
Abstract
PARP (poly(ADP-ribose) polymerase) inhibitors represent a novel class of anti-cancer therapy; they take advantage of synthetic lethality and induce cell death by exploiting a defect in DNA repair. This class of medication was initially evaluated in patients with BRCA-associated tumors, but efficacy was also demonstrated in other populations. Since 2014, four PARP inhibitors have been approved in various indications: olaparib, niraparib, and rucaparib in high-grade serous ovarian cancer, and olaparib and talazoparib in metastatic breast cancer. The exact indications and study populations vary slightly between the different approvals in both disease states but there is significant overlap. PARP inhibitors continue to be investigated in ongoing clinical trials. In line with other targeted therapies, benefit appears to be strongest in a distinct population of patients with BRCA mutations or other defects in homologous recombination repair. Combination therapies, which include anti-angiogenesis agents and immunotherapy, show promise as a strategy to broaden efficacy for unselected patients. Initial studies of PARP inhibitors in combination with chemotherapy were limited by toxicity, but further studies are underway. To date, head-to-head trials comparing various PARP inhibitors have not been conducted, so questions remain in terms of choosing a PARP inhibitor to administer when indications overlap, as well as how to sequence these medications. Here we review both completed and ongoing clinical trials involving PARP inhibitors and mechanisms of resistance to this class of drugs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
PARPs are a family of enzymes that play a role in DNA repair. |
PARP inhibitors have shown promising anti-cancer activity in a number of studies, and FDA approvals were granted in advanced ovarian cancer and metastatic breast cancer. |
Tumors carrying mutations in BRCA1 or BRCA2 genes and other genes implicated in homologous repair deficiency are particularly sensitive to PARP inhibition. |
Combination therapy is increasingly explored as a means to augment efficacy of PARP inhibition and overcome resistance mechanisms. |
1 Introduction
DNA is continually damaged through various endogenous and exogenous stressors. Mechanisms of damage include single-strand breaks, double-strand breaks, base modifications, and inter-strand and intra-strand cross-links. In the absence of adequate repair mechanisms, DNA damage results in cell death.
Poly(ADP-ribose) polymerase-1 (PARP1) and poly(ADP-ribose) polymerase-2 (PARP2), members of the PARP superfamily of enzymes, play a key role in DNA damage response (DDR) by acting as DNA damage sensors and signal transducers. PARP1 is the most extensively studied of these enzymes. In a process known as poly-ADP-ribosylation (PARylation), PARP1transfers ADP-ribose from nicotinamide adenine dinucleotide (NAD+) to target proteins [1], which in turn enables recruitment of DNA repair proteins [2, 3]. PARP1 eventually PARylates itself and releases itself from repaired DNA [4, 5]. Development of PARP inhibitors was fueled by the observation of small molecule nicotinamide analogs suppressing PARylation and thus enhancing cytotoxicity of DNA-damaging agents [6].
The mechanism of action of PARP inhibitors is complex and involves two separate methods of cytotoxicity. First, PARP inhibitors interact with the binding site of the PARP enzyme cofactor, β-NAD+, in the catalytic domain of PARP1 and PARP2, thus inhibiting enzymatic activity. Errors in repair increase, which leads to cell death. The second action involves “trapping” of PARP1, whereby the prevention of autoPARylation and the release of PARP1 from the damaged DNA ultimately leads to cell death in double-strand repair-deficient cells [7,8,9,10].
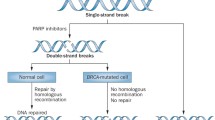
In most cell lines PARP inhibition alone was insufficient to cause cell death [11]. Lack of lethality imparted by PARP inhibition alone was demonstrated by animal models, as mice engineered to lack PARP1 enzyme (PARP1-/-) were both viable and fertile [12]. However, in 2005, two seminal papers demonstrated that breast cancer cells with mutations in BRCA1 or BRCA2 genes were extremely sensitive to PARP inhibition [13, 14]. BRCA1 and BRCA2 proteins are important in homologous recombination for repair of double-stranded breaks. When BRCA1 and BRCA2 genes are mutated, their subsequent proteins are unable to repair DNA damage, ultimately leading to cell death (Fig. 1) [15, 16]. This idea can be more broadly applied; cells with defects in the double-strand repair pathway appear to be more dependent on PARP1 to maintain genomic integrity, thus are more sensitive to the cytotoxic effects of PARP inhibition [17].

Synthetic lethality with PARP inhibition and BRCA mutation. Single stranded breaks in DNA can be repaired by PARP pathway with NAD + as a cofactor. Treatment with a PARP inhibitor prevents repair of single stranded breaks and instead leads to double stranded breaks which then undergo homologous recombination. BRCA mutated cells are unable to undergo DNA repair via the homologous recombination pathway thus leading to cell death. This illustrates the concept of synthetic lethality with PARP inhibition and BRCA mutation, as both are required for cell death
Deleterious mutations of BRCA1 and BRCA2 lead to an increased risk of cancer, including breast, ovarian, prostate, and pancreatic cancers, because of the impaired homologous recombination repair pathways [18,19,20,21]. BRCA-deficient cells rely on the single-strand annealing sub-pathway of homologous recombination or non-homologous end-joining to repair damaged DNA. However, both these repair pathways are prone to errors leading to an increased rate of mutations and cell death. Defects in homologous recombination repair can be caused by other mechanisms aside from mutations in BRCA1 and BRCA2. Loss of function of other proteins such as RecA homologue RAD51, ataxia telangiectasia-mutated (ATM), ataxia telangiectasia and Rad3-related (ATR) proteins also demonstrate impaired homologous recombination [22]. Tumors with these abnormalities, described as “BRCA-like”, are often sensitive to similar therapies [23]. Phenotypically, triple-negative breast cancer and high-grade serous ovarian cancer have “BRCA-like” features [24, 25].
In the era of targeted therapy, there have been many instances of successful targeting of driver mutations of tumor growth. The development of PARP inhibitors represents a departure from this paradigm of cancer drug development. A unique aspect of PARP inhibition is the reliance on the tumor’s limitations, in this case the inability to repair DNA, in order to cause tumor cell death [26, 27]. PARP inhibition relies on the concept of synthetic lethality, a phenomenon that arises when combined mutation or blockade of two genes leads to cell death, whereas mutation or blockade of only one of the genes does not. In this case, PARP inhibition induces DNA damage that cannot be repaired by tumors with deficient homologous recombination repair. Cells with intact homologous recombination remain unaffected, thus minimizing off-target effects and toxicities.
Another treatment strategy focuses on using PARP inhibitors in combination with cytotoxic chemotherapy, antiangiogenesis agents, radiotherapy, immune therapy, or by inhibiting proteins in the DNA damage-response pathway. Rationale for this approach is discussed later in this review; as of now combination therapy with PARP inhibition remains investigational. As monotherapy, several PARP inhibitors have been approved by the US Food and Drug Administration (FDA) (Table 1) and more are being investigated in clinical trials. PARP inhibitors target the same binding site, but have differences in their chemical structures translating into different pharmacokinetic properties. For instance, talazoparib is more potent in PARP1 trapping when compared to niraparib, but niraparib is more potent at PARP1 trapping than olaparib and rucaparib [9]. Veliparib on the other hand is more effective at inhibiting PARylation than in trapping PARP1 [7]. These differences translate into distinct side-effect profiles, and it remains to be seen if they have implications for efficacy, biomarker development, etc. A summary of approved and investigational PARP inhibitors is presented below.
2 Olaparib (AZD2281)
Olaparib, initially known as AZD2281, is an oral PARP inhibitor. Pre-clinical studies showed impressive cytotoxicity of PARP inhibition in cell lines with BRCA1/2 mutations [14], a finding that was confirmed by animal studies using a genetically engineered mouse model for BRCA1-associated breast cancer [28]. In clinical studies, olaparib was mostly evaluated in BRCA-associated tumors. To date it is FDA approved in advanced BRCA-mutated ovarian cancer, advanced BRCA-mutated, HER2-negative breast cancer, and as maintenance therapy for platinum-sensitive ovarian cancer regardless of BRCA mutation status (Tables 1 and 2). More recently it was granted an orphan drug designation for maintenance therapy in BRCA-mutated metastatic pancreatic cancer. Studies for other indications are ongoing.
2.1 Phase I Studies
Olaparib was the first PARP inhibitor tested in clinical trials as a single agent. Results from the phase I study of olaparib were published by Fong et al. in 2009 [29]. A total of 60 patients with advanced solid tumors with tumor types including ovarian, breast, colorectal, melanoma, sarcoma, and prostate were enrolled. Initially, BRCA mutation was not required for eligibility; however, in the expansion phase only BRCA mutation carriers were enrolled to allow enrichment of this population. Ultimately, 23 of the 60 patients carried BRCA1 or BRCA2 mutations. Durable objective antitumor activity was observed only in BRCA mutation carriers, whereas no objective antitumor response was seen in the non-BRCA carriers. Two BRCA carriers had tumors not typically associated with BRCA mutation status, including one with small-cell lung cancer and one with vaginal adenocarcinoma. Both of these patients progressed rapidly after initiating olaparib. Of the 19 patients with BRCA mutations, 63% had a clinical benefit with olaparib. These patients had ovarian, breast, or prostate cancer. Olaparib 400 mg twice daily (BID) was determined to be the maximum tolerated dose (MTD), although antitumor efficacy was observed with dosing as low as 60 mg BID. Generally, olaparib was well tolerated. Most adverse effects were grade 1 or 2 and these included nausea (32%), fatigue (30%), taste alteration (13%), and anorexia (3%). Grade 3–4 thrombocytopenia, lymphopenia, nausea, vomiting, and dizziness were each seen in one patient at the 400-mg twice-daily (BID) dosing [29]. Given the lack of response in non-BRCA mutation carriers, future studies focused mostly on patients with BRCA mutations or homologous recombination deficiency in typical BRCA-associated tumors.
Other phase I studies examined olaparib in combination with chemotherapy. Two trials looking at olaparib in conjunction with cisplatin and gemcitabine in adults with solid tumors observed significant myelosuppression, even with relatively low doses of olaparib [30, 31]. Excess myelosuppression was also noted when olaparib was given in combination with dacarbazine [32] and topotecan [33]. Thus, further trials with these particular combinations are not being pursued.
BRCA mutations and other forms of homologous recombination deficiency have been reported in a small but significant proportion of pancreatic cancers [34], and mice bearing human pancreatic tumor xenografts showed tumor response to PARP inhibition in combination with gemcitabine [35]. As a result, a phase I clinical trial examined this combination in patients with solid tumors, including 22 patients in a dose-expansion cohort with chemotherapy-naïve advanced pancreatic cancer. Presence of a BRCA mutation was not required for enrollment. Dose-limiting toxicities were noted in four patients of the dose-escalation cohorts, all of which resolved with gemcitabine dose modification. Overall, 38 of the 47 patients treated with olaparib and gemcitabine reported grade 3 or 4 adverse events, the most common of which were hematologic toxicities, occurring in 55% of patients. Other common side effects included nausea, vomiting, and an increase in alanine aminotransferase. Intermittent olaparib dosing was explored, and ultimately olaparib 100 mg BID on days 1–14 in combination with gemcitabine 600 mg/m2 on day 1 was selected for the expansion phase based on acceptable tolerability. Overall response rates (ORRs) were 10% (n = 4/41) in the dose-escalation phase. In the dose-expansion phase the ORR was 27% (n = 4/14) for the combination compared with 14% (n = 1/7) with gemcitabine alone, though the difference was not significant. Analysis by BRCA mutation status was not possible given the small number of patients with known BRCA mutation status [36]. Though the numbers were small, the response rates were encouraging, and olaparib is being further investigated with pancreatic cancer patients with BRCA mutations both as monotherapy (NCT01078662) and in combination with chemotherapy (NCT01296763).
Olaparib in combination with pegylated liposomal doxorubicin was examined in a phase I study in patients with solid cancers, the majority having breast or ovarian cancer. The maximum tolerated dose was reached using continuous olaparib 400 mg BID and pegylated liposomal doxorubicin 40 mg/m2 on day 1 every 28 days. Fourteen of 44 patients (33%) had an objective response; 13 of the responders had ovarian cancer, of which 10 were platinum sensitive and 11 had a germline BRCA mutation [37]. A phase II trial further examining this combination in patients with platinum-resistant ovarian cancer is ongoing (NCT03161132).
