
Abstract
The layered Li1.2Mn0.54Ni0.13Co0.13O2 lithium-rich manganese-based solid solution cathode material has been synthesized by a simple solid-state method. The as-prepared material has a typical layered structure with R-3m and C2/m space group. The synthesized Li1.2Mn0.54Ni0.13Co0.13O2 has an irregular shape with the size range from 200 to 500 nm, and the primary particle of Li1.2Mn0.54Ni0.13Co0.13O2 has regular sphere morphology with a diameter of 320 nm. Electrochemical performances also have been investigated. The results show that the cathode material Li1.2Mn0.54Ni0.13Co0.13O2 prepared at 900 °C for 12 h has a good electrochemical performance, which can deliver a high initial discharge capacity of 233.5, 214.2, 199.3, and 168.1 mAh g−1 at 0.1, 0.2, 0.5, and 1 C, respectively. After 50 cycles, the capacity retains 178.0, 166.3, 162.1, and 155.9 mAh g−1 at 0.1, 0.2, 0.5, and 1 C, respectively. The results indicate that the simple method has a great potential in synthesizing manganese-based cathode materials for Li-ion batteries.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recently, lithium-ion batteries have been considered as the dominant power sources for large-scale applications, such as electric vehicles (EV) and hybrid electric vehicles (HEV) [1–3]. While conventional cathode materials, such as olivine LiFePO4 [4], spinel LiMn2O4 [5], and layered LiMO2 (M = Ni, Co, Mn, etc.) [6–8], cannot meet the increasing demands for high-energy and high-power batteries due to limited theoretical capacity. Searching for high-capacity and safe alternatives has become one of the most important subjects. The material system based on the layered-layered compositions of xLi2MnO3 · (1−x)LiMO2 (M = Ni, Mn, etc.) has recently attracted more and more attention due to low cost, environmental friendliness, and higher capacity [9–11]. Lithium-excess layered oxides xLi2MnO3 · (1−x)LiMO2 (M = Ni, Mn, etc.) are composed of the intergrowth of LiMO2 (R3m) and Li2MnO3 (C2/m) phases and can deliver higher capacity (>250 mA h g−1) with relatively high operating voltage (3.5 V versus Li/Li + on average). However, there are several disadvantages such as high first cycle irreversible capacity and inherently low rate capability, which limit its application. Thus, various strategies have been used to improve its electrochemical performances, most of which involves reducing particle size [12, 13] or coating with other materials [14–16] and doping with different elements [10, 17, 18]. Compared with doping and coating methods, reducing particle size is an effective way to improve the properties of as-prepared materials without degrading the intrinsic properties [19]. Meanwhile, synthesis method has significant effects on the morphology and particle size of materials [19, 20]. Solid-state approach is considered to be one of the simplest methods, which has been applied widely in the synthesis of electrode materials [21–24].
During conventional synthesis of Li2MnO3-LiMO2, the raw materials usually have micrometer scale with irregular morphology and mixed by grinding or ball-milling methods. After high-temperature sintering process, the resultant products are usually composed of irregular nano- or microparticles due to undesirable particle growth, although the undesirable particle growth can be partly inhibited by adding growth inhibitors [25, 26]. However, morphology and size control of Li2MnO3-LiMO2 nanostructures remain a great challenge. Herein, we present a simple and feasible synthesis of Li1.2Mn0.54Ni0.13Co0.13O2 microspheres with nano-sized subunits by a precipitation method followed by a simple solid-state reaction. As the distance for atomic migration is shortened significantly to several nanometers, the undesired particle growth during the annealing is effectively suppressed in the present synthesis. The resultant Li1.2Mn0.54Ni0.13Co0.13O2 exhibit a reasonable discharge capacity and fairly good cycling stability.
