
Abstract
In this study cannabidiol (CBD) was administered orally to determine its effects and mechanisms in the experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis (MS). We hypothesized that 75 mg/kg of oral CBD given for 5 days after initiation of disease would reduce EAE severity through suppression of either the early peripheral immune or late neuroimmune response. EAE was induced in C57BL/6 mice at two different magnitudes, and peripheral inflammatory and neuroinflammatory responses were measured at days 3, 10, and 18. Th1, Th17, Tc1, Tc17, Tregs, and myeloid derived suppressor cells (MDSC) were identified from the lymph nodes and spleens of each mouse to determine if CBD altered the suppressor cell or inflammatory cell populations in secondary lymphoid tissues. Additionally, neuroinflammation was identified in brain and spinal cord tissues using various immunohistochemical techniques and flow cytometry. Early treatment of EAE with oral CBD reduced clinical disease at the day 18 timepoint which correlated with a significant decrease in the percentage of MOG35–55 specific IFN-γ producing CD8+ T cells in the spleen at day 10. Analysis of both T cell infiltration and lesion size within the spinal cord also showed a moderate reduction in neuroinflammation within the central nervous system (CNS). These results provide evidence that oral CBD suppressed the peripheral immune response that precedes neuroinflammation; however, analysis of the neuroinflammatory endpoints also suggest that the modest reduction in neuroinflammation was only partially responsible for CBD’s neuroprotective capability.

CBD was administered orally for the first 5 days following initiation of EAE. CBD attenuated clinical disease, and we found that CBD suppressed IFN-γ producing CD8+ T cells in the spleen at day 10. There was also modest suppression of neuroinflammation. Together these data demonstrate that early, oral administration of CBD protected mice from disease, but the modest effects on neuroinflammation suggest other mechanisms participate in CBD’s neuroprotective effect in EAE.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
MS is a severely debilitating neurodegenerative autoimmune disease that results in the demyelination of neurons and eventual destruction of axons within the CNS. Onset of symptoms is usually in young adults, occurs more frequently in women, and results in progressively worsening neurologic dysfunction that can be debilitating. Despite much research in this area, we still do not understand the etiology behind this disease, but current evidence suggests the involvement of both genetic and environmental factors in triggering the immune system to attack myelin sheaths (Mallucci et al. 2015). EAE is a murine model commonly used to study MS that has been instrumental in our understanding of MS pathophysiology and development of treatments for MS patients due to its ability to imitate the neuroinflammation seen in MS. EAE can be induced by either immunizing the mice with antigens that are present in the CNS such as myelin basic protein (MBP), proteolipid protein (PLP), or myelin oligodendrocyte protein (MOG), or by adoptive transfer of autoreactive T cells (Rao and Segal 2012).
Most early studies of MS and EAE focused on Th1 cells as the most important cell type in pathogenesis; however more recent studies have shown that Th17 cells have a more important role in disease initiation than Th1 cells (Miller and Karpus 1994; Bongioanni et al. 1999; Langrish et al. 2005; Tzartos et al. 2008; Brucklacher-Waldert et al. 2009; Fletcher et al. 2010). Typically, Th1 cells and Th17 cells are identified by their ability to produce IFN-γ and IL-17 respectively, but the plasticity of T cells can also result in cells that are capable of producing both cytokines (Annunziato et al. 2007; Sallusto et al. 2012). Furthermore, in addition to CD4+ T cells, there are CD8+ T cells that produce IFN-γ and IL-17, known as Tc1 and Tc17 cells respectively, and although CD4+ T cells are currently the primary focus of most MS and EAE studies, the Tc1 and Tc17 cells also appear to play a role in disease progression (Huseby et al. 2001; Gravano and Hoyer 2013; Sinha et al. 2015). In early disease autoreactive T cells enter the CNS, recognize autoantigens, and begin activating local microglia and macrophages, which in turn recruit more leukocytes from the periphery and begin the demyelination process seen in both EAE and MS (Mallucci et al. 2015). Then, in response to this overwhelming autoimmunity, the immune system begins to launch a concurrent anti-inflammatory response in several different cell types. Examples of this include increases in myeloid derived suppressor cells (MDSCs) in MS and EAE, microglia and macrophages that switch to immunosuppressive phenotypes after phagocytizing myelin, and suppressor B cells that have been identified in both MS and EAE (Boven et al. 2006; Bogie et al. 2014; Crook and Liu 2014; Li et al. 2015). Based on the current body of literature available it is clear that MS and EAE involve an integrated systemic immune response that ranges across both pro-inflammatory and anti-inflammatory branches of the immune system, and therefore investigations of new therapies should examine systemic inflammation as well as neuroinflammation.