A phase I/II multicenter trial evaluated the tolerability of olaparib in combination with weekly paclitaxel in women with triple-negative breast cancer. A total of 19 patients were enrolled. Although over a third of patients experienced grade 3 or 4 adverse events, the most common being myelosuppression, response rates were promising. Despite 15 patients having received prior taxane chemotherapy, 37% had confirmed partial response (PR) and another 16% had unconfirmed response. One patient who was not a BRCA mutation carrier even enjoyed a complete radiologic remission and had continued on olaparib 4 years after enrollment began [38].
There are ongoing studies examining the role of various PARP inhibitors with radiotherapy. Preclinical studies demonstrated activity of PARP inhibition in combination with radiation in head and neck squamous cell carcinoma cell lines. A phase I study evaluated escalating doses of olaparib with fixed doses of cetuximab and radiation in heavy smokers with locally advanced head and neck cancer. Of the 13 patients enrolled, three progressed at 14 months. With a starting dose of olaparib 50 mg BID, toxicities mainly included grade 3 dermatitis and acneiform rash [39]. Results with longer follow-up are awaited.
In summary, olaparib monotherapy was found to be fairly well tolerated at the maximum tolerated dose of 400 mg BID, whereas in combination with chemotherapy, response rates are promising but at the cost of excess myelosuppression. When given in combination, a dose reduction of 100 mg BID is generally more tolerable with either intermittent or continuous dosing. The greatest efficacy of olaparib is noted in BRCA-mutated tumors.
2.2 Phase II/III Studies
2.2.1 Ovarian Cancer
A phase II proof-of-concept study examined olaparib in BRCA-mutated ovarian cancer. Patients received olaparib initially at the MTD of 400 mg BID, and subsequently at 100 mg BID. Tumor response rates were 33% with the higher dose compared to 12.5% at 100 mg BID dose. This study confirmed olaparib is active in BRCA-deficient ovarian cancer and is more effective at the 400 mg BID dose, thus paving the way for further studies [40].
To answer the question of whether PARP inhibition could be effective in BRCA-like cancers, a phase II study was conducted with women with BRCA-mutated breast or ovarian cancer as well as non-BRCA-mutated triple-negative breast and high-grade serous ovarian cancers, phenotypes typically seen with BRCA1 mutations. A total of 91 patients, 65 with ovarian and 26 with breast cancer, were treated with olaparib 400 mg BID. Responses were seen exclusively in the ovarian cancer cohort, with response rates of 41% in the BRCA-mutated patients and 24% in the non-BRCA-mutated patients [41]. Though BRCA-mutated patients demonstrated better responses, there did seem to be activity in BRCA wild-type patients, providing rationale for further study in this high-grade serous ovarian population.
A larger phase II single-arm trial conducted by Kaufman et al. confirmed activity in BRCA-mutated ovarian cancer. Patients with germline BRCA 1 or 2 mutations and recurrent metastatic cancer were enrolled, including 193 women with platinum-resistant ovarian cancer. Patients were administered olaparib 400 mg BID. Among the patients with ovarian cancer, ORR was about 30% in a notably heavily pretreated population who had received a median of four prior therapies [42]. Based on these results, in 2014 the FDA granted approval for olaparib in women with germline BRCA-mutated ovarian cancer that received three or more prior lines of chemotherapy.
In 2012, Ledermann et al. published a phase II randomized, double-blind, placebo-controlled trial, Study 19, examining the role of olaparib in maintenance therapy. A total of 264 women with recurrent platinum-sensitive high-grade ovarian cancer that responded to most recent therapy, were randomized to olaparib 400 mg daily or placebo as maintenance therapy. Over half of the patients had unknown BRCA mutation status, 15% of patients in each arm were BRCA negative, with the remainder having BRCA1 or BRCA2 mutations. Patients on olaparib demonstrated improved progression-free survival (PFS) of 8 months with olaparib versus 5 months with placebo. In subgroup analysis, olaparib showed superiority in both BRCA-positive and BRCA-status unknown patients, though PFS benefit was greater in BRCA-mutated patients. BRCA-negative women were not included in the subgroup analysis due to the small sample size [43]. The initial study demonstrated a non-statistically significant trend towards survival benefit. In an update published in 2018 with more mature data, a survival benefit was observed, though it did not meet the predefined threshold for statistical significance. Cross-over was not permitted in the trial, but 13% of patients in the placebo group went on to receive PARP inhibitors outside of the trial, thus confounding the survival data. A proportion of patients enjoyed a long-term response, with nearly a quarter of patients on treatment for 2 years and 11% of patients on treatment for over 6 years. The long-term responders were made up of both BRCA-mutated and BRCA wild-type patients [44].
A rare but important safety signal arose in this trial: three cases of myelodysplastic syndrome (MDS) or acute myelogenous leukemia (AML) were observed, with two cases in patients randomized to olaparib and one in a patient randomized to placebo.
The phase III SOLO2/ENGOT-Ov21 (NCT01874353) evaluated olaparib maintenance in patients with recurrent high-grade ovarian cancer, but restricted enrollment to women with a known germline or sporadic BRCA mutation. The trial randomized 295 patients that had received two lines of previous chemotherapy in a 2:1 ratio to olaparib maintenance or placebo. Those on the olaparib arm had an improved PFS of 19.1 months versus 5.5 months with placebo (HR 0.30, 95% CI 0.22–0.41) [45]. In a historical comparison to Study 19, PFS was about 10 months longer in the olaparib arm in this trial, presumably due to selection of BRCA-mutated tumors. A follow-up study examining quality-of-life metrics showed no significant detriment on health-related quality of life compared to placebo [46], thus indicating it is an overall well-tolerated maintenance strategy. The results of Study 19 and SOLO2 led to FDA approval for maintenance therapy after response to platinum-based chemotherapy in patients with recurrent ovarian cancer regardless of BRCA status in 2017.
Most recently, the phase III SOLO1 (NCT01844986) trial led to the newest FDA approval for olaparib; maintenance therapy in the first-line setting for ovarian cancer. This trial randomized 391 women with newly diagnosed advanced ovarian cancer, almost all of whom were germline BRCA mutation carriers, in a 2:1 ratio to olaparib versus placebo maintenance. At 41-month follow-up, this placebo-controlled trial achieved its primary end point of improvement in PFS. Median PFS was not reached in the olaparib group and was 13.8 months with placebo. Survival data were not mature at time of publication, but a non-significant trend towards improved overall survival (OS) was seen with olaparib. Despite a longer duration of treatment when compared to maintenance therapy in the relapsed setting, the toxicity profile was fairly similar. A 1% incidence of MDS or AML occurred in the olaparib group, compared to none in the placebo group [47]; whether this increases with longer follow-up will need to be assessed. The question of whether PARP inhibitor maintenance can be expanded to all comers will need to be answered. Bevacizumab, a VEFG inhibitor, already has FDA approval in advanced platinum-sensitive ovarian cancer in combination with chemotherapy and as maintenance therapy. An ongoing phase III study, PAOLA-1 (NCT02477644), is evaluating the efficacy of the combination of these two maintenance options in the first-line setting, with results expected in late 2019. Patients with newly diagnosed ovarian cancer will be randomized to bevacizumab versus bevacizumab plus olaparib maintenance following platinum therapy, and results will be stratified by BRCA mutation status [48].
Olaparib in combination with chemotherapy for recurrent platinum-sensitive ovarian cancer was examined in a randomized phase II trial [49]. Patients received up to three prior courses of platinum-based chemotherapy with a progression-free interval of at least 6 months. The study randomized 162 patients to either olaparib 200 mg BID on days 1–10 of each 21-day cycle plus paclitaxel 175 mg/m2 and carboplatin AUC 4 intravenously on day 1 or to carboplatin AUC 6 and paclitaxel 175 mg/m2 intravenously on day 1 of each 21-day cycle. Median PFS was significantly longer in the olaparib plus chemotherapy group, 12.2 months, compared to the chemotherapy-alone group, 9.6 months. This difference was more pronounced in the BRCA-mutated patients. Not surprisingly, slight increased toxicity occurred in the combination group, with 65% of patients in the combination group experiencing grade 3 or 4 toxicity, compared to 57% patients in the control arm. A late separation of the PFS curves suggests the maintenance phase was likely the main contributor to the benefit seen. Overall, the authors concluded the combination of chemotherapy and olaparib did not seem to offer a benefit over olaparib maintenance therapy alone [49].
Olaparib and cediranib, a VEGF-inhibitor, was studied in a randomized open-label phase II study for patients with platinum-sensitive, relapsed ovarian cancer or those with germline BRCA mutations. The combination of cediranib plus olaparib led to an improvement in PFS, 17.7 months with the combination compared to 9 months with olaparib monotherapy, in women with recurrent, platinum-sensitive ovarian cancer. Grade 3 or 4 adverse events were more common with the combination [50]. Two large phase III studies have been conducted comparing the combination of olaparib and cediranib to chemotherapy, NRG-GY004 in platinum-sensitive ovarian cancer and NRG-GY005 in platinum-resistant ovarian cancer. Enrollment is complete, but data are not yet mature. ICON 9 (NCT03278717) is a planned phase III follow-up study also looking at this combination; however, recruitment has not started.
Though combination therapy appears to be promising, toxicity may be a limiting factor. Currently, only olaparib monotherapy is FDA approved for women who have progressed after three or more prior lines of treatment irrespective of BRCA-mutation status, and as maintenance therapy in women with BRCA-mutated newly diagnosed and relapsed ovarian cancer.
2.2.2 Breast Cancer
Following the results of the initial phase I study of olaparib, the phase II ICEBERG1 trial (NCT00494234) examining olaparib in BRCA-mutated breast cancer was published. This trial contained two patient cohorts; the first cohort of patients was treated at the phase I MTD of 400 mg BID and the second sequential cohort was given the lowest inhibitory dose of 100 mg BID. The ORR of 46% was higher in cohort 1 compared to cohort 2 with an ORR of 22%. Exploratory analysis showed responses in traditionally treatment-resistant populations, including women with triple-negative breast cancer and heavily pre-treated patients. Toxicity was slightly increased with the higher dose, although overall therapy was well tolerated. In total, grade 3 or 4 adverse events were reported in 41% of patients in the olaparib 400 mg BID cohort and in 33% of patients in the olaparib 100 mg BID cohort. There were no grade 5 events [51].
OlympiAD (NCT02000622) further examined the role of olaparib in BRCA-mutated breast cancer. This phase III, open-label study randomized 302 women with BRCA-mutated, Her2-negative, metastatic breast cancer, who had received no more than two prior lines of chemotherapy, to olaparib versus physician’s choice. The study met its primary endpoint of improved PFS, 7 months with olaparib versus 4.2 months with standard therapy. Olaparib was better tolerated with 36.6% grade 3 or higher toxicity compared to 50.5% with standard therapy [52]. Based on these results, the FDA approved olaparib for metastatic, germline-mutated, Her2-negative breast cancer in early 2018, representing the first non-cytotoxic approval for this indication. In 2019, a published final analysis of OS did not detect a statistically significant survival benefit with olaparib compared to chemotherapy, with median OS 19.3 months with olaparib versus 17.1 months with chemotherapy, p value of 0.513. There was a suggestion of greater benefit among treatment-naïve patients, though this was not statistically significant. Crossover upon progression was not permitted and cannot explain the lack of survival benefit. It is important to note, however, the study was not powered to identify a difference in this endpoint, and olaparib had less overall toxicity compared to chemotherapy [53].
Olaparib’ s potential role as adjuvant therapy in early-stage disease is being examined in OlympiA (NCT02032823), a phase III trial enrolling patients with high-risk, BRCA-mutated, Her2-negative breast cancer who have completed local therapy and at least six cycles of chemotherapy. Patients will be randomized to olaparib 300 mg BID versus placebo for 12 months. Estimated enrollment is 1800 patients and interim results are eagerly awaited [54].
