
Abstract
Sn-substituted α-Ni(OH)2 for Ni-MH battery is prepared by supersonic coprecipitating method, using NiSO4 as nickel ion source and SnSO4 as doping material. The crystal structure and morphology of the samples were characterized with X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FT-IR), and scanning electron microscopy (SEM). The results demonstrate that the crystal structure of all samples changes from α/β mixed phase to pure α phase and the interlayer spacing becomes larger with more Sn2+ substitution for Ni2+, while the agglomeration particle size decreases initially and then increases. The electrochemical performances of the prepared nickel hydroxide were characterized by cyclic voltammetry (CV) and charge-discharge tests. The results reveal that 15 % Sn-substituted α-Ni(OH)2 has best electrochemical performance, such as best reaction reversibility, highest proton diffusion coefficient, the maximum discharge capacity (303.6 mAh/g at 0.2 C), and the maximum capacity retention (maintaining 90.5 % after 30 cycles at 0.2 C).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ni-MH battery is widely used in power tools to electric vehicles or hybrid electric vehicles, portable electronics, UPS, because of high power, high energy density, good safety [1, 2]. As is well-known, there are two polymorphs known as α- and β-phase, which in the oxidation process convert to γ-NiOOH and β-NiOOH, respectively [3, 4]. Traditionally, β-Ni(OH)2 is widely used in commercial nickel-metal hydride (Ni/MH) battery because of its high tap density and good stability in strong alkaline electrolyte [5–8]. However, the theoretical specific capacity of β-NiOOH (289 mAh/g) is much less than that of γ-NiOOH (433 mAh/g), because the average nickel oxidation state of β-NiOOH is +3, while that of γ-NiOOH is +3.67 or higher [8–12]. And, compared to β-Ni(OH)2, α-Ni(OH)2 behaves no noticeable volume change during charge/discharge cycling due to the similar lattice of α-Ni(OH)2/γ-NiOOH, which is beneficial to keeping the capacity retention during overcharging [8, 12–14]. But, α-Ni(OH)2 is unstable in an alkaline electrolyte and easily converts to β-Ni(OH)2, leading to the rapid degradation of the nickel hydroxide [15–18]. In order to prepare stabilized α-Ni(OH)2, many works have been focused on the preparation of α-Ni(OH)2 by partial substitution of nickel ion in the nickel hydroxide lattice by other metal ions for enhancing the stability of α-Ni(OH)2, such as Al [8, 10, 16], Cu [19], Zn [20], Yb [17], Fe [21], and Mn [22] ions.
According to the crystallography, β-Ni(OH)2 is isostructural with brucite and shows closely stacked two-dimensional (2D) Ni(OH)2 principle layers, while α-Ni(OH)2 is isostructural with hydrotalcite and shows hydroxyl-deficient Ni(OH)2-x host layers and interlayer species, such as anions and water molecules [23, 24]. So, when Ni2+ is substituted by other metal ions, closely stacked two-dimensional (2D) Ni(OH)2 principle layers convert to hydroxyl-deficient Ni(OH)2-x host layers, which lead to the intercalation of species such as anions (e.g., SO4 2−, CO3 2−) and water molecules into the interlayer. Meanwhile, the intercalation of species can enlarge the interlayer spacing and enhance the electrostatic force, which result in improving proton diffusion coefficient and increasing the stability of α-Ni(OH)2 [9, 20].
In previous studies, metal cations with closed shell-like ion strutures (such as Ca2+-1s22s22p63s23p6; Zn2+-1s22s22p63s23p63d10; Lu3+-1s22s22p63s23p63d104s24p64d104f145s25p6; Y3+-1s22s22p63s23p63d104s24p6) can react with NiOOH leading to the inhibition of electron transfer and the increase of the voltage gap between the oxygen evolution potential and the oxidation potential [25–28]. Sn2+ with 1s22s22p63s23p63d104s24p65s2 electron configuration is also closed shell-like ion struture, which is similar to Ca2+, Zn2+, Lu3+, and Y3+. And, there is no related report about Sn-substituted α-Ni(OH)2. So, Sn2+ maybe can enhance the electrochemical performance by increasing charging efficient. In the present work, Sn-substituted α-Ni(OH)2 is prepared by supersonic coprecipitation method. The effect of Sn content on the structure, morphology, crystallinity, and electrochemical performance of α-Ni(OH)2 samples is studied in detail.