Experiments and characterization
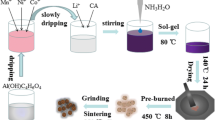
Preparation of MnCO3 and MnO2 microspheres
The MnCO3 microspheres were prepared by a precipitation method. 7.5 mmol of MnSO4·H2O (A.R., Guangdong Guanghua Sci-Tech Co., Ltd.), and 75 mmol of NH4HCO3 (A.R., Guangdong Guanghua Sci-Tech Co., Ltd.) was separately dissolved in 500 mL of distilled water. Fifty milliliters of ethanol and the NH4HCO3 solution was then added to the MnSO4·H2O solution in sequence under stirring. The mixture was kept under stirring for 1 h at room temperature, then centrifuged, and washed with water for several times. The as-obtained MnCO3 microspheres were dried at 80 °C. The Mn2O3 microspheres were synthesized by thermal decomposition of the MnCO3 microspheres at 400 °C for 5 h at air atmosphere.
Preparation of Li1.2Mn0.54Ni0.13Co0.13O2
To obtain Li1.2Mn0.54Ni0.13Co0.13O2 microspheres, stoichiometric amounts of Mn2O3 microspheres, Ni(NO3)2·6H2O (A.R., Guangdong Guanghua Sci-Tech Co., Ltd.), LiOH·H2O (A.R., Guangdong Guanghua Sci-Tech Co., Ltd.), and Co(NO3)2·6H2O (A.R., Guangdong Guanghua Sci-Tech Co., Ltd.) were mixed and ball-milled for 4 h at a speed of 300 rpm. The Li1.2Mn0.54Ni0.13Co0.13O2 sample was formed by heating the milled precursors at 900 °C for different times in air atmosphere in a muffle furnace.
Material characterization
The crystal structures of the Li1.2Mn0.54Ni0.13Co0.13O2 samples were characterized employing an X-ray diffractometer (Rigaku RINT2000) with Cu Ka radiation and recorded between 10 and 80° with a scanning speed of 3° min−1. The morphologies of the precursors and as-prepared materials were observed using scanning electron microscope (SEM) (Hitachi S4700). Transmission electron microscopy studies of the samples were conducted using a JEM-2100F transmission electron microscope operated at 200 kV.
The electrochemical tests were performed with CR2032 coin-type cells. To fabricate the cathodes, 80 wt% active material, 10 wt% Super P, and 10 wt% polyvinylidene difluoride (PVDF) binder in N-methylpyrrolidone (NMP) solution were mixed homogeneously. The resulting slurry was coated on aluminum foil, dried at 120 °C, and pressed with 10 MPa. The resulting cathodes had an active material loading of about 2.5∼2.7 mg cm−2. The cells were assembled in an argon-filled box with the moisture content and oxygen levels less than 5 ppm. A metallic lithium foil served as counter and reference electrodes. The electrolyte was 1 M LiPF6 in a mixture of ethylene carbonate and dimethyl carbonate (1:1 in volume). Galvanostatic charge and discharge tests were performed between 2.0 and 4.8 V at room temperature on a battery test system (Neware BTS-5V5mA, China). Cyclic voltammetry tests were carried out at a scanning rate of 0.1 mV s−1 between 2.0 and 4.8 V on an electrochemical workstation (ChenHua CHI1040B, China). The electrochemical impedance spectroscopy (EIS) analysis of the fresh cell with the open circuit voltage of the cell about 3.0 V was carried out by applying an AC voltage of 5 mV over the frequency range from 100 kHz to 10 MHz on an electrochemical workstation (Gamery PC14-750, USA). All electrical measurements have been performed at room temperature.
Results and discussion
Figure 1 shows the XRD patterns of the synthesized microsphere MnCO3 and Mn2O3 precursor. As shown in Fig. 1a, the diffraction peaks of the precursor MnCO3 are matched well with MnCO3 (JCPDF No. 44-1472) and no impurity is detected, which indicates the MnCO3 microspheres were prepared by a precipitation method. As shown in Fig. 1b, the diffraction peaks of the precursor Mn2O3 are matched well with Mn2O3 (JCPDF No. 71-0636), which indicates that the Mn2O3 microspheres were synthesized by thermal decomposition of the MnCO3 microsphere at 400 °C for 5 h. Thus, the MnCO3 microspheres are converted into Mn2O3 by thermal decomposition at 400 °C according to 4MnCO3 + O2→2Mn2O3 + 4CO2. Besides, Fig. 1b shows that a little impurity phases are indexed as Mn3O4 (JCPDF No. 75-0765), which responds with the side reaction 6MnCO3 + 3O2→3Mn3O4 + 6CO2.