CBD is a phytocannabinoid found in Cannabis sp. with immunosuppressive properties and structural similarity to Δ9-tetrahydrocannabinol (THC). However, slight differences in CBD’s structure cause it to have lower affinity for cannabinoid receptors (CB1 and CB2) than THC, so it does not exhibit the negative cognitive effects associated with cannabis use (Kozela et al. 2011; Burstein 2015; M. Mecha et al. 2017). CBD’s lack of psychotropic effects makes it an attractive compound for development as an immunosuppressive drug. One emerging mechanism of immunosuppression that is of particular interest for work being done in the field of MS is CBD’s ability to suppress IFN-γ and IL-2 production in response to in vitro stimulation of T cells with phorbol 12-myristate-13-acetate and ionomycin (PMA/Io) (Kaplan et al. 2003; Kaplan et al. 2008). If this mechanism of T cell suppression holds true in vivo, it could have broad reaching implications for MS and other T cell-driven diseases. Moreover, while some studies have shown CBD’s ability to reduce clinical disease and neuroinflammation in EAE, none have examined CBD as an oral medication in this model or the effects of CBD on the initial peripheral immune response which precedes neuroinflammation (Kozela et al. 2011; Mecha et al. 2013). We hypothesized that administration of oral CBD for 5 days after initiation of disease would reduce EAE severity through suppression of either the early peripheral immune or late neuroimmune response. In this study, two degrees of severity of EAE were induced in female C57BL/6 mice and 75 mg/kg of CBD was dosed for 5 days after initiation of disease to determine if CBD had different effects at varying levels of neuroinflammation. Various tissues were collected from mice at three time points to examine the effects of CBD on EAE as disease progressed from a peripheral immune response to a neuroinflammatory response. The systemic approach used in this study provides a more complete understanding of how early use of oral CBD reduces clinical severity in the EAE model, and that initial attenuation of the peripheral immune response by CBD precedes modest suppression of neuroinflammation later in disease.
Materials
Reagents
CBD was provided by the National Institute on Drug Abuse. MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) was obtained from Biosynthesis (Lewisville, TX). Heat-killed Mycobacterium tuberculosis H37Ra (HKMT) was purchased from Difco/BD Biosciences (Detroit, MI). Complete Freund’s Adjuvant was obtained from Sigma (St. Louis, MO). All other chemicals/reagents were obtained from Fisher Scientific unless otherwise noted.
Antibodies and dyes used for flow cytometry analysis included Pacific Blue Zombie Fixable Viability Dye (FVD; BioLegend, San Diego, CA), Near IR (NIR) FVD (BioLegend) CD4-FITC (BioLegend Clone Gk1.5), CD8α-PE/Cy7 (BioLegend Clone 53–6.7), IL-17A-APC (BioLegend Clone TC11-18H10.1) IFN-γ-PE (BioLegend Clone XMG1.2), CD11b-APC (BioLegend Clone M1/70), Ly6G-PE/Cy7 (BioLegend Clone 1A8), Ly6C-PE (BioLegend Clone HK1.4), CD4-PE/Cy7 (BioLegend Clone GK1.5), CD25-FITC (eBioscience Clone PC61.5), FoxP3-APC (eBioscience Clone FJK-16 s), CD3 Pacific Blue (BioLegend Clone 17A2), CD45 Briliant Violet 605 (BV605; BioLegend Clone 30-F11). Other reagents used include flow cytometry buffer (FC buffer, 1X Hank’s Buffered Saline Solution with 1% bovine serum albumin, pH 7.4), Fc Block (purified mouse CD16/CD32, BD Biosciences, Billerica, MA), fixation/permeabilization solution (eBioscience/ThermoFisher, San Diego, CA), and 1X permeabilization solution (eBioscience).
The reagents used for the ELISA procedure outlined below were IL-17A antibodies (BioLegend), IFN-γ antibodies (BioLegend), IL-17A (BioLegend) and IFN-γ (BioLegend) standards, biotinylated IL-17A antibodies (BioLegend), biotinylated IFN-γ antibodies (BioLegend), HRP-Avidin (BioLegend), and TMB substrate (BioLegend),
The antibodies used for histologic preperations were polyclonal rabbit anti-human CD3 antibody (Dako), biotinylated goat anti-rabbit secondary antibody (Vector Laboratories), biotinylated monoclonal rat CD4 antibody (ThermoFisher Clone 4SM95), unconjugated monoclonal rabbit CD8 antibody (Abcam, Cambridge, MA Clone EPR21769), Alexa Fluor 488 conjugated goat anti-rabbit IgG antibody (ThermoFisher), and unconjugated polyclonal rabbit GFAP antibody (Millipore, Burlington, MA). Other reagents used were 4% goat serum (Vector Laboratories, Burlingame, CA), Vectastain Elite ABC Kit (Vector Laboratories), diaminobenzidine (DAB; Dako), Vectashield (Vector Laboratories), and 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI; ThermoFisher).