Experiment
Preparation of nickel hydroxide
The α-Ni(OH)2 are prepared by supersonic coprecipitation method at 50 °C. An aqueous solution of NaOH containing appropriate amount of anhydrous sodium carbonate and ammonia as buffer solution and an aqueous solution of mixed NiSO4 · 6H2O, SnSO4 with a certain molar ratio dissolved dilute sulfuric acid, which is to prevent Sn2+ from converting to Sn(OH)2 by hydrolyzing as Eq. (1), are dropped into a mother liquid synchronously under stirring. The drop rates are adjusted to control the pH value at 9.00 ± 0.10 by pH meter. The agitation lasted 5 h after dropping is finished; supersonic is employed in all above process. The suspension is aged for 12 h at 50 °C, followed by filtering and washing three times to neutral and then dried to constant weight at 80 °C (sample C obtained). The samples prepared by the Ni2+/Sn2+ molar ratios of 1:0.05, 1:0.10, 1:0.15, 1:0.20, and 1:0 are denoted as sample A, sample B, sample C, sample D, and Sample E, respectively, which is listed as followed in Table 1.
Preparation of nickel hydroxide electrodes
The nickel electrodes are prepared as follows: 8 wt.% samples A, B, C, and D, 86 wt.% commercial micro-size spherical nickel, 3 wt.% cobalt oxide, and 2 wt.% CMC are mixed thoroughly with 1 wt.% PTFE solution. The paste obtained is incorporated into nickel foam substrates with the geometrical size of 2.5 cm × 2.5 cm. The obtained nickel electrodes a, b, c, d listed in Table 1 are dried at 80 °C and then mechanically pressed under a pressure of 8 Mpa for 3 min to a thickness of about 0.5 mm.
Physical characterization
The phase structure of the prepared samples is determined by X-ray diffraction (XRD) using a D/max-IA X-ray diffractometer with Cu Kα radiation (k = 1.54 Å) at 36 kV and 20 mA. IR spectrum of the prepared samples are obtained by Nicol ET6700 Fourier transform infrared spectrometer (KBr, resolution of 4 cm−1). The surface morphology of the samples is identified by S-3400 N scanning electron microscope (SEM).
Electrochemical measurements
Electrochemical characterization is carried out in a standard three electrode system: the prepared positive electrode, Hg/HgO reference electrode, and nickel foam are used as work electrode, reference electrode, and counter electrode. The electrolyte is 6.0 mol/L KOH and 1.5 g/L LiOH solution. The cyclic voltammetry (CV) is carried out using an electrochemical work station (CHI760D) in the potential range of 0 to 0.7 V with the scanning speed of 0.005 to 0.04 V/s. Electrochemical impedance spectroscopy (EIS) measurements are carried out using an electrochemical work station (CHI760D) at open circuit potential with a superimposed 5-mV sinusoidal voltage in the frequency range of 10 kHz–10 Hz.
The positive electrode is placed between the two negative electrodes to form simulated batteries with polypropylene as separator. Tests of galvanostatic charge/discharge and cycle performance are carried out at 0.2 C rate using a Neware BTS-51800 Battery testing system.
