
Abstract
Pemigatinib (Pemazyre®), a selective, potent, reversible, oral inhibitor of fibroblast growth factor receptor (FGFR) 1–3, has received conditional (in the EU) or accelerated (in the USA) approval for the treatment of adults with previously treated, unresectable locally-advanced or metastatic cholangiocarcinoma (CCA) with an FGFR2 gene fusion or rearrangement. Over the course of a single-arm, phase 2 study (FIGHT-202), just over a third of patients with pretreated, advanced CCA [almost exclusively intrahepatic CCA (iCCA)] harbouring an FGFR2 fusion or rearrangement who received pemigatinib once daily (2 weeks on, 1 week off) had an objective response; nearly half had stable disease. Median progression-free survival and overall survival at the time of the final analysis were 7.0 months and 17.5 months, respectively. Pemigatinib was generally well tolerated and had a manageable safety profile. The most common treatment-related adverse event, hyperphosphataemia, was exclusively grade 1–2 in severity and, similarly, observed ocular and nail toxicities were rarely grade ≥ 3 in severity. Pending confirmation of its clinical benefits in an ongoing cisplatin plus gemcitabine-controlled, phase 3 study (FIGHT-302), pemigatinib provides a valuable targeted therapy for pretreated patients with advanced (i)CCA harbouring a FGFR2 fusion or rearrangement.
Plain Language Summary
Bile duct cancer or cholangiocarcinoma (CCA) has a very poor prognosis, partly because the majority of patients are first diagnosed at an advanced stage when they are no longer eligible for potentially curative surgery and are therefore limited to receiving systemic (palliative) chemotherapy, which results in only modest survival gains. 10–20% of CCAs arise inside the liver [intrahepatic CCAs (iCCAs)]; ≈ 10–20% of patients with advanced iCCAs are eligible to receive fibroblast growth factor receptors (FGFR) inhibitors, as the development of their tumours depends, in part, on FGFRs that have been inappropriately activated due to underlying genetic abnormalities. Pemigatinib (Pemazyre®) is a selective, potent, once-daily oral FGFR 1–3 inhibitor. In a phase 2 trial in patients with advanced CCA (almost exclusively iCCA) containing an abnormal FGFR2 fusion or rearrangement who had already received systemic chemotherapy, more than a third receiving pemigatinib experienced partial or complete shrinkage of their tumours, while almost half had neither growth nor shrinkage of their tumours. Pemigatinib was generally well tolerated with a manageable safety profile. Pending completion of a phase 3 study designed to confirm its clinical benefits, pemigatinib represents a valuable targeted therapy for pretreated patients with advanced (i)CCA harbouring a FGFR2 fusion or rearrangement.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Digital Features for this Adis Drug Evaluation can be found at https://doi.org/10.6084/m9.figshare.24710424. |
Selective, potent, reversible oral FGFR1–3 inhibitor. |
Administered once daily (2 weeks on, 1 week off). |
Produces durable responses in more than a third of patients. |
Generally well tolerated with a manageable safety profile. |
1 Introduction
Bile duct cancers or cholangiocarcinomas (CCAs) are relatively infrequent (< 1% of all cancers) [1, 2]. CCAs are, however, highly heterogeneous and aggressive tumours that represent ≈ 2% of all annual cancer-related deaths, globally [1, 2]. The age-standardized incidence of CCA in the Western world (including Europe and North America) is 0.3–3.5 cases per 100,000 population, with tumours arising from epithelial cells lining intrahepatic and extrahepatic bile ducts [i.e. intrahepatic CCAs (iCCAs) and extrahepatic CCAs (eCCAs)] accounting for 10–20% and 80–90% of all CCAs, respectively [1, 2]. Over recent decades, the incidence of iCCA has increased more than that of eCCA in many countries (e.g. the USA [3, 4]); mortality from iCCA has also increased and/or remained higher than that from eCCA, which has decreased [3, 5, 6].
Late presentation and treatment refractoriness also contribute to the very poor prognosis of CCA (an estimated 5-year survival rate of 7–20%) [7, 8]. As these tumours are usually asymptomatic in early stages, the majority (around two-thirds) of patients are first diagnosed with advanced disease, such that they are no longer eligible for potentially curative surgery or transplantation and are limited to receiving palliative treatment, particularly systemic chemotherapy [7, 8]. The survival benefits with historical first-line and current second-line regimens are, however, modest and, accordingly, the search for more effective chemotherapy-based (and other) treatment options for patients with advanced or unresectable CCA has continued [8, 9].