XRD patterns of the MnCO3 (a) and Mn2O3 (b) and cathode material Li1.2Mn0.54Ni0.13Co0.13O2 synthesized at 900° C for different time
In Fig. 2, the SEM images illustrate the morphology of the precursor MnCO3 and Mn2O3. As shown in Fig. 2a, c, the precursors MnCO3 exhibited spherical morphology with the size of about 3–6 μm. It is clearly observed from Fig. 2c that the spherical MnCO3 particles have a rough surface and are composed of well-crystalized nanoparticles. Figure 2b, d show that precursor Mn2O3 obtained by thermal decomposition of spherical MnCO3 particles retains the microsphere structure with the size of about 6–7 μm. Thus, the use of the pre-grown MnCO3 as the precursor allows for the shape control of the resultant Li1.2Mn0.54Ni0.13Co0.13O2.

SEM images of the precursors MnCO3 (a, c) and Mn2O3 (b, d)
Figure 3 shows the diffraction peaks of Li1.2Mn0.54Ni0.13Co0.13O2 synthesized at 900 °C for various times. All the diffraction peaks are indexed by two-phase system consisting of rhombohedral LiMO2 (R-3m, a-NaFeO2 structure) and layered monoclinic Li2MnO3 (C2/m). The results were in good agreement with the previous reports [27]. The reflection peaks between 20° and 25° originated from the ordering of lithium ions with transition metal ions in the transition metal layers, corresponding to Li2MnO3 phase. Both the (006)/(102) and (108)/(110) doublets are clearly separated, indicating that the well-crystallized layered structures have been formed.

XRD patterns and Rietveld refinement of cathode material Li1.2Mn0.54Ni0.13Co0.13O2 synthesized at 900° C for different time
The XRD patterns have been refined by the Rietveld method with General Structure Analysis Software (GSAS Los Alamos National Laboratory, USA) using the approach outlined in Ref. [28]. The refined lattice parameters are listed in Table 1. Because the crystallographic sites of Li1.2Mn0.54Ni0.13Co0.13O2 are shared by more than two cations, it is hard to obtain reliable results on atomic occupancies from the Rietveld refinement. As shown in Ref. [28], the XRD diffraction patterns are refined by the sets of diffraction data: aNaFeO2 type with R-3m and monoclinic Li2MnO3 type with C2/m. As listed in Table 1, the cell parameters firstly increase from t = 8 h and then reach the maximum till t = 12 h and finally decrease with the increasing of calcination time. The increasing lattice parameters can enhance lithium-ion migration in the crystal lattice, which improve its electrochemical performances. These results indicate that the sample synthesized at 900 °C for 12 h has a better electrochemical properties.
In Fig. 4, the SEM images illustrate the morphology of cathode material Li1.2Mn0.54Ni0.13Co0.13O2. As shown in Fig. 4, the as-prepared Li1.2Mn0.54Ni0.13Co0.13O2 is composed of well-crystallized particles with size range from 200 to 500 nm. These nanoparticles have irregular shape. As the calcining time increases, the particles grow up and aggregate more seriously. In order to investigate the detailed morphology, transmission electron microscopy studies on the material synthesized at 900 °C for 12 h are shown in Fig. 5. As shown in Fig. 5a, the micrograph shows that the primary particle of Li1.2Mn0.54Ni0.13Co0.13O2 has regular sphere morphology with a diameter of 320 nm. The Fig. 5b shows that the particles are high crystalline, and the lattice fringes are found to be about 0.204 nm, corresponding to the d104 and d202 spacing in the XRD patterns of rhombohedral LiMO2 (R-3m) and layered monoclinic Li2MnO3 (C2/m), respectively. Furthermore, as shown in the Fig. 5c, the Fourier transform images exhibit spot patterns suggesting highly crystalline structures, which are in accordance with XRD results.