Animals
Experiments were approved by Mississippi State University Institutional Animal Care and Use Committee (IACUC). Wild type female C57BL/6 mice obtained from Envigo (Indianapolis, IN) at 8 weeks of age were used for this experiment. Mice were housed three to five in a cage in a temperature (22 °C ± 1 °C), humidity (40–60%), and light (12 h light:12 h dark) controlled room. Food and water were provided ad libitum. As disease progressed, access to water was ensured with use of long sipper tubes and food pellets were placed on the floor of the cage after day 7.
Methods
EAE Induction and Clinical Assessment
Two different levels of disease were initiated in order to address whether CBD would produce similar results; thus, the relatively more severe disease is indicated as “EAE” as opposed to “mild EAE”. Moreover, pertussis toxin (PTX) was not used in this study to avoid possible confounding factors that are associated with PTX, such as its ability to inhibit G protein coupled receptors like CB1 or CB2 (Mangmool and Kurose 2011). This is important because although CBD does not exhibit high affinity for CB1 or CB2 (Matsuda et al. 1990; Munro et al. 1993), the exact receptor(s) mediating its effects have not been identified and these cannabinoid receptors may still play a role in CBD’s effects despite their low affinity (Nichols and Kaplan 2020). C57BL/6 mice were immunized with 100 μl of CFA containing either 100 μg of MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) with 0.5 mg of HKMT or 20 μg of MOG35–55 peptide with 0.1 mg HKMT to induce EAE and Mild EAE, respectively. Control mice were injected with saline. To perform the injections, mice were anesthetized with isoflurane, and immunizations were given subcutaneously in tissues over the shoulders and hips (25 μl at each site), after which the mice were allowed to recover in a portion of the cage cleared of bedding. Starting 24 h after the injections were administered, mice were dosed each morning with either 75 mg/kg of CBD in 100 μl of corn oil (CO) or 100 μl of CO for 5 days via oral gavage. This dose of CBD has been used in previous studies and results in serum concentrations of CBD similar to that found in humans (Karmaus et al. 2013). Clinical scores were recorded for the duration of the experiment using the following clinical scale: 0 – Healthy; 0.5 – Flaccid tail; 1 – Hind limb paresis/waddling gait; 2 – Unable to prevent being placed in dorsal recumbency; 3 – Single hind limb paralysis/Leg dragging; 4 – Complete hind limb paralysis; 5 – Moribund/involvement of forelimbs (modified from Rao et al.) (Rao and Segal 2012). This study is a repeat of previous studies our lab has done to examine CBD effects on late disease (day 18), but also includes immune function assessments at days 3 and 10.
Ex Vivo Restimulation with MOG Peptide
Cells were isolated from the secondary lymphoid tissues by mechanical disruption of individual spleens and lymph nodes in 1X RPMI, followed by filtration using a 70 μm filter to obtain a single cell suspension. A portion of these isolates were set aside to stain for the presence of Tregs and MDSCs as described below. 1 × 106 cells were seeded into 96 well U-bottom plates in 200 μl of 1X RPMI supplemented with 5% bovine calf serum (HyClone, Logan, UT), 1% penicillin (Gibco, Gaithersburg, MD), and 50 μM 2-mercaptoethanol (2-ME, Gibco) and restimulated with 100 μg/ml of MOG peptide for 48 h at 37 °C. At 44 h, supernatants were collected for ELISAs, and cells were resuspended in 1x RPMI containing 5 μg/ml of Brefeldin A (BioLegend, San Diego, CA) for the last 4 h. Cells were then stained for extracellular markers (CD4 and CD8) and intracellular cytokine production (IFN-γ and IL-17A) and analyzed using flow cytometry.
Isolation of Mononuclear Cells from the Spinal Cords
To isolate cells from the spinal cord from each mouse, the spinal column was first removed and PBS was injected into the caudal aspect of the spinal canal. This caused extrusion of the spinal cord from the cranial spinal canal. The spinal cord of each mouse was then cut into small pieces and digested with 1 mg/ml collagenase type 4 (Worthington) and 20 μg/ml DNase I for 45 min at 37 °C with agitation. To isolate mononuclear cells the digestion solution was then pressed through a 70-μm cell strainer and resuspended in 30% Percoll. The 30% Percoll was then underlaid with 70% Percoll to form a gradient and centrifuged at 500 x g on low speed with no brake for 20 min. The mononuclear cells were then gently removed from the buffy coat layer, and washed in PBS twice before proceeding to staining for flow cytometry. To be sure enough cells were obtained for analysis, cells from two mice were pooled before staining for flow cytometry.