Against this background, the advent of comprehensive genomic profiling has led to a number of potentially actionable molecular alterations involved in CCA tumourigenesis being identified; these targetable oncogenic drivers include fibroblast growth factor receptor (FGFR) 2 gene fusions or rearrangements, which are estimated to occur in ≈ 10–20% of patients with CCAs (almost exclusively the small duct subtype of iCCA) [8, 10, 11]. Pemigatinib (Pemazyre®), a selective, potent, oral FGFR1–3 inhibitor [12], has received conditional (in the EU [13]) or accelerated (in the USA [14]) approval for the treatment of adults with pretreated, unresectable, locally-advanced or metastatic CCA who are harbouring an FGFR2 fusion or rearrangement (Sect. 6). Additionally, in Japan, pemigatinib has been approved for the treatment of patients with unresectable biliary tract cancer with a FGFR2 fusion, worsening after cancer chemotherapy [15].
This article summarizes the pharmacological properties of pemigatinib and reviews its efficacy and tolerability in the treatment of advanced CCA with FGFR2 fusion or rearrangement. Pemigatinib is also approved in the USA [14] and Japan [16] for the treatment of myeloid/lymphoid neoplasms with FGFR1 rearrangement. However, discussion of this indication is beyond the scope of this review.
2 Pharmacodynamic Properties of Pemigatinib
Pemigatinib is an oral small-molecule, adenosine triphosphate (ATP)-competitive, reversible tyrosine kinase inhibitor of FGFR1–3 [12]. Like other FGFR inhibitors, it inhibits FGFR phosphorylation and signaling and hence the viability of cells expressing oncogenic FGFRs that have been aberrantly activated due to genetic alterations, including mutations and fusions or rearrangements [12, 14, 17, 18].
In vitro, pemigatinib potently inhibited recombinant human FGFR1, FGFR2 and FGFR3 with mean half-maximal inhibitory concentration (IC50) values of 0.4, 0.5 and 1 nmol/L, respectively, while demonstrating weaker activity against FGFR4 (mean IC50 of 30 nmol/L). The high selectivity of the drug was established on the basis that it only inhibited two out of a total of 56 non-FGFR kinases with an IC50 value < 1 μmol/L, namely vascular endothelial growth factor receptor-2/kinase insert domain containing receptor (mean IC50 of 182 nmol/L) and c-KIT (mean IC50 of 266 nmol/L). Pemigatinib also selectively inhibited the growth of tumour cell lines with FGFR1, FGFR2 or FGFR3 alterations compared with cell lines lacking such FGFR aberrations. In vivo, pemigatinib exhibited anti-tumour activity in mouse xenograft models of human tumours with FGFR1, FGFR2 or FGFR3 alterations, including a patient-derived xenograft model of chemorefractory CCA harbouring the FGFR2-Transformer-2 beta homolog (TRA2b) fusion protein [17].
Given the central role of FGFR1 (and its ligand FGF23) in the homeostatic regulation of phosphate, increases in serum phosphate levels are an expected pharmacodynamic effect of FGFR inhibitors, such as pemigatinib. [11, 19]. In FIGHT-101, a first-in-human phase 1/2 dose-escalation/dose-expansion study of pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies, serum phosphate concentrations increased with increasing pemigatinib exposure across the dosage range of 1–20 mg once daily (i.e. 0.07- to 1.5-times the recommended dosage; Sect. 6), with increased risk of hyperphosphatemia with higher pemigatinib exposure [14, 17].
3 Pharmacokinetic Properties of Pemigatinib
The pharmacokinetics of pemigatinib in patients with cancer can be adequately described by a two-compartment disposition model with first-order absorption and linear elimination [20]. Following oral administration of pemigatinib at the recommended dosage of 13.5 mg once daily (Sect. 6) in patients with advanced malignancies, steady state was reached by day 4, with a geometric mean accumulation ratio of 1.6 [13, 14, 21]. At steady state, the median time to peak plasma concentration (Cmax) was 1.13 h. Steady-state pemigatinib concentrations increased proportionally over the dose range of 1–20 mg. Administration of pemigatinib with a high-fat, high-calorie meal had no clinically meaningful effect on the pharmacokinetics of the drug, which may be taken with or without food [13, 14, 21] (Sect. 6).
Over the concentration range 1–10 μmol/L pemigatinib was 90.6% bound to human plasma proteins in vitro [13, 14]. The estimated apparent volume of distribution was 235 L following oral administration of pemigatinib 13.5 mg once daily in patients with advanced malignancies [13, 14].
Extensive hepatic metabolism (primarily by CYP3A4) followed by biliary excretion is the major clearance/elimination pathway for pemigatinib; renal clearance/elimination of the drug is low [13, 14]. In a human mass balance study, 82.4 % of a radiolabeled pemigatinib dose was recovered in faeces (1.4 % as unchanged drug); 12.6 % was recovered in urine (1 % as unchanged drug) [13, 14]. M2 (O-desmethyl pemigatinib) and its secondary metabolites (M7, M8, and M9) accounted for ≈ 77% of the metabolite burden in faeces and urine [20]. Among patients with advanced malignancies receiving pemigatinib 13.5 mg once daily, the geometric mean apparent clearance at steady state was 10.6 L/h; the geometric mean elimination half-life was 15.4 h [13, 14, 21].