Results and discussion
Physical properties of the prepared nickel hydroxide
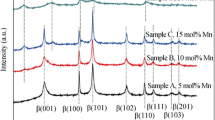
XRD patterns, which is used to study the phase purity of the prepared nickel hydroxide samples with different contents of Sn2+, are shown in Fig. 1. For comparison, the standard XRD pattern of α-Ni(OH)2 · 0.75H2O (ICDD-JCPDS 38-0715) is also given in Fig. 1. The general features of the diffraction patterns for samples B, C, and D are similar to the standard XRD pattern of α-Ni(OH)2 · 0.75H2O, which prove that samples B, C, and D are α-Ni(OH)2 phase. [15, 16]. The general feature of the diffraction patterns for sample A is similar to the standard XRD pattern of α-Ni(OH)2 · 0.75H2O, while a peak at 17.5° is ascribed to β-Ni(OH)2 phase. This phenomenon illustrates that sample A is α/β-Ni(OH)2 [6]. Meanwhile, there is no characteristic peak for Sn(OH)2, which reveals that the doped ions have successfully embedded into each sample crystal lattice. Futhermore, the characteristic peaks of sample E are ascribed to β-Ni(OH)2 according to standard card (JCPDS-14-0117), which indicate that the crystalline phases of sample E without doping are β-Ni(OH)2 phase.

XRD patterns of Sn-substituted α-Ni(OH)2 samples A, B, C, D, and E
The value of d (003) for all samples, which is related to the lattice parameter c (=3 d (003)), is shown in Table 2. As shown in Table 2, the d (003) value increases with Sn-doping, that is to say, D > C > B > A. Moreover, the d (003) value of samples B, C, and D is much larger than that (7.27–7.32 Å) for the pure α-Ni(OH)2, while that of sample A is almost equal to that (7.27–7.32 Å) for the pure α-Ni(OH)2 [5, 14]. The increase of the interlayer spacing can be attributed to an increase in the intercalated anions in the nickel hydroxide lattice, which plays an important role on enhancing the proton diffusion coefficient of nickel hydroxide. The intensity ratio z of I(003) to I(110) is a good measure to estimate the crystal growth/stacking speed [29]. The value z for sample C is largest in the XRD pattern, which is different from others. This reveals that the crystal growth/stacking speed of sample C in the direction of “c-axis” of layered structure is faster than that in “a-axis”, which may influence the electrochemical properties. Meanwhile, the intense of the (003) diffraction lines is largest among all samples, which indicate that sample C has a better crystallinity. The FWHMs for respective sample, which can reveal the primary crystallite size perpendicular to (003) diffraction plane, are listed in Table 1. The FWHMs of the (003) diffraction lines decrease initially and then increase with Sn-doping. This result reveals that sample C has a bigger primary crystalline grain than others, which may influence the electrochemical properties. This phenomenon illustrates that the content of Sn2+ can change the crystal structure of nickel hydroxide and has a significant effect on enhancing interlayer spacing [15–19].
To complement the XRD measurements, FT-IR spectra for the prepared nickel hydroxide samples are made, and the results are shown in Fig. 2. The broad band at about 3455 and 1630 cm−1 are the characteristic of the stretching vibration of hydroxyl groups hydrogen-bonded and the bending mode of interlayer and absorbing water molecules, which are ascribed to α-Ni(OH)2[16], while the broad band at about 3640 cm−1 is the characteristic of the stretching vibration of hydroxyl groups in the nickel hydroxide lattice, which is ascribed to β-Ni(OH)2 [5, 30]. In Fig. 2, the absorbed peaks at about 3455 and 1630 cm−1 are seen for samples B, C, and D, while the low absorbed peak at about 3640 cm−1 and the strong absorbed peaks at about 3455 and 1630 cm−1 are seen for sample A. Meanwhile, the intense of absorbed peaks at about 3455 and 1630 cm−1 becomes stronger with the increase in doped Sn2+. The results demonstrate that sample A is α/β-Ni(OH)2 phase structure, samples B, C, and D are α-Ni(OH)2 phase structure, and water molecules in the nickel hydroxide lattice increase with the increase in doped Sn2+, which plays an important role in improving the rate capacity performance and decreasing the polarization effect because they provide the passage of proton diffusion along the molecule chain between layers. For all samples, the absorbed peaks at 1110 cm−1 can be obviously observed and the intense of absorbed peaks becomes stronger, indicating that the existence of SO4 2− in the nickel hydroxide lattice and the content of SO4 2− becomes larger [16]. The weak absorbed peaks at 1380 cm−1 can be also observed, indicating the existence of CO3 2− in the nickel hydroxide lattice [17]. At low wave numbers, the bands for all samples at about 630 cm−1 in all IR spectra can be assigned to the bending vibration Ni-O-H [16].