The data for olaparib’s efficacy in non-BRCA-mutated breast cancer is less convincing. The aforementioned Study 19, which included women with BRCA-mutated and “BRCA-like” triple-negative breast cancer, demonstrated disappointing results with no objective response among the 23 patients in the trial. However, of the BRCA-mutated breast cancers, the disease control rate at 8 weeks was 70% compared to 19% in the mutation-negative cohorts [55].
Rationale exists for combining cytotoxic chemotherapy with PARP inhibition, based on the assumption that chemotherapy damages the DNA and PARP inhibition prevents repair of this damage. This strategy is being examined in triple-negative breast cancer. The PARTNER trial (NCT03150576) is an ongoing phase II/III trial examining whether addition of olaparib to platinum-based neoadjuvant chemotherapy increases the pathologic response rate amongst women with localized triple-negative breast cancer [56].
The combination of olaparib and immunotherapy may also be effective, based on the idea that olaparib can induce DNA damage leading to neo-antigen formation and increased efficacy of checkpoint inhibition. The phase II MEDIOLA trial (NCT02734004) examining the combination of olaparib and PDL-1 inhibitor durvalumab in women with metastatic, Her2-negative breast cancer and germline BRCA mutations demonstrated favorable results on interim analysis. Of 30 evaluable patients, a disease control rate of 80% was noted with an ORR of 63% and duration of response of about 9 months. When compared to olaparib monotherapy data from OlympiAD, response rates are roughly similar, but duration of response appears prolonged with the combination by about 2.5 months [57].
The phase II DORA trial ((NCT03167619) is evaluating the role of durvalumab maintenance in patients with advanced triple-negative breast cancer. DORA is a non-comparator randomized trial in which patients who respond to four cycles of platinum-based chemotherapy will be randomized to olaparib maintenance versus olaparib plus durvalumab maintenance [58]. In the future, PARP inhibitor monotherapy or in combination may be explored further for breast cancer patients with somatic BRCA mutations and patients with other pathways of homologous recombination deficiency.
2.2.3 Prostate Cancer
BRCA mutations are associated with other types of malignancies aside from breast and ovarian cancers. A phase II trial was conducted to examine the efficacy of olaparib as monotherapy in patients with germline BRCA1 or BRCA2 mutations irrespective of tumor type. Patients with advanced solid tumors and confirmed germline mutations in BRCA1/2 were enrolled and treated with olaparib 400 mg BID. Of the eight patients with prostate cancer, tumor response rate was 50% [59].
Though BRCA mutations significantly increase the risk of prostate cancer, they are responsible for a minority of prostate cancer cases; germline BRCA2 mutations are present in 1.2% of prostate cancer cases [60] while germline BRCA1 mutations are present in 0.44% of prostate cancer [61]. However, inactivating mutations in homologous repair genes including CHEK2, BRIPI/FANCJ, NSB1, and ATM are more common, occurring in 20–25% of prostate cancers [62].
In the Trial of PARP Inhibition in Prostate Cancer (TOPARP-A) (NCT01682772), men with metastatic castration-resistant prostate cancer that progressed through one or two lines of chemotherapy were treated with olaparib 400 mg BID. A total of 50 patients were enrolled and 49 patients were evaluable for response. The tumor response rate was 33%, with 16/49 showing radiographic response. Tumor aberrations in DNA repair genes were found in 16 patients, including mutations in BRCA2, ATM, FANCA, CHEK2, and PALB2. Of this cohort, tumor response rates were higher at 88% [63].
A trial investigating olaparib monotherapy in men with high-risk biochemically recurrent prostate cancer is currently ongoing (NCT03047135). An exploratory analysis will include biomarker discovery, including somatic DNA mutation analysis, RNA expression analysis, and immunohistochemistry for DNA damage markers. The study initially started enrolling in unselected patients, but if deemed appropriate, enrichment with biomarker-selected patients will occur [64].
PARP combination therapy is also being investigated in prostate cancer. At GU ASCO in 2018 data were presented from a phase II trial of olaparib and abiraterone in metastatic castrate-resistant prostate cancer in unselected patients (NCT01972217). Men were randomized post-docetaxel to olaparib plus abiraterone versus olaparib plus placebo. Improved radiographic PFS was noted with the combination versus the comparator, 13.8 months versus 8.2 months, irrespective of presence of homologous recombination deficiency. However, toxicity increased with the combination with nearly double grade 3 or greater adverse events, 54% versus 28% [65]. A phase III trial (NCT03732820) is planned to further evaluate this approach.
The idea of PARP inhibition augmenting checkpoint inhibition is undergoing evaluation in prostate cancer. Preliminary data from a single-arm pilot study with accrual of 25 patients (NCT02484404) with metastatic, castrate-resistant prostate cancer showed the combination of olaparib and durvalumab yielded a dramatic PSA response of > 50% in eight of 17 patients, six of whom possessed mutations in DNA repair pathways. Accrual for this study is ongoing [66].
To date there are no FDA approvals for PARP inhibitors in prostate cancer, but if further studies do yield an approval, the target population would likely expand beyond BRCA mutation to include any mechanism of homologous recombination deficiency, thus potentially impacting a broader patient base. Proof of clinical benefit of combination therapy with PARP inhibition in unselected patients requires further study.
2.2.4 Gastrointestinal Cancers
Preclinical data suggest gastric cancer lines are more responsive to olaparib when ATM protein levels are low [67]. A phase II study enriched for patients with low ATM tumors randomized patients with advanced gastric cancer to either olaparib 100 mg BID plus paclitaxel 80 mg/m2 on days 1, 8, and 15 per 28-day cycles versus placebo plus paclitaxel 80 mg/m2 on days 1, 8, and 15 per 28-day cycles. A total of 123 patients underwent treatment. The most common grade 3 or 4 toxicity in the combination arm was neutropenia, which occurred in 59% of patients, and anemia, which occurred in 11% of patients. Though PFS was not significantly improved with the combination, OS was 13.1 months with olaparib plus paclitaxel versus 8.3 months with paclitaxel alone [68].
These findings prompted the phase III GOLD trial, recruiting Asian patients with advanced gastric cancer that progressed on first-line therapy. Treatment arms were identical to the preceding phase II trial. The study did not meet its primary endpoint of improved OS. Further exploration is being conducted on the data set to see if markers of DNA damage repair may be associated with sensitivity to PARP inhibitors [69]. Another phase I/II study (NCT03008278) is investigating olaparib in combination with ramucirumab in patients with advanced gastric cancer, irrespective of gene mutations. Similarly, an open phase II trial testing olaparib 400 mg BID in patients with advanced colorectal cancer did not observe any anti-tumor response [70].
Results from the phase III, randomized, double-blind POLO study (nct 02184195) looking at olaparib maintenance for BRCA-mutated pancreatic cancer were reported at ASCO 2019. The trial randomized 152 patients with metastatic pancreatic cancer with germline BRCA1 or BRCA2 mutations to olaparib maintenance or placebo after completion of first-line platinum-based chemotherapy. The primary endpoint of PFS was significantly longer in the olaparib group at 7.4 months, compared to 3.8 months with placebo. Median duration of response was 25 months with olaparib versus 4 months with placebo. At the time of interim analysis, no survival benefit was seen, though these data were not yet mature. Additionally, there were no differences in quality of life between the two groups despite a higher percentage of grade 3 adverse events in the olaparib group [71]. This practice-changing study was the first to target a specific subpopulation of patients with metastatic pancreatic cancer.
3 Rucaparib (AG014699)
Rucaparib is an oral, potent, small molecule inhibitor of PARP-1, PARP-2, and PARP-3. In preclinical studies, rucaparib displayed preferential cytotoxicity to cells with BRCA mutations or those lacking components of the homologous recombination repair pathway [72], but also has demonstrated activity in ovarian cancer cell lines lacking BRCA mutations [73]. It is FDA approved as maintenance treatment in patients with platinum-sensitive recurrent ovarian cancer irrespective of BRCA mutation status (Table 2). Rucaparib also demonstrates synergistic activity with radiation and chemotherapy, including platinum agents and topoisomerase inhibitors, in various human cancer cell lines [73,74,75].
3.1 Phase I/II Trials
Rucaparib has shown promising results in ovarian cancer. The Study 10 trial (NCT01482715) was a multiphase trial assessing rucaparib in patients with germline BRCA1/2-mutated ovarian cancer and other solid tumors [76]. In the phase I portion of the trial, one patient with breast cancer and one patient with ovarian cancer, both of whom had germline BRCA1 mutations, had a complete response at a dose of 360 mg BID and 300 mg daily, respectively. A PR was seen in three breast cancer patients, two ovarian cancer patients, and one pancreatic cancer patient, all of whom had either germline BRCA1/2 or tumor BRCA1 mutations [76]. In the phase II part of the Study 10 trial, 42 females, all of whom contained germline BRCA1- or BRCA2-mutated, platinum-sensitive ovarian cancer received rucaparib 600 mg BID. About a third of the patients received at least three prior lines of platinum-based chemotherapy and the majority had between a 6- and a 12-month disease-free interval after their last platinum-based therapy. The ORR was 59.5% by RECIST criteria and 83.3% when evaluated by RECIST criteria and CA-125 response. The median duration of response was 7.8 months [76].
The ARIEL2 study (NCT01891344) is a two-part, multicenter, phase II trial evaluating efficacy of rucaparib in patients with relapsed, high-grade, platinum-sensitive ovarian cancer. In part 1, patients had to have received at least one or more chemotherapy regimens and the primary endpoint was PFS. The 204 patients enrolled were classified into three groups including BRCA mutated, BRCA wildtype with loss of heterozygosity high, and BRCA wildtype with loss of heterozygosity low. In contrast to the previously discussed ovarian cancer trials with olaparib, the BRCA-mutated group included patients with both germline or somatic BRCA mutations. Genomic loss of heterozygosity (LOH) was considered a biomarker of homologous recombination deficiency regardless of BRCA mutation status. The cut-off for classification as LOH high was at least 14%. Patients in the BRCA-mutated and BRCA wildtype with LOH high group had significantly longer PFS after treatment with rucaparib compared to the BRCA wildtype with LOH low group, HR 0.27 and 0.62, respectively. PFS was 12.8 months, 5.7 months, and 5.2 months, respectively [77].
In part 2, 134 patients were stratified into four groups including if they were platinum sensitive with platinum given as the immediate prior treatment, platinum sensitive with non-platinum treatment given as the immediate prior treatment, platinum resistant, and platinum refractory. ORR and PFS were highest in the platinum-sensitive group with platinum being immediate prior treatment. The ORR was 70% in the platinum-sensitive receiving platinum immediately prior group, 43% in the platinum-sensitive with non-platinum therapy immediately prior group, 25% in the platinum-resistant group, and 0% in the platinum-refractory group. Similarly, PFS was 12.7 months, 7.4 months, 7.3 months, and 5 months, respectively [78]. Based on both the Study 10 and ARIEL2 trials, the FDA granted rucaparib expedited approval for women with either germline or somatic BRCA-mutated, advanced ovarian cancer who have received at least two prior lines of chemotherapy.
Rucaparib has also been given in combination with chemotherapy with positive results. Preclinical studies showed that AG14447, the prodrug of rucaparib, had the most potent enhancing effect of temozolomide in vitro [74]. Thus rucaparib, which is the phosphate salt of AG14447, was selected for a phase I trial in combination with temozolomide. Additionally, the methylated breakdown compound of temozolomide in the plasma induces DNA damage that is repaired by the DNA base excision repair pathway. Therefore, the rationale for using a PARP inhibitor with temozolomide is to inhibit DNA repair triggered by temozolomide-induced DNA damage [79]. A two-part phase I study evaluated the pharmacokinetics and pharmacodynamics of rucaparib in combination with temozolomide and then specifically in those with melanoma [80]. Interestingly, there was some clinical benefit from the combination in melanoma patients, which led to a phase II study evaluating the response rate in chemotherapy-naïve patients with metastatic melanoma. Of the 46 patients recruited, 17.4% had a PR and 17.4% had stable disease [79]. No phase III trial is underway for further assessment of this combination in melanoma.