Systemic exposure to pemigatinib was not affected to a clinically relevant extent by age (21–79 years), sex, ethnicity/race or bodyweight (39.8–156 kg) [14, 20]. Similarly, systemic exposure to pemigatinib was not affected to a clinically significant extent by mild to moderate hepatic impairment, mild to moderate renal impairment or end-stage renal disease maintained on haemodialysis [14, 20, 22]. However, compared to healthy matched controls with normal hepatic or renal function, the geometric mean pemigatinib area under the plasma concentration-time curve from time zero to infinity (AUC0–∞) increased by 136% in patients with severe hepatic impairment (total bilirubin > 3 × ULN with any aspartate aminotransferase), and by 59% in patients with severe renal impairment [estimated glomerular filtration rate (eGFR) 15–29 mL/min/1.73 m2] [14]. In both the EU [13] and USA [14], therefore, the pemigatinib dose should be reduced from 13.5 mg to 9 mg in patients with severe hepatic or renal impairment.
In dedicated drug-drug interaction (DDI) studies [23], coadministration of pemigatinib with a strong CYP3A inhibitor (itraconazole 200 mg once daily) resulted in an 88% increase in the geometric mean pemigatinib AUC0–∞ while, conversely, coadministration of pemigatinib with a strong CYP3A inducer (rifampin 600 mg once daily) resulted in an 85% decrease in the geometric mean pemigatinib AUC0–∞. Moreover, clinical DDI data-validated and physiologically-based pharmacokinetic modeling predicted a > 50% increase and > 50% decrease in pemigatinib AUC when coadministered with a strong or moderate CYP3A4 inhibitor or inducer, respectively [24]. Accordingly, in the EU [13] and USA [14], concomitant use of pemigatinib with moderate [14] or strong [13, 14] CYP3A inhibitors should be avoided; if coadministration is unavoidable, the pemigatinib dose should be reduced [13, 14]. Additionally, concomitant use of pemigatinib with strong or moderate CYP3A4 inducers should be avoided [14] or is not recommended [13]; coadministration of the drug with St John’s wort is contraindicated in the EU [13]. Consult local prescribing information for further details of potential DDIs involving pemigatinib.
4 Therapeutic Efficacy of Pemigatinib
The efficacy of pemigatinib as monotherapy in previously-treated adults with locally advanced or metastatic CCA harbouring an FGFR2 fusion or rearrangement has been evaluated in FIGHT-202, an open-label, single-arm, multinational, phase 2 study [25,26,27]. The initiation of FIGHT-202 was prompted by the results of the FIGHT-101 study in pan-cancer patients with FGF/FGFR alterations and advanced malignancies (Sect. 2), in which pemigatinib showed the most encouraging anti-tumour activity in individuals with CCA harbouring FGFR2 fusions or rearrangements [21]. Of note, FIGHT-202 was a multicohort study that not only enrolled patients with pretreated, advanced CCA who had FGFR2 fusions or rearrangements (cohort A; n = 107), but also those who had other FGF/FGFR alterations (cohort B; n = 20) or no FGF/FGFR alterations (cohort C; n = 18). Consideration of efficacy outcomes in the off-label patient populations enrolled in cohorts B and C is beyond the scope of this review; however, all 38 individuals were included in the overall safety population of FIGHT-202 (n = 146) (Sect. 5).
Eligible patients in cohort A of FIGHT-202 were aged ≥18 years with: a histological or cytological diagnosis of locally advanced or metastatic CCA; documented disease progression following ≥1 prior systemic cancer therapy (previous treatment with selective FGFR inhibitors was not permitted); and a centrally-confirmed FGFR2 fusion or rearrangement [25]. Additional entry requirements included: an Eastern Cooperative Oncology Group performance status (ECOG PS) score of ≤ 2; radiologically measurable disease according to Response Evaluation Criteria in Solid Tumours (RECIST) version 1.1; serum phosphate less than or equal to institutional upper limit of normal (ULN); and no clinically significant corneal or retinal disorders [25].
All enrolled patients self-administered oral pemigatinib at a starting dose of 13.5 mg once daily according to an intermittent 21-day cycle (i.e. 2 weeks on/1 week off) until radiological disease progression, unacceptable toxicity, withdrawal of consent, or physician choice [25]. The primary efficacy endpoint was the objective response rate [ORR; i.e. the proportion of patients with a best overall response (BOR) of a complete response (CR) or a partial response (PR)], as assessed by independent central review according to RECIST 1.1 [25].