FT-IR spectra of Sn-substituted α-Ni(OH)2 samples A, B, C, and D
Figure 3 illustrates the morphology of the Sn-substituted α-Ni(OH)2 samples. As shown in Fig. 3, all samples appear as aggregates of irregular shape, and their crystal edge is blur. The irregular shape of the sample may result from the different K sp value between Sn(OH)2 (5.45 × 10−27) and Ni(OH)2 (5.5 × 10−16), which is similar to Al-substituted α-Ni(OH)2 [6]. Due to the K sp of Sn(OH)2 is much smaller than that of Ni(OH)2, the deposition of Sn2+ in Sn(OH)2 lattice may be quicker than that of Ni(OH)2, which may form a net structure and promote the agglomerations of colloidal nickel hydroxide particles. The agglomeration particle size of all samples, which influences the activity of nickel hydroxide, decreases initially and then increases with the increase in doped Sn2+. And, the agglomeration particle size of sample C is more uniform than others. The more uniform and smaller agglomeration particle size can improve the activity of alpha nickel hydroxide [26]. So, sample C may show better electrochemical performance in the future study.

SEM morgraphs of a sample A; b sample B; c sample C; d sample D
Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements of nickel electrode
Figure 4 displays typical CV curves at various scan rate and the relationship between the anodic peak current and the square root of scan rate for electrodes a, b, c, and d [6, 16]. As is given in Fig. 4 (1) ~ (4), at the scan rate of 0.005 V/s, both oxidation peak and one reduction peak are observed in the CV curves. As the scan rate increase from 0.005 to 0.04 V/s, oxidation and reduction peaks can be observed clearly and move to the positive and the negative, which shows typical diffusion control phenomenon. The relationship between the anodic peak current I p and the scan rate v 1/2 is given in Fig. 4 (5). The linear relationship between I p and v 1/2 reveals that the electrode reaction is controlled by proton diffusion [19]. According to Eq. (2) [18], the anodic peak current I p and the scan rate v of the electrode system controlled by diffusion process follow the following relationship at 25 °C:
where n is the electronic number of reaction (the value is about 1 for nickel hydroxide), S is the real surface area of the electrode (cm2), D is the diffusion coefficient (cm2/s), and C 0 is the proton concentration (mol/cm3).

Cyclic voltammograms and relationship between the anodic peak current and the square root of scan rate for electrodes a, b, c, and d
If \( \frac{I_P}{\sqrt{\upsilon }}=k \), Eq. (2) becomes
The diffusion coefficient D′ of each electrode can be calculated from S, C 0 and the slope of I p and v 1/2 line by Eq. (3). The results are tabulated in Table 3. Electrode c has the largest proton diffusion coefficient of 3.54 × 10−11 cm2/s, which is 86.82 % higher than that of electrode a. The variation in D′ is ascribed to the interlayer spacing, and the content of water molecules in the nickel hydroxide lattice of samples A ~ D increases gradually. However, the proton diffusion coefficient of electrode d decreases which may be caused by the bad crystallinity of sample D and more defects in the nickel hydroxide lattice [31].