The phase I study by Wilson et al. assigned 85 patients to four arms giving rucaparib in combination with a different chemotherapy regimen [81]. All the arms administered rucaparib intravenously except for arm A, which administered rucaparib orally to 33 patients. The four arms (A through D) consisted of rucaparib administered in combination with carboplatin, paclitaxel and carboplatin, pemetrexed and cisplatin, and cyclophosphamide, respectively. The MTD for arm A was carboplatin AUC 5 and rucaparib 240 mg given orally daily. The study discontinued recruitment for rucaparib given intravenously, thus MTD was not determined for the other three arms. Across all arms, the disease-control rate, defined as complete response (CR), PR, or stable disease (SD) of at least 12 months, was 68.8%. This includes one patient with CR, who was a patient with breast cancer, nine patients achieving a PR, and 43 patients having SD. In analyzing the 33 patients who received oral rucaparib with carboplatin, two ovarian cancer patients achieved PR, one of whom had BRCA1 mutation, and one was a BRCA1-mutated breast cancer patient [81].
The BRE09-146 phase II study (NCT01074970) did not show a difference in 2-year disease-free survival in triple-negative breast cancer patients, with residual disease after neoadjuvant chemotherapy, assigned to receive either cisplatin or cisplatin in combination with rucaparib intravenously [82]. These women were considered high risk for recurrence. However, the dose used for rucaparib was much lower than the 600-mg BID dose used in the monotherapy trials.
Overall, rucaparib is well tolerated at the recommended dose of 600 mg BID as monotherapy. The most common adverse effects experienced were nausea, fatigue, anemia, and vomiting. Anemia was the most common grade 3/4 adverse event reported; however, it was managed by treatment modification [83].
Rucaparib is also well tolerated when administered with chemotherapy. In the temozolomide combination phase II study, the two agents were well tolerated, with myelosuppression being the dose-limiting toxicity. However, rucaparib alone was not responsible for any toxicity. Myelosuppression resolved with dose reduction in temozolomide and cumulative myelosuppression was not present [79]. In the phase I study of rucaparib in combination with various chemotherapy regimens, grade 3 or higher toxicities occurred in 75.3%. These were due to myelosuppression, nausea, and fatigue [81].
3.2 Phase III Trials
3.2.1 Ovarian/Fallopian/Primary Peritoneal Cancer
Based on the promising results from the Study 10 and ARIEL2 trials, rucaparib was evaluated in a double-blind, randomized, placebo-controlled, phase III trial called ARIEL3 (NCT01968213) [84]. This study randomized 564 patients with high-grade serous or endometrioid ovarian, primary peritoneal, or fallopian tube carcinoma to receive either rucaparib 600 mg BID or placebo. All patients received at least two prior lines of platinum-based therapy and had to obtain a complete or PR to their most recent platinum-based regimen. Similar to the ARIEL2 trial, patients were grouped into one of the following: BRCA-mutated including germline or somatic mutations, non-BRCA-mutated with high LOH, and no mutations in BRCA or LOH. The primary endpoint was PFS. Rucaparib significantly improved median PFS over placebo in all three groups, with median PFS of the overall study population being 10.8 months in the rucaparib group versus 5.4 months in the placebo group. The greatest benefit in PFS was observed in the BRCA-mutated population with median PFS 16.6 months in the rucaparib group versus 5.4 months in the placebo group [84]. Based on this study, the FDA approved rucaparib as maintenance treatment in patients with platinum-sensitive recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma.
Multiple trials looking at rucaparib in various tumor types are currently recruiting. Of interest is the ARIEL4 trial (NCT02855944) comparing rucaparib versus chemotherapy in women with BRCA-mutated ovarian, fallopian, or primary peritoneal cancers. The RUBY (NCT02505048) and RIO (NCT02395536) trials are phase II studies looking at rucaparib in breast cancer patients. The RUBY trial is recruiting women with BRCA1/2 somatic mutated metastatic breast cancer. The RIO trial is studying rucaparib in triple-negative or BRCA-mutated breast cancers. The TRITON trials (NCT0295234, NCT02975934) consist of looking at rucaparib in men with metastatic castrate-resistant prostate cancer and homologous recombination gene deficiency. There is also the ATLAS trial (NCt03397394) looking at the efficacy of rucaparib in metastatic urothelial carcinoma and another trial for patients with pancreatic cancer (NCT03140670).
4 Niraparib (MK-4827)
Niraparib is an oral, highly selective, small molecule that inhibits both PARP-1 and PARP-2. Preclinical studies show activity of niraparib against BRCA1- and BRCA2-mutated cell lines as monotherapy and in combination therapy [85]. Based on phase III data, FDA gave approval in March 2017 for maintenance treatment of patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who had either a complete or a PR to platinum-based chemotherapy (Table 2). Similar to the previously discussed PARP inhibitors, niraparib is effective in preclinical models with platinum agents, topoisomerase inhibitors, methylation agents, alkylating agents, and also sensitizes cells to radiation [85].
4.1 Phase I Trials
The first phase I study involving niraparib was a dose-escalation study in which niraparib showed activity against ovarian, breast, and castrate-resistant prostate cancer. Out of the 20 patients with assessable BRCA-mutated ovarian cancer, eight patients achieved a PR with variable doses of niraparib. This included three out of nine patients with platinum-resistant disease and five out of 10 patients with platinum-sensitive disease. For the 22 evaluable patients with high-grade serious ovarian cancer, two out of three patients with platinum-sensitive disease demonstrated a PR and only three out of 19 patients with platinum-resistant disease had a PR. Additionally, two out of four patients with BRCA-mutated breast cancer had a PR to niraparib at doses of 150 mg and 210 mg daily. Lastly, nine out of 21 patients with castrate-resistant prostate cancer had stable disease and one patient had biochemical regression by over 50% decrease in PSA. This phase I dose-escalation trial found the optimal dose of niraparib to be 300 mg daily [86]. Niraparib showed promise against BRCA-mutated breast cancer, with two out of four patients obtaining a PR; histological subtypes were not reported [86].
The TOPACIO trial, or Keynote-162 (NCT02657889), is a phase I/II study assessing the combination of niraparib with pembrolizumab. There were two groups of women; the first included women with metastatic triple-negative breast cancer treated with four or less prior lines of chemotherapy and the second group included women with recurrent, platinum-resistant ovarian cancer treated with five or less previous lines of therapy and a response greater than 6 months to the first line of platinum-based chemotherapy. Preliminary data from January 2018 in 54 patients with TNBC show an ORR 29% [87]. Twelve of the 54 women had a BRCA mutation. In this subgroup seven patients had PR, one patient had CR, and one patient had SD. In the women with tumors expressing at least 1% PD-L1 positivity, the ORR was 33% as compared to 15% in those not expressing at least 1% PD-L1 positivity [87]. Preliminary data in the recurrent ovarian cancer cohort show that ORR was 25% out of 60 women evaluated [88]. When further assessing the 11 patients with BRCA mutations, the ORR was 45% [88]. The phase II portion of the trial is currently recruiting.
Given both bevacizumab and PARP inhibitors have efficacy in platinum-sensitive recurrent ovarian cancer, the combination of niraparib with bevacizumab was evaluated in the ANANOVA/ENGOT-OV24 trial (NCT02354131) trial and presented at ASCO 2019. In this proof-of-concept, phase II trial, 97 women with measurable high-grade serous or endometroid platinum-sensitive recurrent ovarian cancer were randomized to receive niraparib monotherapy or niraparib in combination with bevacizumab. The primary endpoint of median PFS was significantly longer in the combination arm, 11.9 months versus 5.5 months with an HR of 0.35. On subgroup analysis, the benefit with the combination was seen in those with and without homologous recombination deficiency [89]. This study was the first to show such promising results in recurrent ovarian cancer with a non-chemotherapy-based combination.
Phase I studies have demonstrated that as single agent, higher doses can be tolerated and more effective. However, as expected in combination therapy, the MTD is lower, as limited by toxicities when adding a second agent. In a phase I combination trial of niraparib and temozolomide in patients with advanced cancer, the MTD of niraparib was 40 mg daily in combination with the temozolomide 150 mg/m2 for the first 5 days of each 28-day cycle [90]. Of the 19 patients enrolled, one patient with glioblastoma had a PR with six cycles of niraparib 40 mg. Two patients, one with malignant melanoma and the other with serous ovarian cancer, demonstrated stable disease at a dose of 40 mg and 30 mg, respectively [90].
The most common adverse effect that has been reported with niraparib monotherapy is myelosuppression [86, 91]. Similarly, in the combination trials, hematologic toxicities were also the most common [87, 88, 90]. Niraparib appears to have increased incidences of hematologic toxicity compared to other PARP inhibitors.
4.2 Phase III Trials
4.2.1 Ovarian/Fallopian/Primary Peritoneal Cancer
The phase III NOVA trial (NCT01847274) was designed based on the phase I/II trial that showed some efficacy of niraparib ovarian cancer, especially in platinum-sensitive disease. The NOVA trial is a double-blind, phase III trial randomizing 553 patients with platinum-sensitive recurrent ovarian cancer, fallopian tube cancer, or primary peritoneal cancer to receive either niraparib 300 mg daily or placebo for maintenance therapy. Patients had received at least two prior lines of platinum-based therapy and had either complete or PR to the most recent line of platinum therapy. Patients were divided into two cohorts based on whether they had a deleterious germline BRCA mutation. The cohort without germline BRCA mutation was further stratified during the statistical analysis to whether they had homologous recombination deficiency. The primary endpoint of median PFS was met for those taking niraparib in both cohorts. In those with a germline BRCA mutation, median PFS was significantly longer in those randomized to niraparib compared to placebo, 21 months versus 5.5 months (HR 0.27; 95% CI 0.17–0.41; p < 0.001), respectively. In those without a germline BRCA mutation, the median PFS was still significantly longer in the niraparib group, 9.3 months versus 3.9 months (HR 0.45; 95% CI 0.34–0.61; p < 0.001), respectively. The difference in median PFS was even more pronounced in those without germline BRCA mutation and with homologous recombination deficiency, 12.9 months in the niraparib group compared to 3.8 months in the placebo group (HR 0.38; 95% CI 0.24–0.59; p < 0.001) [91]. Overall survival data are not mature. Based on these results, niraparib gained FDA approval in March 2017 for maintenance treatment of patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who had either a complete or a PR to platinum-based chemotherapy.
Similar to that seen in the phase I/II trials with niraparib, myelosuppression was the most common adverse event. The incidence of grade 3 or 4 thrombocytopenia, anemia, and neutropenia was 33.8%, 25.3%, and 19.6%, respectively, in the phase III NOVA trial [91]. These toxicities could be managed with dose modifications.
There are multiple ongoing and recruiting trials with niraparib as monotherapy and in combination therapy. The BRAVO trial (NCT01905592) is an ongoing phase III trial randomizing patients with previously treated BRCA-mutated, Her2-negative breast cancer to niraparib versus physician’s choice. Final results of the TOPACIO trial are also eagerly awaited. Some other combination therapy trials include niraparib combination therapy for prostate cancer (QUEST, NCT03431350) and niraparib with carboplatin for homologous recombination deficiency advanced solid malignancies (NCT03209401). Other trials are looking at niraparib in prostate cancer (NCT03553004), pancreatic cancer (NCT03601923), and endometrial cancer (NCT03016338).
5 Talazoparib (MDV3800 or BMN 673)
Talazoparib is a novel, selective inhibitor of PARP 1 and PARP 2 that has been shown to achieve antitumor response at lower concentrations than earlier generation PARP inhibitors [92]. Though it is comparable at inhibiting PARP catalytic activity to olaparib and rucaparib, it is 100 times more potent at trapping PARP-DNA complexes [9]. Preclinical studies demonstrate activity in osteosarcoma [93], chronic lymphocytic leukemia [94], and ovarian cancer cell lines [95]. It was the second drug to be FDA approved for BRCA-mutated, Her2-negative breast cancer, after olaparib (Table 2).