The median age of the study population was 56 years, 61% of patients were female, 74% were Caucasian, 98% had iCCA, 82% had metastatic disease, 95% had an ECOG PS score of ≤ 1, and 61, 27 and 12% had received 1, 2 and ≥ 3 prior lines of therapy, respectively [25]. Of the 56 different FGFR2 fusion partners identified [most commonly BICC1; in 31 (29%) of the 107 patients], 42 (75%) were unique to individual patients [25].
More than a third of the FIGHT-202 participants achieved a durable objective response; nearly half had stable disease (SD) [25,26,27] (Table 1). In this regard, the results of the primary analysis [25] were substantiated by those of both the updated [26] and final [27] analyses, with an ORR of 37% and a disease control rate (DCR) of 82% observed on each occasion (Table 1). The majority of objective responses were PRs (Table 1); the median best percentage change from baseline in the sum of target lesion diameters at the time of the final analysis was −28.4% (range − 100% to + 55%) [n = 104 evaluable patients] [27]. The median time to first response was 2.7 (range 1.4–3.9) months [25].
A reduction in tumour burden was apparent across all patient subgroups assessed in exploratory analyses performed at the time of the primary analysis, including those based on gender, age, ECOG PS, presence or absence of metastatic disease, FGFR2 rearrangement partner, and number of prior lines of therapy [25].
Median progression-free survival (PFS) and overall survival (OS) values were unchanged from the time of the updated analysis to the time of the final analysis, being 7.0 and 17.5 months, respectively (Table 1). PFS values were generally consistent across the same subgroups in which ORR was assessed [25]. Interestingly, the results of a post hoc analysis suggested that median PFS in patients with advanced CCA and FGFR2 fusion or rearrangement who enrolled in FIGHT-202 having received only one prior line of therapy (i.e. those receiving pemigatinib as a second-line treatment) was longer than that seen in patients who, immediately before enroling in the trial, had received two prior lines of therapy (i.e. those receiving a second-line treatment other than pemigatinib) [7.0 months (n = 65) vs 4.2 months (n = 39)] [28]. Similarly, median PFS in patients receiving third-line treatment with pemigatinib during FIGHT-202 was seemingly longer than that observed in patients receiving third-line treatment with systemic therapy before study enrolment [8.9 months (n = 30) vs 6.6 months (n = 13)] [29]. According to another post hoc analysis, median OS was 30.1 months in ‘responders’ (patients with either a CR or PR; n = 40) versus 13.7 months in ‘nonresponders’ (patients with either SD or progressive disease; n = 68) [26].
5 Tolerability of Pemigatinib
Oral pemigatinib was generally well tolerated by, and had a manageable safety profile in, adults with previously-treated, locally-advanced or metastatic CCA in the FIGHT-202 trial, almost three-quarters (73%) of whom had FGFR2 fusions or rearrangements [25, 26] (Sect. 4). This section largely focuses on the results of the safety analysis of FIGHT-202, which was conducted at the time of the primary efficacy analysis [25]. The median duration of pemigatinib exposure for the overall safety population (n = 146) was 181 (range 7–730) days [14]. These findings are supported and/or supplemented by those from a recently updated pooled analysis of clinical trials of pemigatinib (including FIGHT-202), in which a total of 635 patients with advanced malignancies received the drug at a starting dose of 13.5 mg once daily, either on an intermittent or continuous basis (hereafter referred to as the ‘pooled cancer population’) [14].
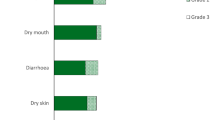
The most common grade 1–2 treatment-related adverse events (TRAEs) in FIGHT-202 were hyperphosphataemia, alopecia, dysgeusia, diarrhoea, fatigue, stomatitis, dry mouth, nausea, decreased appetite, dry eye, dry skin, arthralgia, constipation, palmar-plantar erythrodysaesthesia (PPE) and pain in extremity [25] (Fig.1). The most frequent grade 3 TRAE was hypophosphataemia (in 7% of patients); the next most frequent grade 3 TRAEs were stomatitis, arthralgia, PPE, diarrhoea and hyponatraemia (Fig. 1). There were no grade 4 TRAEs, with the exception of a single case of hyponatraemia (1%) [25].

Most common (incidence ≥ 10%) grade 1–3 treatment-related adverse events in the FIGHT-202 trial [25]. PPE palmar-plantar erythrodysaesthesia, Ǿ = zero incidence of grade 3 treatment-related adverse events
Overall, 14%, 42% and 9% of FIGHT-202 participants had pemigatinib dose reduction, dose interruption and permanent discontinuation due to treatment-emergent adverse events (TEAEs), respectively [25]. Serious TEAEs occurred in 45% of FIGHT-202 participants, the most common (n ≥ 3 patients) being: abdominal pain and pyrexia (each n = 7); cholangitis and pleural effusion (each n = 5); and acute kidney injury, cholangitis infective, failure to thrive, hypercalcemia, hyponatremia, small intestinal obstruction and urinary tract infection (each n = 3) [14, 25]. Fatal TEAEs occurred in 4.1% of FIGHT-202 participants; however, none were deemed to be related to the drug [25].