The CV curves of all electrodes at the same scan rate of 0.04 V/s are given in Fig. 5, and the results are calculated in Table 4. The potential difference (E O -E R) between E O and E R and the potential difference (E OR –E O) between E OR and E O can be used to characterize the reversibility of redox reaction and the charge efficient, respectively [6–8]. Compared to all electrodes, it is obviously seen that the potential difference (E O –E R) decreases and the potential difference (E OR –E O) increases from electrode a to electrode c. However, the potential difference (E O –E R) increases and the potential difference (E OR –E O) decreases for electrode d. This phenomenon may be caused by the variation of proton diffusion coefficient. Electrode c shows the smallest the potential difference (E O –E R) and the highest potential difference (E OR –E O), which may result from the better crystalline, larger interlayer spacing, bigger content of water molecule, and smaller agglomeration particle size [16, 17, 26, 32]. So, electrode c may display good electrochemical performance.

Cyclic voltammograms at the same scan rate (0.04 V/s) for electrodes a, b, c, and d
Figure 6 shows the electrochemical impedance spectra for all electrodes at a steady state. It can be seen that the Nyquist plots of both electrodes exhibit a semicircle resulting from transfer resistance in the high-frequency region. The calculated charge-transfer resistances (Rct) of electrodes a, b, c, and d, which are shown in Table 5, are 0.533, 0.4692, 0.3014, and 0.3854 Ω, respectively. This result demonstrates that the electrochemical reaction of electrode c is more easily than others, because sample C has a smaller particle size than others, which can enhance the proton diffusion in the electrode. Thus, the solid-state proton diffusion and intercalation/deintercalation processes are remarkably improved, resulting in a higher electrochemical activity and lower charge-transfer resistance.

Nyquist plots of all electrodes
Charging/discharging test of nickel electrode
The charging curves obtained at 0.2 C rate to 120 % state of charge at 25 °C for electrodes a, b, c, and d are shown in Fig. 7, and the results are presented in Table 6. The MPV is taken to be the voltage value at 50 % state of charge, measured after 2.5 h of charge at 0.2 C, while the OEP is taken to be the voltage value measured at the end of charge. As expected (Fig. 7 and Table 6), electrodes exhibit important electrochemical differences, particularly in MPV and VG. Electrode c presents the largest value of voltage gap (97.9 mV), which illustrates that electrode c has the high charging efficiency. This phenomenon may be caused by the largest proton diffusion coefficient, which is in accordance with the CV results.

Charging curves obtained at 0.2 C rate to 120 % state of charge at 25 °C for electrodes a, b, c and d
The discharging curves of electrodes at 0.2 C rate at 25 °C are shown in Fig. 8. As can be seen clearly in Fig. 8, all electrodes exhibit similar discharge platform, the discharge capacity increases initially and the decrease with Sn-doping increasing, and electrode c has the largest specific discharge capacity (303.6 mAh/g). This phenomenon illustrates that Sn2+ has no significant effect on improving discharge potential, but enhancing discharge capacity dramatically.

Discharging curves obtained at 0.2 C rate at 25 °C for electrodes a, b, c, and d
The cycle performance of electrodes at 0.2 C rate at 25 °C is given in Fig. 9. It can be seen that the maximum discharge capacity of electrodes a, b, c, and d are 277.2, 281.9, 306.2, and 297.2 mAh/g, respectively, and the decay rate R d values of electrodes a, b, c, and d after 60 cycles are 16.41, 15.35, 10.45, and 14.23 %, respectively. This indicates that electrode c shows good cycling stability and higher specific discharge capacity dramatically, which may be caused by better crystallinity, smaller agglomeration particle size, larger interlayer spacing, better reversibility of redox reaction, and higher charging efficient [17, 26, 32].

Cyclic performance of electrodes at 0.2 C discharge rate
Conclusions
The better cyclic stability and higher specific discharge capacity of 15 % Sn-substituted alpha nickel hydroxide make it a promising candidate as battery materials in Ni-MH battery. With the increase of Sn content, the crystallinity becomes better initially and then worse, and the primary crystal size becomes bigger initially and then smaller, while the trend of agglomeration particle size in contrast. The 15 % Sn-doped alpha nickel hydroxide exhibits a maximum specific discharge capacity of 306.2 mAh/g (at 0.2 C rate) and better cycle performance, which are directly related to better crystallinity, smaller agglomeration particle size, better reversibility of redox reaction, and higher charging efficient.