SEM images of cathode material Li1.2Mn0.54Ni0.13Co0.13O2 synthesized at 900° C for different time: 8 h (a, e), 10 h (b, f), 12 h (c, g), and 14 h (d, h)

TEM (a), HRTEM (b), and Fourier transform (c) images of cathode material Li1.2Mn0.54Ni0.13Co0.13O2 synthesized at 900° C for 12 h
Figure 6 exhibits the charge-discharge curves of the Li1.2Mn0.54Ni0.13Co0.13O2 synthesized at 900 °C for various times operated at 0.1 C (1 C = 180 mA g−1). Similar charge-discharge profiles can be observed in Fig. 6. All the initial charge curves clearly displayed two plateaus located at 3.7–4.5 V and above 4.5 V. The first plateau is ascribed to lithium ion extraction from the layered LiCoO2 and LiNiO2 structure [28]. The second plateau could be attributed to the oxygen loss process from the Li2MnO3 component, which results in high irreversible capacity of Li1.2Mn0.54Ni0.13Co0.13O2 electrode [28]. The initial discharge capacities are 181.4, 213.5, 233.6, and 229.4 mAh g−1 for 8, 10, 12, and 14 h, respectively. After 10 cycles, the discharge capacities retain 143.3, 167.4, 214.3, 189.8 mAh g−1, respectively. The capacity retentions are 78.4, 79.2, 91.7, and 82.7 %, respectively. The results indicate the sample synthesized at 900 °C for 12 h has the best electrochemical performances.

The charge/discharge curves of cathode material Li1.2Mn0.54Ni0.13Co0.13O2 synthesized at 900° C for different time. The charge rate for fixed at a current of 0.1 C
To further investigate the electrochemical behaviors of the Li1.2Mn0.54Ni0.13Co0.13O2 synthesized at 900 °C for various time, cyclic voltammetry (CV) was conducted on the electrode at the scan rate of 0.1 mV s−1 between 2.0 and 4.8 V and the results are shown in Fig. 7. The two oxidation peaks at 4.06 and 4.75 V in the first cycle corresponded to the oxidation of the Ni2+ and Co3+ ions and the release of oxygen, respectively. The reduction peaks at 4.45 and 3.79 V in the first cycle could be ascribed to the reduction of Ni4+ and Co4+ ions. In the second and third cycles, the CV profiles are significantly different from that observed in the first cycle. A new weak oxidation peak emerges at 4.6 V with the disappearance of the strong oxidation peak at 4.7 V. The two oxidation peaks at 3.9 and 4.6 V could be attributed to the oxidation of Ni2+ and Co3+, respectively. The negative shift of Mn4+/Mn3+ reduction peak with cycles evidenced the structure transformation from layered to a layered-spinel intergrowth structure [29].

Cyclic voltammetry curves of cathode material Li1.2Mn0.54Ni0.13Co0.13O2 synthesized at 900° C for different time
Furthermore, electrochemical impedance spectroscopy (EIS) is used to analyze the electrochemical properties of Li1.2Mn0.54Ni0.13Co0.13O2 synthesized at 900 °C for various time. As shown in Fig. 8a, the Nyquist plots of the cells consist of barely visible semicircles alike in appearance of a half ellipse (in the high frequency ranges) and a straight line with changing slope to the real axes (in the lower frequency range). The study of the EIS results has been performed by using the approach outlined in Ref. [19, 30, 31] by using an equivalent circuit model shown in Fig. 8b. The fitting results derived from the equivalent circuit are presented in Table 2. Generally, an intercept at the Zreal axis at a high frequency corresponds to the ohmic resistance (Rs), which represents the total resistance of the electrolyte, separator, and electrical contacts. The depressed semicircle in the high frequency range indicates the charge transfer resistance (Rct). The inclined line in the lower frequency range represents the Warburg impedance and corresponds to the lithium diffusion kinetics towards the electrodes. As shown in Table 2, the electrolyte resistance remained almost constant, which was expected since the variation of an electrolyte concentration was not so large as to affect the electrolyte conductivity. The transfer resistance (Rct) of the sample synthesized at 12 h is smaller than the other ones, indicating that the Li1.2Mn0.54Ni0.13Co0.13O2 synthesized at 900 °C 12 h has better electrochemical performances. These results are in accordance with above results.