Phosphorus imbalances were evaluated as notable, drug-related adverse events of special interest (AESIs) in clinical trials of pemigatinib [30]. Hyperphosphataemia is an expected pharmacodynamic effect of FGFR inhibitors, such as pemigatinib (Sect. 2). In FIGHT-202, 60.3% of pemigatinib recipients experienced hyperphosphataemia, regardless of cause [25]; the median time to onset was 15 days. Of note, all episodes of hyperphosphataemia were mild-to-moderate (grade 1–2) in severity, and were managed using several strategies, including dietary phosphate restriction, phosphate-lowering therapy and pemigatinib dose modifications [25]. Hyperphosphataemia (defined as a laboratory value > ULN) was reported in 93% of patients in the updated pooled cancer population and had a median time to onset of 8 days [14]. One-third (33%) of patients required phosphate-lowering therapy [14].
Whereas hyperphosphataemia is a known on-target side effect of FGFR inhibitors, such as pemigatinib, hypophosphataemia reported in clinical trials of these agents, such as FIGHT-202, may reflect the overcorrection of elevated phosphorous levels through the use of phosphate-lowering therapy and/or decreased nutrient intake as a result of other adverse events, such as stomatitis [19]. In FIGHT-202, 23% of pemigatinib recipients experienced hypophosphataemia, regardless of cause; 12% experienced grade 3 hypophosphataemia, irrespective of causality. There were no cases of grade 4 hypophosphataemia [25].
Serous retinal detachment (SRD) encompassing retinal pigment epithelial detachment (RPED) and nail toxicity have also been evaluated as AESIs in clinical trials of pemigatinib [30]. In FIGHT-202, 4% of pemigatinib-treated patients experienced SRD-related TEAEs. All such events were mild-to-moderate (grade 1–2) in severity, with the exception of a single grade 3 event that was classified as being of rhegmatogenous origin and unrelated to treatment [25]. RPED was observed in 11% of patients in the updated pooled cancer population, with grade 3–4 RPED reported in 1.3% of patients [14]. The median time to first onset of RPED was 56 days; it led to pemigatinib dose reduction, dose interruption and permanent discontinuation in 1.3%, 3.1% and 0.2% of patients, respectively. Among patients requiring pemigatinib dose modification for RPED, 76% had their RPED resolved or improved to a grade 1 level of severity [14]. Nail toxicities were seen in 62 (42%) of FIGHT-202 participants, with the most common (incidence > 3%) being nail discolouration (9.6%), onychomadesis (9.6%), onycholysis (8.9%), nail dystrophy (7.5%), paronychia (6.8%), onychoclasis (6.2%) and nail disorder (3.4%) [25]. Grade ≥ 3 nail toxicities occurred in three (2%) of patients [paronychia, onychoclasis and nail disorder (each n = 1)] [25, 30]. The median time to onset of nail toxicity was 6.0 months. Nail toxicities leading to pemigatinib dose reductions and interruptions occurred in five (3%) and six (4%) of the 146 patients, respectively [25].
The longer-term tolerability profile of pemigatinib as assessed at the time of both the updated [26] and final [27] efficacy analyses, was consistent with that in the shorter-term, as assessed at the time of the primary efficacy analysis [25]; no new safety signals were identified [26, 27].
6 Dosage and Administration of Pemigatinib
In the EU [13], oral pemigatinib is conditionally approved as monotherapy for the treatment of adults with locally advanced or metastatic CCA with a FGFR2 fusion or rearrangement that have progressed after ≥ 1 prior line of systemic therapy. Similarly, in the USA [14], pemigatinib received accelerated approval for the treatment of adults with previously treated, unresectable locally advanced or metastatic CCA with a FGFR2 fusion or other rearrangement.
The recommended intermittent dosing schedule of pemigatinib is 13.5 mg orally once daily for 2 consecutive weeks followed by 1 week off therapy in 21-day cycles; treatment should be continued until disease progression or unacceptable toxicity occurs [13, 14]. Tablets should be swallowed whole, with or without food [13, 14].
Consult local prescribing information for further details pertaining to the use of pemigatinib, including: monitoring and management strategies for hyperphosphataemia, hypophosphataemia and SRD/RPED; recommended dose modifications for managing potential DDIs; and its use in special populations.