5.1 Phase I Trials
The first phase I trial of talazoparib enrolled over 100 patients with advanced solid tumors, with an expansion cohort for patients with tumors predicted to be potentially sensitive to PARP inhibition. This expansion cohort included patients with germline BRCA mutations, triple-negative breast cancer, ovarian cancer, prostate cancer, and pancreatic cancer. Those in the dose-escalation cohort received doses ranging from 0.025 mg/day to 1.1 mg/day, and those in the expansion cohort received a dose of 1.0 mg/day with continuous dosing. The most common toxicities included fatigue (37%), anemia (35%), nausea (32%), thrombocytopenia (21%), alopecia (20%), and neutropenia (15%). The most frequent grade 3 or 4 adverse events were cytopenias. Though there were eight deaths in the study, none were attributed to talazoparib. In the 14 BRCA-mutated breast cancer patients, the ORR was 50% and the clinical benefit rate was 86%, with a median PFS of about 35 weeks. For the 25 patients with BRCA-mutated ovarian cancer, ORR was 48% and clinical benefit rate was 76%, though response rates were much lower for platinum-resistant patients at 20%. Twenty-three patients with small-cell lung cancer were also enrolled, with poor response rates of 9%. Finally, of the 13 patients with pancreatic cancer, four patients responded with ORR 31%, one of whom had a BRCA2 mutation and another a PALB2 mutation. In summary, talazoparib was well tolerated and active mostly in BRCA-mutated breast and ovarian cancers [96].
Another phase I study of talazoparib in combination with carboplatin was conducted in 24 patients with solid tumors. A total of 24 patients with solid tumors were enrolled in four cohorts at 0.75 mg and 1 mg daily of talazoparib and weekly carboplatin AUC 1 and 1.5 at every two weeks or every 3 weeks, respectively. Grade 3 or 4 toxicities included fatigue (13%), neutropenia (63%), thrombocytopenia (29%), and anemia (38%). One complete and two PRs occurred in patients with germline BRCA1 and BRCA2 mutations, and of the four patients with stable disease beyond 4 months, three had somatic BRCA mutations and one had a BRIP1 germline mutation, suggesting this combination is active in tumors with DNA repair mutations [97]. Response by tumor type was not specified, but overall this combination was deemed to be well tolerated.
5.2 Phase II/III Trials
5.2.1 Solid Tumors
A phase II study was performed to determine if the benefit seen with PARP inhibition in BRCA-mutated breast, ovarian, and prostate cancers could be extended to other tumor types and other mutations. Thirty-five patients with solid tumors, excluding breast and ovarian cancer, with either germline BRCA mutations and not breast or ovarian cancer, somatic BRCA mutations, mutations/deletions of PTEN or mutations/deletions in other BRCA pathway genes were treated with talazoparib 1 mg/day. The clinical benefit rate was 29% for patients with germline BRCA mutations not including breast or ovarian cancer, 0% for somatic BRCA mutations, 44% for mutations in other BRCA pathway genes, and 8% for patients with PTEN mutations. In terms of safety, grade 3–4 events occurred in 37% of patients, the most common of which was thrombocytopenia. Overall, patients with PTEN mutations had a disappointing response, while there was some activity in non-breast and non-ovarian cancer patients with germline BRCA mutations, or mutations in other BRCA pathway genes [98].
5.2.2 Breast Cancer
Another phase II trial, ABRAZO (NCT02034916), enrolled 84 patients with advanced breast cancer and germline BRCA1 and BRCA2 mutations that either were previously treated with platinum therapy or had three or more platinum-free cytotoxic-based regimens. ORR was 24% for BRCA1 patients, 34% for BRCA2, 26% for triple-negative breast cancer patients, and 29% for hormone receptor-positive breast cancer patients. The most common grade 3 or 4 toxicity was anemia, which occurred in about a third of patients, followed by thrombocytopenia and neutropenia [99].
Based on encouraging efficacy and safety data from the ABRAZO trial, the phase III, open-label EMBRACA study (NCT01945775) was initiated. EMBRACA randomized 431 women with locally advanced or metastatic HER2-negative breast cancer and germline BRCA1/2 mutations who had received no more than three prior lines of therapy to receive talazoparib 1 mg daily versus investigator’s choice of conventional chemotherapy. The primary endpoint of median PFS was significantly higher in the talazoparib group versus the chemotherapy group, 8.6 months versus 5.6 months, respectively, with an HR of 0.54. Response rate was higher in the talazoparib group, reaching 63% versus 27% in the chemotherapy group, with 5.5% of patients in the talazoparib group having a CR as compared to none of the patients in the chemotherapy group. The majority of adverse events noted with talazoparib were hematologic events, with grade 3–4 hematologic events reaching 55%. However, only 6% patients discontinued talazoparib due to adverse events compared to 9% in the chemotherapy group. The majority of the patients in both groups required dose reduction or interruption, 66% in talazoparib group versus 60% in the chemotherapy group. Lastly, this trial demonstrated a significant delay in the time to clinical deterioration in women receiving talazoparib as compared to those receiving chemotherapy [100]. Based on the EMBRACA trial, the FDA approved talazoparib for patients with germline BRCA-mutated, HER2-negative, locally advanced or metastatic breast cancer. Investigators are also assessing whether talazoparib is active in BRCA wild-type patients with triple-negative breast cancer with a phase II trial that is actively enrolling patients (NCT02401347).
In terms of quality of life, a follow-up of EMBRACA showed that quality of life was significantly improved with talazoparib compared to chemotherapy, based on the observation of greater delay to clinically meaningful deterioration with talazoparib compared to chemotherapy, 24 months and 6 months, respectively [101]. Quality-of-life data from the ABRAZO trial demonstrated deterioration in role functioning and dyspnea, but improvement in breast and arm symptoms [102].
Ongoing efforts are evaluating whether talazoparib may be effective when moved into the curative setting. A pilot study was conducted at the MD Anderson Cancer Center to test the efficacy of single-agent talazoparib as neoadjuvant therapy in untreated BRCA mutation carriers. In this trial, patients were treated with 2 months of talazoparib followed by anthracycline- and taxane-based chemotherapy with or without carboplatin. A total of 13 patients were enrolled; eight patients were hormone receptor negative and four were hormone receptor positive. All patients experienced tumor shrinkage with the 2 months of talazoparib, with average tumor loss of 78% [103]. In ASCO 2018, Litton et al. presented updated data showing nine of 20 patients achieving no residual tumor burden after 6 months of neoadjuvant talazoparib [104]. If these findings are confirmed with larger trials, talazoparib could become the first targeted drug to be approved as monotherapy for this indication.
5.2.3 Prostate Cancer
Though there are no published phase II or III studies evaluating talazoparib in prostate cancer, several trials are underway. A phase II study (NCT03148795) evaluating efficacy and safety of talazoparib in men with metastatic, castration-resistant prostate cancer with DNA repair defects is enrolling patients, as is another phase III study TALAPRO-2 (NCT03395197), comparing talazoparib plus enzalutamide versus placebo plus enzalutamide in DNA damage repair-deficient metastatic castration-resistant prostate cancer.
6 Veliparib (ABT-888)
Veliparib is an orally bioavailable PARP inhibitor that crosses the blood–brain barrier. Preclinical trials in mice demonstrated veliparib augmented apoptotic response and potentiated the effects of chemotherapy [105].
6.1 Phase I Trials
Various phase I clinical trials established the safety and toxicity profile for veliparib as monotherapy as well as in combination with chemotherapy. A phase I dose-escalation study treated Japanese patients with veliparib BID. Out of the 16 enrolled patients, 14 had high-grade serous ovarian cancer, one had primary peritoneal cancer, and one had BRCA-mutated breast cancer. A PR was the best response achieved and was seen in two patients with ovarian cancer. Grade 3 or 4 adverse events were observed in 50% of the patients, and the most common toxicities included nausea, vomiting, decreased appetite, abdominal pain, diarrhea, and malaise. The recommended phase II dose from this study was 400 mg BID [106].
Preclinical studies demonstrate enhanced activity when veliparib is combined with chemotherapy, thus prompting phase I trials combining velaparib with conventional chemotherapy. A dose-escalation trial evaluated veliparib in combination with carboplatin and paclitaxel for newly diagnosed advanced ovarian cancer. An optimal dose of 150 mg BID was established. Response was assessed in five out of the nine enrolled patients, with PR seen in four patients and a CR seen in one patient [107]. A separate study established a tolerable dose of velapirib 400 mg BID in combination with cisplatin and paclitaxel for recurrent or persistent cervical cancer not amenable to curative chemotherapy. Out of the 29 patients with measurable disease, the ORR was 34% [108]. In non-small-cell lung cancer, veliparib in combination with carboplatin and paclitaxel yielded a PR for six out of 12 patients. The recommended phase II dose of veliparib was 120 mg BID when given with carboplatin and paclitaxel [109]. An ORR of 69% was observed in a study of patients with advanced ovarian and breast cancers treated with veliparib plus carboplatin and gemcitabine. The MTD of veliparib in this trial was 250 mg BID with carboplatin AUC 4 on day 1 and gemcitabine 800 mg/m2 on days 1 and 8 of a 21-day cycle. Myelosuppression was the most common toxicity limiting the dose of veliparib [110].
Another phase I study evaluated velapirib in combination with bendamustine with an expansion cohort also including rituximab for patients with B-cell lymphomas. Response was seen in five out of seven patients given veliparib with bendamustine alone and in six out of seven patients given veliparib, bendamustine, and rituximab. When given with bendamustine, the MTD of bendamustine is reduced to 300 mg BID [111].
Further studies measured the bioavailability of an extended-release formulation and established that veliparib did not alter the pharmacokinetics of temozolomide [112, 113]. A pilot study of velapirib plus temozolomide for metastatic castration-resistant prostate cancer showed PSA response in two out of 25 evaluable patients and a stable PSA in 13 patients [114]. This combination could represent a viable option for patients who have exhausted endocrine and chemotherapy options.
In the neoadjuvant setting, veliparib was given with capecitabine and radiotherapy for stage II or stage III locally advanced rectal adenocarcinoma. Out of the 31 evaluable patients, 71% demonstrated downstaging of their tumor and 29% achieved a complete pathological response. No dose-limiting toxicities occurred, and importantly the addition of veliparib did not alter the pharmacokinetics of capecitabine. The recommended phase II dose was veliparib 400 mg BID [115], although it does not appear as though any further clinical trials incorporating veliparib into rectal cancer therapy are ongoing.
The most common treatment-related adverse effects in the above combination studies were nausea and cytopenia, and dose-limiting toxicities were mostly cytopenia. After these initial trials established the safety and tolerability of veliparib, larger clinical trials are investigating the drug as monotherapy and as chemosensitizers as well as radiosensitizers.
6.2 Phase II/III Trials
6.2.1 Ovarian/Fallopian/Primary Peritoneal Cancer
Given the promising results seen in gynecological tumors in phase I studies, a phase II NRG Oncology/Gynecologic Oncology Group studied the efficacy of single-agent veliparib, at a dose of 400 mg BID, in 50 women with persistent or recurrent BRCA-mutated epithelial ovarian, fallopian tube, or primary peritoneal cancer. The ORR was 26%, with subgroup analysis revealing an ORR of 20% in platinum-resistant women and 35% in platinum-sensitive women. The median progression-free survival was 8.18 months [116]. A separate phase I/II study evaluated veliparib as monotherapy for BRCA-mutated epithelial ovarian, fallopian tube, or peritoneal cancer patients with platinum-resistant or intermediate sensitive relapse. Among 32 patients enrolled in the phase II trial, ORR, assessed by a combination of RECIST criteria and CA-125 response, was 65%, with 6% of the women having a complete response. Progression-free survival was 5.6 months and OS was 13.7 months [27].
Kummar et al. conducted a phase II trial of veliparib 60 mg daily in combination with low-dose cyclophosphamide compared to cyclophosphamide alone for a 21-day cycle for 75 patients with pretreated high-grade serous or BRCA-mutated ovarian, primary peritoneal, or fallopian tube cancers. The addition of veliparib did not improve response rate or progression-free survival compared to cyclophosphamide monotherapy [117].
A multicenter phase III trial is ongoing to evaluate the efficacy of veliparib in combination with carboplatin and paclitaxel as initial treatment for advanced high-grade serous or epithelial ovarian, fallopian tube, or primary peritoneal cancer (NCT02470585).