References
Kazimirov VY, Smirnov MB, Bourgeois L, Guerlou-Demourgues L, Servant L, Balagurov AM, Natkaniec I, Khasanova NR, Antipov EV (2010) Atomic structure and lattice dynamics of Ni and Mg hydroxides. Solid State Ionics 181:1764–1770
Xu LP, Ding YS, Cheng CH, Zhao LL, Rimkus C, Joesten R, Suib SL (2008) 3D flowerlike alpha-nickel hydroxide with enhanced electrochemical activity synthesized by microwave-assisted hydrothermal method. Chem Mater 20:308–316
Takasaki T, Nishimura K, Mukai T, Iwaki T, Tsutsumi K, Sakai T (2013) Fiber-type Ni(OH)2 electrode with alpha/gamma phase transformation: high-capacity and high-voltage performances of nickel-metal hydride battery. J Electrochem Soc 160:A564–A568
Deki S, Hosokawa A, Béléké AB, Mizuhata M (2009) α-Ni(OH)2 thin films fabricated by liquid phase deposition method. Thin Solid Films 517:1546–1554
Han TA, Tu JP, Wu JB, Li Y, Yuan YF (2006) Electrochemical properties of biphase Ni(OH)2 electrodes for secondary rechargeable Ni/MH batteries. J Electrochem Soc 153:A738–A742
Li YW, Yao JH, Zhu YX, Zou ZG, Wang HB (2012) Synthesis and electrochemical performance of mixed phase alpha/beta nickel hydroxide. J Power Sources 203:177–183
Zhang WK, Xia XH, Huang H, Gan YP, Wu JB, Tu JP (2008) High-rate discharge properties of nickel hydroxide/carbon composite as positive electrode for Ni/MH batteries. J Power Sources 184:646–651
Wu JB, Tu JP, Wang XL, Zhang WK (2007) Synthesis of nanoscale CoO particles and their effect on the positive electrodes of nickel-metal hydride batteries. Int J Hydrog Energy 32:606–610
Bardé F, Palacín MR, Beaudoin B, Tarascon JM (2005) Ozonation: a unique route to prepare nickel oxyhydroxides. Synthesis optimization and reaction mechanism study. Chem Mater 17:470–476
Hu WK, Noréus D (2003) Alpha nickel hydroxides as lightweight nickel electrode materials for alkaline rechargeable cells. Chem Mater 15:974–978
Liu LP, Zhou ZT, Peng CH (2008) Sonochemical intercalation synthesis of nano gamma-nickel oxyhydroxide: structure and electrochemical properties. Electrochim Acta 54:434–441
Caravaggio GA, Christian D, Wronski Z (2001) Synthesis, stability and electrochemical properities of NiAl and NiV layered double hydroxide. J Mater Chem 11:912–921
Guerlou-Demourgues L, Tessier C, Bernard P, Delmas C (2004) Influence of substituted zinc on stacking faults in nickel hydroxide. J Mater Chem 14:2449–2454
Béléké AB, Higuchi E, Inoue H, Mizuhata M (2014) Durability of nickel-metal hydride (Ni-MH) battery cathode using nickel-aluminum layered double hydroxide/carbon (Ni-Al LDH/C) composite. J Power Sources 247:572–578
Jayashree RS, Vishnu Kamath P (2002) Layered double hydroxides of Ni with Cr and Mn candidate electrode materials for alkaline secondary cells. J Power Sources 20:120–124
Li YW, Yao JH, Liu CJ, Zhao WM, Deng WX, Zhong SK (2010) Effect of interlayer anions on the electrochemical performance of Al-substituted α-type nickel hydroxide electrodes. Int J Hydrog Energy 35:2539–2545
Xu QS, Zhu YJ, Han QY, Zhao RD, Zhuang YH, Liu YL, Zhang SJ, Miao CC (2014) Preparation of Yb-substituted α-Ni(OH)2 and its physicochemical properties. J Alloys Compd 584:1–6