7 Current Status of Pemigatinib in Advanced Cholangiocarcinoma
Pemigatinib is a once-daily, oral, small molecule, potent and selective, ATP-competitive, reversible inhibitor of FGFR1–3 (Sects. 2 and 3). Over the course of the open-label, single-arm, multinational, phase 2 FIGHT-202 study, previously treated adults with locally-advanced or metastatic CCA (almost exclusively iCCA) harbouring an FGFR2 fusion or rearrangement who received pemigatinib achieved an ORR of 37%, a DCR of 82%, a median PFS of 7.0 months and a median OS of 17.5 months (Sect. 4). Of note, the mechanisms of acquired resistance to pemigatinib reflected those to other ATP-competitive, reversible FGFR inhibitors in FGFR2-rearranged CCA [31]. Pemigatinib was generally well tolerated and had a manageable safety profile in FIGHT-202; as expected, hyperphosphataemia (exclusively grade 1–2 in severity) was the most common TRAE (Sect. 5).
Based on early (initial) findings from FIGHT-202, pemigatinib became the first molecularly targeted therapy to be approved for use in the setting of pretreated, unresectable or advanced CCA with an FGFR2 fusion or rearrangement [1] (Sect. 1). Continued approval of this agent is dependent upon further demonstration of its clinical benefits in FIGHT-302 [8], an additional (confirmatory) trial that is currently ongoing. Begun in June 2019, FIGHT-302 [8] (NCT03656536) is an open-label, randomized, global, phase 3 study comparing the efficacy and safety of pemigatinib versus that of traditional standard-of-care cisplatin plus gemcitabine combination chemotherapy (CisGem) in the first-line treatment of patients with advanced CCA with FGFR2 rearrangements [8]. The primary endpoint is PFS; secondary endpoints include ORR, DCR and OS. Importantly, because ocular toxicities can occur with FGFR inhibitors, FIGHT-302, unlike previous studies of pemigatinib (including FIGHT-202) [14], will incorporate comprehensive ophthalmic examinations at screening and after every three cycles, or as clinically indicated, during the study [8]. Target enrolment is 432 participants, and the estimated primary completion and study completion dates are October 2027 and July 2028, respectively. Of note, China is one of the participating countries in FIGHT-302, and an encouraging and durable survival benefit of pemigatinib has been seen in Chinese patients in a phase 2 bridging study of FIGHT-202 [32, 33].
The approvals of pemigatinib in the EU and USA are acknowledged in the latest update of the ESMO clinical practice guideline for the diagnosis, treatment and follow-up of biliary tract cancer, as is the approval of futibatinib (an irreversible inhibitor of FGFR1–4) in the USA [1]. The update also acknowledged the approval of infigratinib, a second reversible inhibitor of FGFR1–3, in the USA [1]; however, the manufacturer has since withdrawn this agent from the market in this region [34]). According to the ESMO advice, FGFR inhibitors (where available) are recommended for the treatment of patients with FGFR2 fusions whose (advanced) disease has progressed after ≥ 1 prior line of systemic therapy [1]. In addition, pemigatinib and futibatinib are considered useful as subsequent-line therapies for unresectable and metastatic CCA with FGFR2 fusion or rearrangement that has progressed after primary treatment, according to the current NCCN clinical practice guideline on biliary tract cancer [35]. ESMO and NCCN guidelines both recommend routine molecular profiling of patients with advanced CCA using next-generation sequencing [36]; practical approaches to FGFR2 testing (focusing on which patients to test, when and how) are discussed in detail elsewhere [37].
Future aims for pemigatinib include establishing its efficacy and safety relative to other selective FGFR inhibitors in the intended patient population, and its safety, effectiveness and cost-effectiveness in clinical practice. In terms of currently available data, pemigatinib did not differ significantly from futibatinib with respect to efficacy outcomes, based on an unanchored matching-adjusted indirect comparison (MAIC) of their respective single-arm phase 2 registrational studies (FIGHT-202 and FOENIX-CCA2, respectively [8]) [38]. The results of an anchored MAIC of pemigatinib versus futibatinib using data from their ongoing CisGem-controlled phase 3 confirmatory trials (FIGHT-302 and FOENIX-CCA3) are therefore awaited with interest, as are, ultimately, findings from appropriately designed head-to-head studies. Recently reported data from the global pemigatinib expanded access program (EAP) indicate the safety of the drug in a real-world setting is consistent with that observed in FIGHT-202 [39]. Further studies evaluating not only the safety, but also the effectiveness of pemigatinib in clinical practice are desirable. Regarding its cost-effectiveness, pemigatinib has been approved for use within the National Health Service in England and Wales [40] and Scotland [41]. Pharmacoeconomic studies focusing on other countries where the drug is available are warranted.
Presently, therefore, pending completion of FIGHT-302, pemigatinib provides a valuable targeted therapy for pretreated patients with advanced CCA harbouring a FGFR2 fusion or rearrangement.