6.2.2 Breast Cancer
Kummar et al. also conducted a phase II trial randomizing 45 women with heavily pretreated triple-negative breast cancer to either low-dose cyclophosphamide and veliparib 60 mg daily or low-dose cyclophosphamide alone for 21-day cycles. Similar to the results of the ovarian cancer study, the combination of cyclophosphamide and veliparib did not improve the response rate or median progression-free survival compared to cyclophosphamide [118]. Unfortunately, this combination was not beneficial for a cohort of patients with very limited treatment options and poor prognosis.
In the phase II part of the California Consortium Trial, patients with BRCA-mutated metastatic breast cancer were treated with veliparib 400 mg BID and then on disease progression were given a combination of carboplatin AUC 5 and veliparib 150 mg BID. Out of the 44 women enrolled in the phase II part of the trial, 30 switched to the combination therapy and only one woman responded to the combination of carboplatin and veliparib after progressing on single-agent veliparib. The RR was 14% for BRCA1 patients and 36% or BRCA2 patients, and this difference was not statistically significant. Interestingly there was a significant difference in PFS, 3.6 months in BRCA1 patients and 6.6 months in BRCA2 patients, but no significant differences in OS [119]. This finding is perhaps related to the more aggressive phenotype associated with BRCA1 mutations but also goes against the idea that BRCA-like or triple-negative breast cancer, which is more commonly seen in BRCA1-mutated patients, is more sensitive to PARP inhibition.
The I-SPY2 (NCT01042379) is a multicenter, adaptive, phase II trial evaluating newer agents combined with standard neoadjuvant therapy in women with stage II or stage III breast cancer. The primary endpoint is pathological CR (pCR). One of the experimental groups for women with Her2-negative breast cancer consisted of treatment with veliparib 50 mg BID and carboplatin AUC 6 every 3 weeks for a total of 12 weeks in combination with standard neoadjuvant chemotherapy with paclitaxel for 12 weeks followed by doxorubicin and cyclophosphamide. The comparator arm was standard neoadjuvant therapy with paclitaxel for 12 weeks followed by doxorubicin and cyclophosphamide. Women with Her2-positive cancers were not included because of the lack of safety data for veliparib in combination with trastuzumab. Patients were evaluated in three groups: Her2-negative tumors, Her2-negative but hormone receptor-positive tumors, and triple-negative tumors. For the Her2-negative group pCR was 33% in the veliparib-carboplatin group as opposed to 22% in the standard therapy group, and this difference was largely attributed to the benefit derived in women with triple-negative breast cancers. For women with Her2-negative but hormone receptor-positive breast cancer, the pCR was 14% in the veliparib-carboplatin group compared with pCR of 19% in the control group. Interestingly, women with triple-negative breast cancer had a higher pCR of 51% when given veliparib and carboplatin in addition to standard neoadjuvant therapy as opposed to a pCR rate of 26% in the control group [120].
The promising results of the I-SPY2 trial prompted the BrighTNess study (NCT02032277), a phase III randomized, multicenter trial in women with stage II or stage III triple-negative breast cancer. 634 women were randomized to three groups for segment 1 of neoadjuvant therapy, which included the following: the combination of veliparib, carboplatin, and paclitaxel, the combination of carboplatin, paclitaxel, and veliparib placebo, or paclitaxel with carboplatin placebo and veliparib placebo. The doses included veliparib 50 mg BID, carboplatin AUC 6 every 3 weeks, and paclitaxel 80 mg/m2 weekly for a total of 12 weeks. All patients then went on to segment 2, which included doxorubicin and cyclophosphamide. The primary endpoint was pCR following completion of neoadjuvant therapy. Although a higher proportion of patients in the veliparib-carboplatin-paclitaxel group achieved a pCR compared to the paclitaxel alone group, 53% versus 31%, there was no significant difference in those attaining a pCR in the veliparib-carboplatin-paclitaxel group compared with the carboplatin-paclitaxel group, 53% versus 58%. Additionally, no major toxicities were caused by the addition of veliparib; however, there were increased grade 3 and grade 4 toxicities when carboplatin was added to paclitaxel compared to paclitaxel alone, the most common serious ones being febrile neutropenia and anemia [121].
6.2.3 Other Solid Tumors
In a phase II trial of untreated advanced non-small-cell lung cancer, there was a trend toward improved OS and PFS with veliparib added to carboplatin and paclitaxel, but this was not statistically significant. This benefit appeared to be more pronounced in those with squamous cell carcinoma. Notably, two patients in the veliparib group did have a CR compared to none in the control arm [122]. In a multivariate analysis of the results, recent smokers had a significantly higher PFS and OS compared to former smokers and never smokers, thus implying smoking history predicted benefit from veliparib [123]. In a press release by Abbvie, the phase III randomized trial found that the addition of veliparib to carboplatin and paclitaxel failed to meet the primary endpoint of OS; however, final results have yet to be published.
A phase I study of velapirib plus whole brain radiation for patients with non-small-cell lung cancer brain metastases showed preliminary evidence of efficacy [124]. However, in a follow-up study the addition of veliparib to whole brain radiation did not have an impact on response rate or OS [125].
In a multicenter, double-blind, phase II study, 346 patients with stage III or stage IV unresectable metastatic melanoma were randomized to receive temozolomide with veliparib 20 mg BID, veliparib 40 mg BID, or placebo. Although there was a trend toward increased PFS, this did not reach statistical significance. Additionally, OS was not increased with the addition of veliparib [126].
A single-arm trial followed patients with advanced hepatocellular carcinoma refractory to sorafenib treated with a combination of temozolomide and veliparib. The trial was discontinued due to a poor objective response rate [127].
A phase I/II trial investigated veliparib 10 mg BID in combination with topotecan 0.6 mg/m2 on days 1–5 for 21-day cycles in 27 women with persistent or recurrent uterine cancer. A PR was seen in two women (7%). On subgroup analysis, women with low PARP-1 expression in their cervical cancer had longer PFS and OS, HR 0.25 and HR 0.12, respectively, but this needs to be evaluated with further trials.
7 Mechanisms of Resistance
While PARP inhibitors are being increasingly used, mechanisms of resistance to these agents have also been observed. Studies looking at cell lines of patients with BRCA2-mutated-related cancers resistant to PARP inhibitors or platinum agents found secondary mutations in the BRCA2 gene, thus restoring BRCA2 function leading to repair of DNA from PARP inhibitors [128]. It is possible these reversion mutations occur prior to initiating therapy with PARP inhibitors and that use of PARP inhibitors select for the resistant clone [129]. Alternatively, the BRCA mutation may cause error-prone DNA repair, thus randomly creating a secondary mutation that actually restores BRCA function [130]. Regardless of the timing of these secondary mutations, they most commonly occur in the open reading frame of the gene, therefore permitting translation.
Another proposed mechanism is through the decreased activity of nonhomologous end-joining factor 53BP1, whose presence is critical for promoting nonhomologous end joining and counteracting homologous recombination. Loss of 53BP1 restores homologous recombination in BRCA1-mutated cells by producing single-stranded DNA sufficient for homologous recombination to occur. In preclinical studies, a resistance mechanism to olaparib developed in mice with loss of 53BP1 expression [131] Tumors resistant to PARP by this mechanism were also resistant to topoisomerase II inhibitors but not platinum agents, thought to be due to the fact that platinum agents cause more severe DNA damage through DNA strand crosslinking [128].
Similarly, a recent study showed lack of FAM35A and C20orf196 proteins decreased sensitivity to PARP inhibitors in BRCA1-mutated tumor cell lines but not in BRCA1 wild-type cell lines. FAM35A and C20orf196 proteins together form a complex called Shieldin that interacts with MAD2L2, which is part of the 53BP1 pathway mentioned previously. The interaction of Shieldin with MAD2L2 promotes nonhomologous end joining and antagonizes homologous recombination, thus sensitizing BRCA1-deficient tumor cells to PARP inhibition. The inactivation of either one of the proteins in the Shieldin complex prevents nonhomologous end joining and promotes homologous recombination, thus conferring resistance to PARP inhibition [132].
Other proposed mechanisms include epigenetic changes and upregulation of drug efflux genes. As previously mentioned, PARP inhibitors prevent dissociation of PARP1 and PARP2 enzymes at the site of DNA damage, with the subsequent cytotoxic PARP-DNA complex interfering with the DNA and leading to cell death. When the poly(ADP)ribosylation of PARP occurs, PARP is released from the DNA. PARP1 levels were reduced in human tumor cell lines with acquired resistance to PARP inhibitors, suggesting tumor-induced epigenetic downregulation of PARP and a higher rate of PARP turnover [128]. Additionally, methylation of all BRCA1 copies demonstrates sensitivity to rucaparib in patient-derived xenografts, whereas demethylation or partial methylation confers resistance to rucaparib [133].
Long-term treatment with PARP inhibitors, in particular olaparib, upregulates genes encoding the P-glycoprotein efflux pump, which leads to lower concentrations of the PARP inhibitor in the cells. Newer PARP inhibitors with lower affinity for the P-glycoprotein efflux pump are in development in order to prevent resistance [128].
8 Rationale for Combination Therapies
PARP inhibitors are being investigated in combination with cytotoxic chemotherapy, radiotherapy, antiangiogenesis agents, immune therapy, and with inhibitors of other proteins in the DNA damage response (DDR) pathway. PARP inhibition makes tumor cells less able to repair DNA damage inflicted by cytotoxic chemotherapy or radiation. In preclinical and phase I/II studies, synergy has been observed between PARP inhibitors and various chemotherapy drugs, including alkylating agents and topoisomerase inhibitors. However, myelosuppression is a major concern, such that attenuated dosing of PARP inhibition is needed [79, 134, 135]. PARP inhibitors have also been noted to be effective radio-sensitizers in multiple tumor types in preclinical studies, and this approach is being investigated further.
Combination of antiangiogenesis agents with PARP inhibitors may improve response in non-BRCA-mutated tumors, as suggested by phase 2 data examining the combination of cediranib and olaparib in platinum-sensitive ovarian cancer [50]. Further combinations are under study with niraparib and bevacizumab in recurrent ovarian cancer in AVANOVA, the results of which are expected in late 2019.
Using PARP inhibitors with immunotherapy agents is also under investigation. In preclinical models, PARP inhibitors upregulated PDL1 expression in tumor cell lines, which were then treated with PDL1 blockade. This enhanced T-cell killing of tumor cells in vivo [136]. As mentioned previously, preliminary results from the TOPACIO trial with niraparib and pembrolizumab in women with TNBC showed an ORR 29%. Studies with combining checkpoint inhibitors and PARP inhibitors in the first-line setting are underway, including FIRST (NCT03602859), DUO-O (NCT03737643), JAVELIN (NCT03642132), and ATHENA (NCT03522246).
The DNA damage response (DDR) pathway contains over 400 proteins, including PARP proteins. Simultaneously targeting another protein in this pathway provides a way to overcome resistance to PARP inhibitors. Inhibiting both PARP and another protein in the DDR pathway sequentially instead of concurrently would theoretically allow higher doses of both agents. Currently, combination trials with inhibitors to proteins in the DDR pathway and PARP inhibitors are ongoing. Some of the proteins in the DDR pathway being targeted are ATM, ATR, and WEE1, all of which have a role in cell-cycle checkpoint regulation [137]. One study currently underway is the VIOLETTE study (NCT03330847), a phase II study of either an inhibitor of ATR or WEE1 in combination with olaparib versus olaparib monotherapy in women with triple-negative breast cancer.
9 Conclusion
PARP inhibitors have changed the landscape for treatment of not only BRCA-mutated ovarian and breast malignancies, but also where there is loss of homologous DNA repair. Trials in other types of malignancies with and without BRCA mutation are ongoing, in addition to combination trials with a variety of agents in the hopes of expanding the use of these agents in other non-homologous recombination deficiency-driven tumor types. Although the newer PARP inhibitors, such as talazoparib, demonstrate increased potency, whether this translates into greater efficacy is as yet unknown as PARP inhibitors have not been compared head to head. As more data emerge, hopefully we can identify predictive biomarkers in both monotherapy and drug combinations. Nonetheless, this class of anti-cancer drugs offers incredible promise in conventionally treatment-refractory groups including recurrent ovarian cancer, triple-negative breast cancer, and heavily pre-treated prostate cancer. To date, niraparib, olaparib, rucaparib, and talazoparib are FDA approved, and combination approvals are expected.