Nyquist plots (a) and the equivalent electric circuit (b) of cathode material Li1.2Mn0.54Ni0.13Co0.13O2 synthesized at 900° C for different time
The initial charge-discharge curve and cyclic performance of the sample prepared at 900 °C for 12 h at the rate of 0.1, 0.2, 0.5, and 1 C for 50 cycles are presented in Fig. 9a, b, respectively. As shown in Fig. 9a, the sample has the similar shape and delivers discharge capacities of 233.5, 214.2, 199.3, and 168.1 mAh g−1 at 0.1, 0.2, 0.5, and 1 C, respectively. After 50 cycles, the capacity retains 178.0, 166.3, 162.1, and 155.9 mAh g−1 at 0.1, 0.2, 0.5, and 1 C, respectively. The capacity retentions are 76.2, 77.6, 81.3, and 92.7 %, respectively. These results indicate that the simple and feasible morphology and size control synthesis has a great potential in manganese-based electrode materials. However, the high initial irreversible capacity and poor cycle performances especially in high rate still can be observed. It should be ascribed to the irreversible the oxygen loss process and the side reactions of the cathode surface with electrolyte [32, 33]. Further works, such as doping, surface coating, and electrolyte additive, are necessary to improve the electrochemical performance.

The initial charge/discharge curves and cycle performances of cathode material Li1.2Mn0.54Ni0.13Co0.13O2 synthesized at 900° C for 12 h
Conclusions
Li1.2Mn0.54Ni0.13Co0.13O2 microspheres have been successfully synthesized by the morphology-controlled solid-state method. XRD, SEM results show well-crystallized Li1.2Mn0.54Ni0.13Co0.13O2 microspheres have been obtained. Electrochemical tests show the as-prepared material synthesized at 900 °C for 12 h displays better electrochemical performances. The initial discharge capacities are 233.5, 214.2, 199.3, and 168.1 mAh g−1 at 0.1, 0.2, 0.5, and 1 C, respectively. After 50 cycles, the capacity retains 178.0, 166.3, 162.1, and 155.9 mAh g−1 at 0.1, 0.2, 0.5, and 1 C, respectively. In summary, the results demonstrated that the state-of-art Li1.2Mn0.54Ni0.13Co0.13O2 can be synthesized with the unique solid-state method. The simple method could pave the road for the scale-up production of the advanced manganese-based cathode materials for Li-ion batteries.