Zhang ZJ, Zhu YJ, Bao J, Lin XR, Zheng HZ (2011) Electrochemical performance of multi-element doped α-nickel hydroxide prepared by supersonic co-precipitation method. J Alloys Compd 509:7034–7037
Bao J, Zhu YJ, Zhang ZJ, Xu QS, Zhao WR, Chen J, Zhang W, Han QY (2013) Structure and electrochemical properties of nanometer Cu substituted α-nickel hydroxide. Mater Res Bull 48:422–428
Chen H, Wang JM, Zhao YL, Zhang JQ, Cao CN (2005) Electrochemical performance of Zn-substituted Ni(OH)2 for alkaline rechargeable batteries. J Solid State Electrochem 9:421–428
Guerlou-Demourgues L, Fournès L, Delmas C (1996) In stitu57Fe mössbauer spectroscopy study of the electrochemical behavior of an iron-substituted nickel hydroxide electrode. J Electrochem Soc 143:3083–3088
Guerlou-Demourgues L, Delmas C (1996) Electrochemical behavior of the manganese-substituted nickel hydroxide. J Electrochem Soc 143:561–566
Gao T, Jelle BP (2013) Paraotwayite-type α-Ni(OH)2 nanowires: structural, optical, and electrochemical properties. J Phys Chem C 117:17294–17302
Zhang K, Wang JB, Lu XL, Li LY, Tang YW, Jia ZY (2009) Structural evolution of hydrothermal-synthesized Ni(SO4)0.3(OH)1.4 nanobelts during ex situ heat treatment and in situ electron irradiation. J Phys Chem C 113:142–147
Qin LW, Hu M, Gao XR, Lei LX (2011) Effect of calcium hydroxide on the electrochemical performance of a [Ni4Al(OH)10]OH electrode. J Solid State Electrochem 15:405–412
Re JX, Zhou Z, Gao XP, Yan J (2006) Preparation of porous spherical α-Ni(OH)2 and enhancement of high-temperature electrochemical performances through yttrium addition. Electrochim Acta 52:1120–1126
Ren JX, Yan J, Zhou Z, Wang X, Gao XP (2006) High-temperature electrochemical performance of spherical Ni(OH)2 coated with Lu(OH)3. Int J Hydrog Energy 31:71–76
Gao XH, Lei LX, Hu M, Qin LW, Sun YM (2009) Structure, morphology and electrochemical performance of Zn-doped [Ni4Al(OH)10]OH. J Power Sources 191:662–668
He XM, Li JJ, Cheng HW, Jiang CY, Wan CR (2005) Controlled crystallization and granulation of nano-scale β-Ni(OH)2 cathode materials for high power Ni-MH batteries. J Power Sources 152:285–290
Zhang SM, Zeng CH (2009) Self-assembled hollow spheres of β-Ni(OH)2 and their derived nanomaterials. Chem Mater 21:871–883
Béléké AB, Mizuhata M (2010) Electrochemical properities of nickel-aluminum layered double hydroxide/carbon composite fabricated by liquid phase deposition. J Power Sources 195:7669–7676
Hu WK, Gao XP, Dag N, Trygve B, Nils KN (2006) Evolution of nano-crystal sized α-nickel hydroxide as an electrode material for alkaline rechargeable cells. J Power Sources 160:704–710
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (No. 11304045), the Guangdong Province National Natural Science Foundation of China (No. S2012010009955), and the Science and Technology Program of Guangzhou City of China (No. 2013j4100011).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Miao, C., Zhu, Y., Huang, L. et al. Synthesis, characterization, and electrochemical performances of alpha nickel hydroxide by coprecipitating Sn2+ . Ionics 21, 2295–2302 (2015). https://doi.org/10.1007/s11581-015-1387-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-015-1387-1