient for many electrochemical applications. Several small peaks are observed, which are ascribed to reduction of the low levels of water present in polymer electrolyte or to oxygen impurities. This behavior has been observed for other systems based on PEO blends with lithium salt as well [49].
Electrochemical window
Linear sweep voltammetry curves of the polymer blend electrolytes have been performed for the SS/polymer electrolyte/SS cell couple and are shown in Fig. 10 which are obtained from cathodic and anodic scan rate of 5 mV/s at 30 °C. The polymer electrolyte incorporated with LiN [CF3SO2]2 displays a stable window of −3 to 2.5 V. The anodic limiting current at −3 V indicates the decomposition of the cations and the deposition of the lithium. When compared to other alkali metal systems, the polymer blend electrolyte with LiN [CF3SO2]2 considered in this study has sufficient electrochemical stability to act as the electrolyte material in lithium cells.

Linear sweep voltammetry of the prepared electrolytes: a Pr1, b Pr2, c Pr3, and d Pr4
AFM analysis
The pore size and the root mean square (RMS) value are found from the AFM analysis. The roughness of the prepared sample surface area of 10 μm × 10 μm was also measured. The sample having a maximum ionic conductivity is subjected to atomic force microscopic studies with a scan rate of 1 Hz, and its two- and three-dimensional topographic images are shown in Fig. 10.
It is observed that the width of the chain segments is of the order of 256 nm. The polymer blend electrolyte, PEO/PVdF-HFP/PC/[LiN(CF3SO2)2], shows a clear mountain valley pattern with an increased surface roughness value as 84 nm. The rough surface with numerous humps is due to the monoclinic structure of the salt [LiN(CF3SO2)2]. This confirmed the enhancement of amorphicity of the system that supported the ionic transport. It is observed that the root mean square roughness (RMS) of the sample within the scanned area is found to be 96 nm. The presence of pores is due to solvent removal and by solvent retention ability of the electrolyte system. The topographic images show the pores that help in ensnaring the large volume of the liquid which accounts for the increasing conductivity.
Scanning electron microscope
Comparison of SEM micrographs of PEO/PVdF-HFP blend electrolyte systems with different lithium salts provided the information about the effect of salts on the blend morphology. In particular, the effects of the different types of anion on their miscibility and crystalline morphology were investigated. The PEO/PVdF-HFP/PC/[LiN (CF3SO2)2] complex shows an entirely different surface structure when compared to ClO4 ¯, BF4 ¯, CF3SO3 ¯. When adding the imide salt, dendrite formation occurred, improving the ion conducting pathway between the sites. In addition, the porous structure also improves the ion hopping conduction; hence, the total ionic conductivity is enhanced. The surface of the electrolytes show fine pores in the polymer matrix, which aid in ion mobility. It is well known that the formation of the porous structure is a complex process that depends on the interaction of the solvent with the polymers and is kinetically controlled by the relative rates of evaporation of compounds. The difference in the pore size is related to the difference in the driving force for phase separation. After adding [LiN (CF3SO2)2], the interaction between PEO and PVdF-HFP is appreciable. This interaction produces a more relaxed network in the matrix, and the structure becomes increasingly homogenous (Fig. 11). It is noted that there is no apparent interface between the two polymers, which indicates that PEO and PVdF-HFP have good compatibility (Fig. 12).

Topography image of the sample (Pr4) having maximum conductivity: a 2D image, b 3D image