References
Tarascon JM, Armand M (2001) Nature 414:359–367
Bruce PG, Scrosati B, Tarascon JM (2008) Angew Chem Int Ed 47:2930–2946
Chen J, Cheng FY (2009) Acc Chem Res 42:713–723
Padhi AK, Nanjundaswamy KS, Goodenough JB (1997) J Electrochem Soc 144:1189–1194
Lee HW, Muralidharan P, Ruffo R, Mari CM, Cui Y, Kim DK (2010) Nano Lett 10:3852–3856
Lee ES, Huq A, Chang HY, Manthiram A (2012) Chem Mater 24:600–612
Wu F, Tian J, Su YF, Guan YB, Jin Y, Wang Z, He T, Bao LY, Chen S (2014) J Power Sources 269:747–754
Kang SF, Qin HF, Fang Y, Li X, Wang YG (2014) Electrochim Acta 144:22–30
Wu F, Wang Z, Su YF, Yan N, Bao LY, Chen S (2014) J Power Sources 247:20–25
He F, Wang XQ, Du CQ, Baker AP, Wu JW, Zhang XH (2015) Electrochim Acta 153:484–491
Jiang X, Wang ZH, Rooney D, Zhang XX, Feng J, Qiao JS, Sun W, Sun KN (2015) Electrochim Acta 160:131–138
Zhu Y, Zhu LW (2014) J Power Sources 256:178–182
Yang SY, Huang G, Hua SJ, Hou XH, Huang YY, Yue M, Lei GT (2014) Mater Lett 118:8–11
Ju JH, Cho SW, Hwang SG, Yun SR, Lee Y, Jeong HM, Hwang MJ, Kim KM, Ryu KS (2011) Electrochim Acta 56:8791–8796
Liu XY, Liu JL, Huang T, Yu AS (2013) Electrochim Acta 109:52–58
Shi SJ, Tu JP, Tang YY, Liu XY, Zhang YQ, Wang XL, Gu CD (2013) Electrochim Acta 88:671–679
Song BH, Lai MO, Lu L (2012) J Power Sources 80:187–195
Singh G, Thomas R, Kumar A, Katiyar RS (2012) J Electrochem Soc 159:410–420
Yang M, Du CQ, Tang ZY, Wu JW, Zhang XH (2014) Ionics 20:1039–1046
Sun ST, Du CQ, Wu JW, Tang ZY, Yang M, Zhang XH (2014) Ionics 20:1627–1634
Zhan D, Yang F, Zhang QG, Hu XH, Peng TY (2014) Electrochim Acta 129:364–372
Zhu Z, Yan H, Zhang D, Li W, Lu Q (2013) J Power Sources 224:13–19
Wang JW, Liu J, Yang GL, Zhang XF, Yan XD, Pan XM, Wang RS (2009) Electrochim Acta 54:6451–6454
Yang JG, Cheng FY, Zhang XL, Gao HY, Tao ZL, Chen J (2014) J Mater Chem A 2:1636–1640
Shaju KM, Bruce PG (2008) Dalton Trans, 5471–5475
Dai KH, Mao J, Li ZT, Zhai YC, Wang ZH, Song XY, Battaglia V, Liu G (2014) J Power Sources 248:22–27
Shi SJ, Lou ZR, Xia TF, Wang XL, Gu CD, Tu JP (2014) J Power Sources 257:198–204
Miao XW, Ni H, Zhang H, Wang CG, Fang JH, Yang G (2014) J Power Sources 264:147–154
Johnson CS, Li N, Lefief C, Vaughey JT, Thackeray MM (2008) Chem Mater 20:6095–6106
Jang IC, Son CG, Yang SMG, Lee JW, Cho AR, Aravindan V, Park GJ, Kang KS, Kim WS, Cho WI, Lee YS (2011) J Mater Chem 21:6510
Du CQ, Wu JW, Liu J, Yang M, Xu Q, Tang ZY, Zhang XH (2015) Electrochim Acta 152:473–479
Li GR, Feng X, Ding Y, Ye SH, Gao XP (2012) Electrochim Acta 78:308–315
Fu Q, Du F, Bian XF, Wang YH, Yan X, Zhang YQ, Zhu K, Chen G, Wang CZ, Wei YJ (2014) J Mater Chem A 2:7555–7562
Acknowledgments
This work is supported by the project of Innovative group for high-performance lithium-ion power batteries R&D and industrialization of Guangdong Province (Grant No. 2013N079), State Key Laboratory of Chemical Engineering (No. SKL-ChE-14B0 4), Shenzhen Peacock Plan Program (KQCX20140521144358003), and Fundamental Research Plan of Shenzhen (JCYJ20140417172417144).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Du, C., Zhang, F., Ma, C. et al. Synthesis and electrochemical properties of Li1.2Mn0.54Ni0.13Co0.13O2 cathode material for lithium-ion battery. Ionics 22, 209–218 (2016). https://doi.org/10.1007/s11581-015-1541-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-015-1541-9