For patients with mutations in homologous recombination repair (e.g. BRCA), the use of a PARP inhibitor is likely to become a standard choice as all studies so far suggest that tumors driven by these mutations have high response rates. This brings into the forefront the need for consistent molecular testing to identify these likely responders. Ovarian cancer has up to 26% BRCA mutation, representing an important and clinically meaningful population [136]. Breast, prostate, and even pancreatic cancer all have populations that can benefit from these drugs. Newer areas of interest in tumors with other DNA damage repair mutation, such as endometrial cancer, stress even more the need for careful molecular tumor assessment.
Adverse events associated with PARP inhibitors also require a careful assessment; as already noted, the majority will experience nausea, fatigue, and anemia. Attention to pre-counseling of patients prior to the start of these drugs can help with reducing early discontinuation for side effects. Providing antiemetics in advance, encouraging patients that the nausea appears to decrease after the first 4–8 weeks, and dose reduction as needed can help patients stay on treatment long term. Fatigue may be mitigated by exercise or caffeine. Bone marrow toxicities such as anemia require careful monitoring with weekly cell blood counts in the first 4 weeks of therapy followed by monthly assessment. Dose reduction may be required for bone marrow toxicities.
The opportunity to have patients take an oral agent and have significant durable benefit cannot be understated. PARP inhibitors have changed the landscape of expectations in BRCA-mutated cancers. As rational combinations begin to emerge, more and more patients, even those without a homologous recombination deficiency, will be able to benefit from these revolutionary agents.
References
Pines A, Mullenders LH, van Attikum H, Luijsterburg MS. Touching base with PARPs: moonlighting in the repair of UV lesions and double-strand breaks. Trends Biochem Sci. 2013;38(6):321–30.
Bryant HE, Petermann E, Schultz N, et al. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009;28(17):2601–15.
Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13:411.
Eustermann S, Wu W-F, Langelier M-F, et al. Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Mol Cell. 2015;60(5):742–54.
Dawicki-McKenna JM, Langelier M-F, DeNizio JE, et al. PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol Cell. 2015;60(5):755–68.
Purnell MR, Whish W. Novel inhibitors of poly (ADP-ribose) synthetase. Biochem J. 1980;185(3):775–7.
Lord CJ, Ashworth A. PARP inhibitors: the first synthetic lethal targeted therapy. Science (New York, NY). 2017;355(6330):1152.
Murai J, Shar-yin NH, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Can Res. 2012;72(21):5588–99.
Murai J, Huang SN, Renaud A, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13(2):433–43.
Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8(362):362.
Chalmers AJ. The potential role and application of PARP inhibitors in cancer treatment. Br Med Bull. 2009;89(1):23–40.
Wang Z-Q, Auer B, Stingl L, et al. Mice lacking ADPRT and poly (ADP-ribosyl) ation develop normally but are susceptible to skin disease. Genes Dev. 1995;9(5):509–20.
Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917.
Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymerase. Nature. 2005;434(7035):913.
Tutt A, Ashworth A. The relationship between the roles of BRCA genes in DNA repair and cancer predisposition. Trends Mol Med. 2002;8(12):571–6.
Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25(43):5864.
Saleh-Gohari N, Bryant HE, Schultz N, Parker KM, Cassel TN, Helleday T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol. 2005;25(16):7158–69.
Wooster R, Weber BL. Breast and ovarian cancer. N Engl J Med. 2003;348(23):2339–47.
Thompson D, Easton DF. Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94(18):1358–65.
Gallagher DJ, Gaudet MM, Pal P, et al. Germline BRCA mutations denote a clinicopathologic subset of prostate cancer. Clin Cancer Res. 2010;16(7):2115–21.
Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26(22):3785–90.
Venkitaraman AR. A growing network of cancer-susceptibility genes. N Engl J Med. 2003;348(19):1917–9.
Turner N, Tutt A, Ashworth A. Hallmarks of’BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814.
Mori H, Kubo M, Nishimura R, et al. BRCAness as a biomarker for predicting prognosis and response to anthracycline-based adjuvant chemotherapy for patients with triple-negative breast cancer. PLoS One. 2016;11(12):e0167016.
Rigakos G, Razis E. BRCAness: finding the Achilles heel in ovarian cancer. Oncologist. 2012;17(7):956–62.
Brody LC. Treating cancer by targeting a weakness. N Engl J Med. 2005;353(9):949–50.
Underhill C, Toulmonde M, Bonnefoi H. A review of PARP inhibitors: from bench to bedside. Ann Oncol. 2011;22(2):268–79.
Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci. 2008;105(44):17079–84.
Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-Ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–34.
Giaccone G, Rajan A, Kelly RJ, et al. A phase I combination study of olaparib (AZD2281; KU-0059436) and cisplatin (C) plus gemcitabine (G) in adults with solid tumors. J Clin Oncol. 2010;28(15):3027.
Rajan A, Carter CA, Kelly RJ, et al. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin Cancer Res. 2012;18(8):2344–51.
Khan OA, Gore M, Lorigan P, et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br J Cancer. 2011;104:750.
Samol J, Ranson M, Scott E, et al. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest New Drugs. 2012;30(4):1493–500.
Goggins M, Schutte M, Lu J, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Can Res. 1996;56(23):5360–4.
Jacob DA, Bahra M, Langrehr JM, et al. Combination therapy of poly (ADP-ribose) polymerase inhibitor 3-aminobenzamide and gemcitabine shows strong antitumor activity in pancreatic cancer cells. J Gastroenterol Hepatol. 2007;22(5):738–48.
Bendell J, O’Reilly EM, Middleton MR, et al. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann Oncol. 2015;26(4):804–11.
Del Conte G, Sessa C, von Moos R, et al. Phase I study of olaparib in combination with liposomal doxorubicin in patients with advanced solid tumours. Br J Cancer. 2014;111:651.
Dent RA, Lindeman GJ, Clemons M, et al. Safety and efficacy of the oral PARP inhibitor olaparib (AZD2281) in combination with paclitaxel for the first- or second-line treatment of patients with metastatic triple-negative breast cancer: results from the safety cohort of a phase I/II multicenter trial. J Clin Oncol. 2010;28(15):1018.
Verheij M, De Haan R, Van Triest B, et al. Results of phase I trials combining PARP inhibition and radiotherapy in multiple sites. Radiother Oncol. 2016;119:S138.
Audeh MW, Penson RT, Friedlander M, et al. Phase II trial of the oral PARP inhibitor olaparib (AZD2281) in BRCA-deficient advanced ovarian cancer. J Clin Oncol. 2009;27(15):5500.
Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol 12(9):852–861.
Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244.
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–92.
Friedlander M, Matulonis U, Gourley C, et al. Long-term efficacy, tolerability and overall survival in patients with platinum-sensitive, recurrent high-grade serous ovarian cancer treated with maintenance olaparib capsules following response to chemotherapy. Br J Cancer. 2018;119(9):1075–85.
Pujade-Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(9):1274–84.
Friedlander M, Gebski V, Gibbs E, et al. Health-related quality of life and patient-centred outcomes with olaparib maintenance after chemotherapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT Ov-21): a placebo-controlled, phase 3 randomised trial. Lancet Oncol. 2018;19(8):1126–34.
Moore K, Colombo N, Scambia G, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495–505.
Ray-Coquard I, Selle F, Harter P, et al. PAOLA-1: an ENGOT/GCIG phase III trial of olaparib versus placebo combined with bevacizumab as maintenance treatment in patients with advanced ovarian cancer following first-line platinum-based chemotherapy plus bevacizumab. J Clin Oncol. 2016;34(15):TPS5607.
Oza AM, Cibula D, Benzaquen AO, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015;16(1):87–97.
Liu JF, Barry WT, Birrer M, et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 2014;15(11):1207–14.
Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–44.
Robson M, Im S-A, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523–33.
Robson M, Tung N, Conte P, et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann Oncol. 2019;30(4):558–66.
Tutt A, Kaufman B, Garber J, et al. 216TiPOlympiA: a randomized phase III trial of olaparib as adjuvant therapy in patients with high-risk HER2-negative breast cancer (BC) and a germline BRCA1/2 mutation (gBRCAm). Ann Oncol. 2017;28(suppl_5).
Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–61.
Earl HM, Vallier A-L, Qian W, et al. PARTNER: Randomised, phase II/III trial to evaluate the safety and efficacy of the addition of olaparib to platinum-based neoadjuvant chemotherapy in triple-negative and/or germline BRCA-mutated breast cancer patients. J Clin Oncol. 2017;35(Suppl_15):TPS591.
Drew Y, De Jonge M, Hong S, et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): results in germline BRCA-mutated (gBRCAm) platinum-sensitive relapsed (PSR) ovarian cancer (OC). Gynecol Oncol. 2018;149:246–7.
Dent R, Tan T, Kim S, et al. The DORA trial: a non-comparator randomised phase II multi-center maintenance study of olaparib alone or olaparib in combination with durvalumab in platinum treated advanced triple-negative breast cancer (TNBC). Cancer Res. 2018;78(Suppl_4):OT3-04-02.
Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–50.
Kote-Jarai Z, Leongamornlert D, Saunders E, et al. BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: implications for genetic testing in prostate cancer patients. Br J Cancer. 2011;105(8):1230.
Leongamornlert D, Mahmud N, Tymrakiewicz M, et al. Germline BRCA1 mutations increase prostate cancer risk. Br J Cancer. 2012;106(10):1697.
Beltran H, Yelensky R, Frampton GM, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol 63(5):920–926.
Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–708.
Teply BA, Wang H, Sullivan R, et al. Phase II study of olaparib (without ADT) in men with high-risk biochemically-recurrent prostate cancer following prostatectomy, with integrated biomarker analysis. J Clin Oncol. 2018;36(6):TPS386.
Clarke N, Wiechno P, Alekseev B, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018;19(7):975–86.
Karzai F, Madan RA, Owens H, et al. A phase 2 study of olaparib and durvalumab in metastatic castrate-resistant prostate cancer (mCRPC) in an unselected population. J Clin Oncol. 2018;36(Suppl_6):163.
Kubota E, Williamson CT, Ye R, et al. Low ATM protein expression and depletion of p53 correlates with olaparib sensitivity in gastric cancer cell lines. Cell Cycle. 2014;13(13):2129–37.
Bang Y-J, Im S-A, Lee K-W, et al. Olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer: a randomized, double-blind phase II study. J Clin Oncol. 2013;31(Suppl_15):4013.
Bang Y-J, Xu R-H, Chin K, et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1637–51.
Leichman L, Groshen S, O’Neil BH, et al. Phase II study of olaparib (AZD-2281) after standard systemic therapies for disseminated colorectal cancer. Oncologist. 2016;21(2):172–7.
Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381(4):317–27.
Drew Y, Mulligan EA, Vong WT, et al. Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst. 2011;103(4):334–46.
Ihnen M, Eulenburg C, Kolarova T, et al. Therapeutic potential of the poly(ADP-ribose) polymerase inhibitor rucaparib for the treatment of sporadic human ovarian cancer. Mol Cancer Ther. 2013;12(6):1002–15.
Thomas HD, Calabrese CR, Batey MA, et al. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol Cancer Ther. 2007;6(3):945–56.
Nile DL, Rae C, Hyndman IJ, Gaze MN, Mairs RJ. An evaluation in vitro of PARP-1 inhibitors, rucaparib and olaparib, as radiosensitisers for the treatment of neuroblastoma. BMC Cancer. 2016;16:621.
Kristeleit R, Shapiro GI, Burris HA, et al. A phase I–II study of the oral PARP inhibitor rucaparib in patients with germline BRCA1/2-mutated ovarian carcinoma or other solid tumors. Clin Cancer Res. 2017;23(15):4095–106.
Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(1):75–87.
Dal Molin GZ, Omatsu K, Sood AK, Coleman RL. Rucaparib in ovarian cancer: an update on safety, efficacy and place in therapy. Ther Adv Med Oncol. 2018;10:1758835918778483.
Plummer R, Lorigan P, Steven N, et al. A phase II study of the potent PARP inhibitor, Rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemother Pharmacol. 2013;71(5):1191–9.