SEM micrographs of the cross-sections of the polymer electrolytes: a Pr1, b Pr2, c Pr3, d Pr4
Conclusion
A new polymer electrolyte comprising poly(vinylidene fluoride-co-hexafluoropropylene)/poly(ethylene oxide) complexed with different lithium salts has been prepared for Li-ion battery applications. The prepared polymer electrolytes are free standing and flexible with excellent dimensional stability. Complexation of the polymer matrices has been ascertained by XRD studies and indicates the crystalline nature of the polymer films, showing a decreasing trend of crystallinity due to the addition of imide salt, and FTIR analysis confirms the ion-polymer interaction. The highest ionic conductivity was observed for imide salt at a concentration of 8 wt% for PEO/PVdF-HFP-PC complex (Table 3). The temperature-dependent ionic conductivity shows the Arrhenius behavior, which indicates that the ions transport together with the kinetic movement of polymer chains. Excellent thermal stability has been observed up to 375 °C due to the incorporation of the imide salt. Cyclability and reversibility of the films have been corroborated using cyclic voltammetry. SEM images of the sample showed phase-separated morphology and increase in porosity accounting for increase in conductivity. Electron micrographs showed uniform distribution of one polymer in the other which confirms the compatibility of the blend components. Attainment of smooth surface morphology also suggests the enhancement of degree of amorphicity. Two- and three-dimensional topographic images of the best conducting sample were studied by AFM to observe the roughness parameter of the blended electrolytes. The present imide salt added polymer electrolyte appears to be an excellent material for its use as electrolyte in energy conversion/storage devices.
References
Nunes-Pereira J et al. (2015) J. Power Sources 281:378
Angulakshmi N, Manuel Stephan M (2014) Electrochim Acta 127:167
Gong C et al. (2014) J power sources 246:260
Starkey SR, French R (1997) Electrochim Acta 42:471
Rand DAJ (1979) J Power Sources 4:101
Joykumarsingh I, Bhat SV (2004) J Power Sources 129:280
Johansson P (2001) Polymer 42:4367
Munshi MZA, Owens BB (1988) Solid State Ionics 26:41
Gilies JRM et al. (1987) Polymer 28:1977
Malik P et al. (2006) Polym Degrad Stabil 91:634
Carols M, Costa A, et al. (2013) RSC Adv 3:11404
Xue Z et al. (2015) J Mater Chem A 3:19218
Pradeepa P et al. (2015) Chinese Chem Lett 26:1191
Liu Z et al. (2015) Coordin Chem Rev 292:56
Rajendran S et al. (2004) Solid State Ionics 167:335
Nowinski JL et al. (1994) J Mater Chem 4(10):1579
Hodge RM et al. (1996) Polymer 37:1371
Papke BL et al. (1982) J Electrochem Soc 129:1434
Kesavan K et al. (2014) Polymer Science Ser B 56:520
Ulaganathan M et al. (2011) J Mater Chem Phys 129:471
Stephan AM et al. (1999) J Power Sources 81:752
Pradeepa P, Ramesh Prabhu M (2015) Int J Chem Tech Res 7(4):2077
Jacob MME, Arof AK (2000) Electrochim Acta 45:1701
Chintapalli S, French R (1998) Electrochim Acta 43:1395
Roy I et al. (1998) J Phys Chem A 102:3249
Xu K (2004) Chem Rev 104:4303
Capiglia C et al. (2000) Electrochim Acta 45:1341
Xu JJ, Ye H (2005) Electrochem Commun 7:829
Niitani T et al. (2009) J Electrochem Soc 156:A577
Ali AMM et al. (2007) Mater Lett 61:2026
Abraham KM, Alamgir M (1993) J Power Sources 43:195
Quartarone E et al. (1998) Solid State Ionics 110:14
Jiang G et al. (2005) J Power Sources 141:143
Fan LZ et al. (2007) Adv Funct Mater 17:2800
Saito Y et al. (2002) J Phys Chem B 106:7200
Lalia B et al. (2013) J Solid State Electrochem 17:575
Li M et al. (2011) J Power Sources 196:3355
Fan L et al. (2002) Electrochim Acta 48:205
Costa CM (2014) J Power Sources 245:779
Zhu W et al. (2001) J Polym Sci Part B 39:1246
Immanual Selvaraj I et al. (1995) J Electrochem Soc:142
Stephan AM et al. (2002) Solid State Ionics 148:467
Armstrong RD, Race WP (1976) J Electroanal Chem 74:125
Singh KP, Gupta PN (1998) Eur Polym J 34:1023
Suzhu YU et al. (2000) J Appl Physics 88:398
Simmons JG et al. (1970) J Appl Physics 41:538
Ping P et al. (2010) C Chem J Electrochem Soc 11:157
Lu Z et al. (2006) J Power Sources 156:555
Aurbach D et al. (1991) J Electroanal Chem Interfacial Electrochem 297:225
Acknowledgments
The author P. Pradeepa gratefully acknowledges the UGC-BSR, New Delhi, India, for the financial support to carry out this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Prabakaran, P., Manimuthu, R.P. Enhancement of the electrochemical properties with the effect of alkali metal systems on PEO/PVdF-HFP complex polymer electrolytes. Ionics 22, 827–839 (2016). https://doi.org/10.1007/s11581-015-1618-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-015-1618-5