Data Selection Pemigatinib: 304 Records Identified
Duplicates removed | 4 |
Excluded during initial screening (e.g. press releases; news reports; not relevant drug/indication; preclinical study; reviews; case reports; not randomized trial) | 40 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase 1/2 trials) | 219 |
Cited efficacy/tolerability articles | 8 |
Cited articles not efficacy/tolerability | 33 |
Search Strategy: EMBASE, MEDLINE and PubMed from 1946 to present. Clinical trial registries/databases and websites were also searched for relevant data. Key words were pemigatinib, pemazyre, IBI-375, INCB-054828, Cholangiocarcinoma, cholangiocellular carcinoma, bile duct neoplasms. Records were limited to those in English language. Searches last updated 8 Jan 2024 | |
References
Vogel A, Bridgewater J, Edeline J, et al. Biliary tract cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up*. Ann Oncol. 2023;34(2):127–40.
Banales JM, Marin JJG, Lamarca A, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17(9):557–88.
Javle M, Lee S, Azad NS, et al. Temporal changes in cholangiocarcinoma incidence and mortality in the United States from 2001 to 2017. Oncologist. 2022;27(10):874–83.
Saha SK, Zhu AX, Fuchs CS, et al. Forty-year trends in cholangiocarcinoma incidence in the US: intrahepatic disease on the rise. Oncologist. 2016;21:594–9.
Bertuccio P, Malvezzi M, Carioli G, et al. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J Hepatol. 2019;71(1):104–14.
Florio AA, Ferlay J, Znaor A, et al. Global trends in intrahepatic and extrahepatic cholangiocarcinoma incidence from 1993 to 2012. Cancer. 2020;126(11):2666–78.
Ioffe D, Phull P, Dotan E. Optimal mnagement of patients with advanced or metastatic cholangiocarcinoma: an evidence-based review. Cancer Manag Res. 2021;13:8085–98.
Bekaii-Saab TS, Valle JW, Van Cutsem E, et al. FIGHT-302: first-line pemigatinib vs gemcitabine plus cisplatin for advanced cholangiocarcinoma with FGFR2 rearrangements. Future Oncol. 2020;16(30):2385–99.
Harding JJ, Khalil DN, Fabris L, et al. Rational development of combination therapies for biliary tract cancers. J Hepatol. 2023;78(1):217–28.
Neumann O, Burn TC, Allgäuer M, et al. Genomic architecture of FGFR2 fusions in cholangiocarcinoma and its implication for molecular testing. Br J Cancer. 2022;127(8):1540–9.
Goyal L, Kongpetch S, Crolley VE, et al. Targeting FGFR inhibition in cholangiocarcinoma. Cancer Treat Rev. 2021;95: 102170.
Hoy SM. Pemigatinib: first approval. Drugs. 2020;80(9):923–9.
Incyte Biosciences Distribution B.V., Pemazyre 4.5 mg, 9 mg and 13.5 mg tablets : EU summary of product charcteristics. 2021. https://www.ema.europa.eu. Accessed 1 Nov 2023.
Incyte Corporation. PEMAZYRE™ (pemigatinib) tablets, for oral use: US prescribing information. 2022. https://www.pemazyre.com/pi/pi. Accessed 1 Nov 2023.
Incyte Corporation. Incyte announces approval of Pemazyre® (pemigatinib) in Japan for the treatment of patients with unresectable biliary tract cancer (BTC) with a fibroblast growth factor receptor 2 (FGFR2) fusion gene, worsening after cancer chemotherapy [media release]. 23 Mar 2021. http://www.incyte.com.
Incyte Corporation. Incyte announces Japanese approval of pemazyre® (Pemigatinib) for the treatment of patients with myeloid/lymphoid neoplasms (MLNs) [media release]. 27 Mar 2023. http://www.incyte.com.
Liu PCC, Koblish H, Wu L, et al. INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS ONE. 2020;15(4):1–16.
Rodon J, Damian S, Furqan M, et al. Clinical and translational findings of pemigatinib in previously treated solid tumors with activating FGFR1-3 alterations in the FIGHT-207 study [abstract no. CT016 plus poster]. Cancer Res. 2023;83(8 Suppl):CT016.
Kommalapati A, Tella SH, Borad M, et al. FGFR inhibitors in oncology: insight on the management of toxicities in clinical practice. Cancers (Basel). 2021;13(12):2968.
Ji T, Chen X, Liu X, et al. Population pharmacokinetics analysis of pemigatinib in patients with advanced malignancies. Clin Pharmacol Drug Dev. 2022;11(4):454–66.
Subbiah V, Iannotti NO, Gutierrez M, et al. FIGHT-101, a first-in-human study of potent and selective FGFR 1–3 inhibitor pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies. Ann Oncol. 2022;33(5):522–33.
Ji T, Rockich K, Epstein N, et al. Evaluation of the pharmacokinetics of pemigatinib in patients with impaired hepatic or renal function. Br J Clin Pharmacol. 2022;88(1):237–47.
Ji T, Rockich K, Epstein N, et al. Evaluation of drug-drug interactions of pemigatinib in healthy participants. Eur J Clin Pharmacol. 2021;77(12):1887–97.