Plummer R, Jones C, Middleton M, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14(23):7917–23.
Wilson RH, Evans TJ, Middleton MR, et al. A phase I study of intravenous and oral rucaparib in combination with chemotherapy in patients with advanced solid tumours. Br J Cancer. 2017;116(7):884–92.
Miller K, Tong Y, Jones DR, et al. Cisplatin with or without rucaparib after preoperative chemotherapy in patients with triple-negative breast cancer: final efficacy results of hoosier oncology group BRE09-146. J Clin Oncol. 2015;33(Suppl_15):1082.
Oza AM, Tinker AV, Oaknin A, et al. Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: integrated analysis of data from Study 10 and ARIEL2. Gynecol Oncol. 2017;147(2):267–75.
Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–61.
Jones P, Wilcoxen K, Rowley M, Toniatti C. Niraparib: a poly(ADP-ribose) polymerase (PARP) inhibitor for the treatment of tumors with defective homologous recombination. J Med Chem. 2015;58(8):3302–14.
Sandhu SK, Schelman WR, Wilding G, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14(9):882–92.
Vinayak S, Tolaney SM, Schwartzberg LS, et al. TOPACIO/Keynote-162: Niraparib + pembrolizumab in patients (pts) with metastatic triple-negative breast cancer (TNBC), a phase 2 trial. J Clin Oncol. 2018;36(Suppl_15):1011.
Konstantinopoulos PA, Waggoner SE, Vidal GA, et al. TOPACIO/Keynote-162 (NCT02657889): a phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. J Clin Oncol. 2018;36(Suppl_15):106.
Mirza MR, Avall-Lundqvist E, Birrer MJ, et al. Combination of niraparib and bevacizumab versus niraparib alone as treatment of recurrent platinum-sensitive ovarian cancer: a randomized controlled chemotherapy-free study—NSGO-AVANOVA2/ENGOT-OV24. J Clin Oncol. 2019;37(Suppl_15):5505.
Kurzrock R, Galanis E, Johnson DR, et al. A phase I study of niraparib in combination with temozolomide (TMZ) in patients with advanced cancer. J Clin Oncol. 2014;32(Suppl_15):2092.
Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–64.
Shen Y, Rehman FL, Feng Y, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19(18):5003–15.
Engert F, Kovac M, Baumhoer D, Nathrath M, Fulda S. Osteosarcoma cells with genetic signatures of BRCAness are susceptible to the PARP inhibitor talazoparib alone or in combination with chemotherapeutics. Oncotarget. 2017;8(30):48794.
Herriott A, Tudhope SJ, Junge G, et al. PARP1 expression, activity and ex vivo sensitivity to the PARP inhibitor, talazoparib (BMN 673), in chronic lymphocytic leukaemia. Oncotarget. 2015;6(41):43978.
Pulliam N, Taverna P, Lyons J, Nephew KP. Abstract 2943: novel combination therapy of DNMT inhibitor SGI-110 and PARP inhibitor BMN-673 (talazoparib) for BRCA-proficient ovarian cancer. Can Res. 2015;75(15 Supplement):2943–2943.
de Bono J, Ramanathan RK, Mina L, et al. Phase I, dose-escalation, two-part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov. 2017;7(6):620–9.
Dhawan MS, Bartelink IH, Aggarwal R, et al. Differential toxicity in patients with and without DNA repair mutations: phase I Study of Carboplatin and Talazoparib in advanced solid tumors. Clin Cancer Res. 2017;23(21):6400–10.
Piha-Paul SA, Goldstein JB, Hess KR, et al. Phase II study of the PARP inhibitor talazoparib (BMN-673) in advanced cancer patients with somatic alterations in BRCA1/2, mutations/deletions in PTEN or PTEN loss, a homologous recombination defect, mutations/deletions in other BRCA pathway genes and germline mutation S in BRCA1/2 (not breast or ovarian cancer). J Clin Oncol. 2015;33(Suppl_15):TPS2617.
Turner NC, Telli ML, Rugo HS, et al. Final results of a phase 2 study of talazoparib (TALA) following platinum or multiple cytotoxic regimens in advanced breast cancer patients (pts) with germline BRCA1/2 mutations (ABRAZO). J Clin Oncol. 2017;35(15_suppl):1007.
Litton JK, Rugo HS, Ettl J, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753–63.
Ettl J, Martin M, Roché H, et al. Quality of life with talazoparib versus physician’s choice of chemotherapy in patients with advanced breast cancer and germline BRCA1/2 mutation: patient-reported outcomes from the EMBRACA phase III trial. Ann Oncol. 2018;29(9):1939–47.
Hurvitz S, Quek R, Turner N, et al. Quality of life with talazoparib after platinum or multiple cytotoxic non-platinum regimens in patients with advanced breast cancer and germline BRCA1/2 mutations: patient-reported outcomes from the ABRAZO phase 2 trial. Eur J Cancer. 2018;104:160–8.
Litton J, Scoggins M, Ramirez D, et al. A pilot study of neoadjuvant talazoparib for early-stage breast cancer patients with a BRCA mutation. Ann Oncol. 2016;27(suppl_6).
Litton JK, Scoggins M, Hess KR, et al. Neoadjuvant talazoparib (TALA) for operable breast cancer patients with a BRCA mutation (BRCA +). J Clin Oncol. 2018;36(15):508.
Kummar S, Kinders R, Gutierrez ME, et al. Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol. 2009;27(16):2705–11.
Nishikawa T, Matsumoto K, Tamura K, et al. Phase 1 dose-escalation study of single-agent veliparib in Japanese patients with advanced solid tumors. Cancer Sci. 2017;108(9):1834–42.
Nishio S, Takekuma M, Takeuchi S, et al. Phase 1 study of veliparib with carboplatin and weekly paclitaxel in Japanese patients with newly diagnosed ovarian cancer. Cancer Sci. 2017;108(11):2213–20.
Thaker PH, Salani R, Brady WE, et al. A phase I trial of paclitaxel, cisplatin, and veliparib in the treatment of persistent or recurrent carcinoma of the cervix: an NRG Oncology Study (NCT#01281852). Ann Oncol. 2017;28(3):505–11.
Mizugaki H, Yamamoto N, Nokihara H, et al. A phase 1 study evaluating the pharmacokinetics and preliminary efficacy of veliparib (ABT-888) in combination with carboplatin/paclitaxel in Japanese subjects with non-small cell lung cancer (NSCLC). Cancer Chemother Pharmacol. 2015;76(5):1063–72.
Gray HJ, Bell-McGuinn K, Fleming GF, et al. Phase I combination study of the PARP inhibitor veliparib plus carboplatin and gemcitabine in patients with advanced ovarian cancer and other solid malignancies. Gynecol Oncol. 2018;148(3):507–14.
Soumerai JD, Zelenetz AD, Moskowitz CH, et al. The parp inhibitor veliparib can be safely added to bendamustine and rituximab and has preliminary evidence of activity in B-cell lymphoma. Clin Cancer Res. 2017;23(15):4119–26.
Nuthalapati S, Munasinghe W, Giranda V, Xiong H. Clinical pharmacokinetics and mass balance of veliparib in combination with temozolomide in subjects with nonhematologic malignancies. Clin Pharmacokinet. 2018;57(1):51–8.
Mittapalli RK, Nuthalapati S, Delke DeBord AE, Xiong H. Development of a level a in vitro-in vivo correlation for veliparib (ABT-888) extended release tablet formulation. Pharm Res. 2017;34(6):1187–92.
Hussain M, Carducci MA, Slovin S, et al. Targeting DNA repair with combination veliparib (ABT-888) and temozolomide in patients with metastatic castration-resistant prostate cancer. Invest New Drugs. 2014;32(5):904–12.
Czito BG, Deming DA, Jameson GS, et al. Safety and tolerability of veliparib combined with capecitabine plus radiotherapy in patients with locally advanced rectal cancer: a phase 1b study. Lancet Gastroenterol Hepatol. 2017;2(6):418–26.
Coleman RL, Sill MW, Bell-McGuinn K, et al. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation—An NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol. 2015;137(3):386–91.
Kummar S, Oza AM, Fleming GF, et al. Randomized trial of oral cyclophosphamide and veliparib in high-grade serous ovarian, primary peritoneal, or fallopian tube cancers, or brca-mutant ovarian cancer. Clin Cancer Res. 2015;21(7):1574–82.
Kummar S, Wade JL, Oza AM, et al. Randomized phase II trial of cyclophosphamide and the oral poly (ADP-ribose) polymerase inhibitor veliparib in patients with recurrent, advanced triple-negative breast cancer. Invest New Drugs. 2016;34(3):355–63.
Somlo G, Frankel PH, Arun BK, et al. Efficacy of the PARP inhibitor veliparib with carboplatin or as a single agent in patients with germline BRCA1- or BRCA2-associated metastatic breast cancer: california cancer consortium trial NCT01149083. Clin Cancer Res. 2017;23(15):4066–76.
Rugo HS, Olopade OI, DeMichele A, et al. Adaptive randomization of veliparib-carboplatin treatment in breast cancer. N Engl J Med. 2016;375(1):23–34.
Loibl S, O’Shaughnessy J, Untch M, et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial. Lancet Oncol. 2018;19(4):497–509.
Ramalingam SS, Blais N, Mazieres J, et al. Randomized, Placebo-Controlled, Phase II Study of Veliparib in Combination with Carboplatin and Paclitaxel for Advanced/Metastatic Non-Small Cell Lung Cancer. Clin Cancer Res. 2017;23(8):1937–44.
Reck M, Blais N, Juhasz E, et al. Smoking history predicts sensitivity to parp inhibitor veliparib in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2017;12(7):1098–108.
Mehta MP, Wang D, Wang F, et al. Veliparib in combination with whole brain radiation therapy in patients with brain metastases: results of a phase 1 study. J Neurooncol. 2015;122(2):409–17.
Chabot P, Hsia TC, Ryu JS, et al. Veliparib in combination with whole-brain radiation therapy for patients with brain metastases from non-small cell lung cancer: results of a randomized, global, placebo-controlled study. J Neurooncol. 2017;131(1):105–15.
Middleton MR, Friedlander P, Hamid O, et al. Randomized phase II study evaluating veliparib (ABT-888) with temozolomide in patients with metastatic melanoma. Ann Oncol. 2015;26(10):2173–9.
Gabrielson A, Tesfaye AA, Marshall JL, et al. Phase II study of temozolomide and veliparib combination therapy for sorafenib-refractory advanced hepatocellular carcinoma. Cancer Chemother Pharmacol. 2015;76(5):1073–9.
Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19(11):1381–8.
Barber LJ, Sandhu S, Chen L, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229(3):422–9.
Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–5.
Jaspers JE, Kersbergen A, Boon U, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3(1):68–81.
Dev H, Chiang TW, Lescale C, et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol. 2018;20(8):954–65.
Kondrashova O, Topp M, Nesic K, et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat Commun. 2018;9(1):3970.
Plummer R, Jones C, Middleton M, et al. Phase I study of the poly (ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14(23):7917–23.
Das BB, Huang SN, Murai J, et al. PARP1–TDP1 coupling for the repair of topoisomerase I–induced DNA damage. Nucl Acids Res. 2014;42(7):4435–49.
Jiao S, Xia W, Yamaguchi H, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23(14):3711–20.
Gourley C, Balmana J, Ledermann JA, et al. Moving from poly (ADP-Ribose) polymerase inhibition to targeting DNA repair and DNA damage response in cancer therapy. J Clin Oncol. 2019;37(25):2257–69.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this article.
Conflict of Interest
Esha Sachdev, Roya Tabatabai, Varun Roy, and Monica Mita declare that they have no conflicts of interest that might be relevant to the contents of this article. B.J. Rimel has received consulting fees from Tesaro, Clovis, AstraZeneca, and Genentech.
Rights and permissions
About this article

Cite this article
Sachdev, E., Tabatabai, R., Roy, V. et al. PARP Inhibition in Cancer: An Update on Clinical Development. Targ Oncol 14, 657–679 (2019). https://doi.org/10.1007/s11523-019-00680-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-019-00680-2