Ji T, Chen X, Yeleswaram S. Evaluation of drug-drug interaction potential for pemigatinib using physiologically based pharmacokinetic modeling. CPT Pharmacometrics Syst Pharmacol. 2022;11(7):894–905.
Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21(5):671–84.
Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated locally advanced/metastatic cholangiocarcinoma (CCA): update of FIGHT-202 [abstract no. 4086 plus poster]. J Clin Oncol. 2021;39(15 Suppl):4086.
Vogel A, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated locally advanced or metastatic cholangiocarcinoma: final results from FIGHT-202 [abstract no. O-2 plus poster]. Ann Oncol. 2022;33(S4):S379.
Bibeau K, Féliz L, Lihou CF, et al. Progression-free survival in patients with cholangiocarcinoma with or without FGF/FGFR alterations: a FIGHT-202 post hoc analysis of prior systemic therapy response. JCO Precis Oncol. 2022;6:1–10.
Abou-Alfa G, Sahai V, Hollebecque A, et al. Progression-free survival in patients with cholangiocarcinoma with FGFR2 fusions or rearrangements: a FIGHT-202 post-hoc analysis of prior systemic therapy response [abstract no. SO-4 plus oral presentation]. Ann Oncol. 2021;32(Suppl 3):S203–4.
European Medicines Agency. Pemazyre (pemigatinib): assessment report. https://www.ema.europa.eu. Accessed 17 Nov 2023.
Silverman IM, Hollebecque A, Friboulet L, et al. Clinicogenomic analysis of FGFR2-rearranged cholangiocarcinoma identifies correlates of response and mechanisms of resistance to pemigatinib. Cancer Discov. 2021;11(2):326–39.
Shi G, Huang X, Wen T, et al. Pemigatinib in Chinese patients with advanced/metastatic or surgically unresectable cholangiocarcinoma Including FGFR2 fusion or rearrangement: updated overall survival from an open-label, single-arm, multicenter phase II study [abstract no. CT153]. Cancer Res. 2023;83(8 Suppl):319.
Shi GM, Huang XY, Wen TF, et al. Pemigatinib in previously treated Chinese patients with locally advanced or metastatic cholangiocarcinoma carrying FGFR2 fusions or rearrangements: a phase II study. Cancer Med. 2023;12(4):4137–46.
Subbiah V, Verstovsek S. Clinical development and management of adverse events associated with FGFR inhibitors. Cell Rep Med. 2023;4(10):101204.
NCCN clinical practice guidelines in oncology (NCCN guidelines®) biliary tract cancers version 2.2023—May 10, 2023. https://www.nccn.org. Accessed 1 Nov 2023.
Normanno N, Martinelli E, Melisi D, et al. Role of molecular genetics in the clinical management of cholangiocarcinoma. ESMO Open. 2022;7(3):100505.
Angerilli V, Fornaro L, Pepe F, et al. FGFR2 testing in cholangiocarcinoma: translating molecular studies into clinical practice. Pathologica. 2023;115(2):71–82.
Paine A, Thom H, Wacheck V, et al. Matching-adjusted indirect comparison of futibatinib versus chemotherapy and pemigatinib in cholangiocarcinoma patients with FGFR2 fusions/rearrangements [abstract no. CO78 plus poster]. Value Health. 2022;25(12 Suppl):S33.
Lindley A, Prager G, Bitzer M, et al. Global expanded access program (EAP) to pemigatinib for patients (pts) with previously treated locally advanced or metastatic cholangiocarcinoma (CCA) and FGFR2 fusions or rearrangements [abstract no. 335]. Oncol Res Treat. 2022;45(Suppl 3):81.
National Institute for Health Care Excellence. Pemigatinib for treating relapsed or refractory advanced cholangiocarcinoma with FGFR2 fusion or rearrangement. Technology appraisal guidance [TA722]. https://www.nice.org.uk. Accessed 1 Nov 2023.
Scottish Medicines Consortium. SMC2399. Pemigatinib 4. 5mg, 9 mg, and 13. 5mg tablets (Pemazyre®). https://www.scottishmedicines.org.uk. Accessed 17 Nov 2023.
Acknowledgements
During the peer review process, the manufacturer of pemigatinib was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
James E. Frampton is a salaried employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
The manuscript was reviewed by: C. Hierro, Medical Oncology Department. Catalan Institute of Oncology-Badalona, Badalona-Applied Research Group in Oncology (B-ARGO), Badalona, Barcelona, Spain; M. A. Morse, Department of Medicine, Duke University Medical Center, Durham, North Carolina, USA.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Frampton, J.E. Pemigatinib: A Review in Advanced Cholangiocarcinoma. Targ Oncol 19, 107–114 (2024). https://doi.org/10.1007/s11523-023-01024-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-023-01024-x