Extracellular and Intracellular Staining
IL-17A and IFN-γ Producing T Cells
After treatment of cells with Brefeldin A, the 96-well U-bottom plates were centrifuged at 500 x g for 5 min and the media was removed. The cells were then rinsed by suspending them in 200 μl of 1X phosphate buffered saline (PBS), centrifuged at 500 x g for 5 min, and the PBS was removed. To measure viability 0.5 μl Pacific Blue Zombie FVD in 200 μl of PBS was added to each well and allowed to incubate for 30 min at 4 °C. The cells were centrifuged again at 500 x g for 5 min and the FVD solution was removed. To prevent non-specific Ab binding, cells were incubated at room temperature (RT) for 15 min in 50 μl of FC buffer containing 0.5 μl of Fc Block. Without removing the Fc Block from the wells, 50 μl of FC buffer containing 0.3 μl of CD4-FITC and CD8α-PE/Cy7 was added to each well and allowed to incubate at RT for 30 min. The plate was then centrifuged at 500 x g for 5 min, the supernatants containing the antibodies were removed, and a rinse step was performed with FC buffer. 200 μl of fixation/permeabilization solution was added to each well in preparation for intracellular staining, allowed to incubate for 30 min at RT, and removed by centrifuging the plate and removing the supernatants. For intracellular staining 50 μl of 1X permeabilization solution containing 0.5 μl IL-17A-APC and 0.5 μl IFN-γ-PE was added to each well, and allowed to incubate at RT for 1 h. The antibodies were then removed as before and the cells were resuspended in 200 μl of FC buffer for analysis by flow cytometry.
Tregs and MDSC
5 × 105 cells isolated directly from the lymph nodes and spleens were suspended in 200 μl of FC buffer, and centrifuged at 500 x g for 5 min to rinse the cells. As before, cells were first treated with FC block, and then extracellular antibodies specific for each cell type were added and allowed to incubate for 30 min at room temperature. For MDSC, 50 μl of FC buffer containing 0.3 μl of CD11b-APC, Ly6G-PE/Cy7 and Ly6C-PE was added. For Tregs, 50 μl of FC buffer containing 0.3 μl of CD4-PE/Cy7 and CD25-FITC was added. The antibodies were then removed and cells were washed with FC buffer. At this point the MDSCs were fixed and suspended in 200 μl of FC buffer for analysis by flow cytometry, and the Tregs were stained for intracellular FoxP3 expression. For intracellular staining, cells were incubated with 200 μl of fixation/permeabilization solution for 30 min at RT. Then the fixation/permeabilization solution was removed, the cells were incubated with 50 μl of 1X permeabilization solution containing 0.5 μl FoxP3-APC for 1 h, and then suspended in 200 μl of FC buffer for analysis by flow cytometry.
Spinal Cord Infiltrates
The cells isolated from the spinal cord were rinsed two times with PBS, as previously described, and then incubated for 30 min at 4 °C in 50 μl of PBS containing 0.5 μl NIR FVD. The cells were then rinsed twice with PBS and incubated for 15 min in 50 μl of FC buffer containing 0.5 μl of Fc Block. Without removing the Fc Block from the wells, 50 μl of FC buffer containing 0.3 μl of CD45-BV605, CD4-FITC, CD3-Pacific Blue, CD8α-PE/Cy7, and CD25-PE was added to each well and allowed to incubate at RT for 30 min. After this incubation the cells were rinsed with FCM and 50 μl of fixation/permeabilization solution was added and allowed to incubate for 30 min at RT. Intracellular staining for FoxP3 was performed as described in the previous section.
Flow Cytometry
Cells stained with intracellular and extracellular florescent antibodies were analyzed using an ACEA Novocyte flow cytometer (ACEA Biosciences, San Diego, CA).
IL-17A and IFN-γ Producing T Cells
Dead cells were excluded from analysis by selecting cells that were not stained by Pacific Blue FVD. Doublets were then eliminated based on side scatter (SSC) forward scatter (FSC) area/height characteristics. Lymphocytes were then selected based on FSC versus SSC characteristics, and CD4+ and CD8+ positive populations were selected by expression of CD4 and CD8, respectively. Once the CD4+ and CD8+ lymphocytes had been identified, gates were set up to determine the percentage of these cells that contained intracellular IL-17A and IFN-γ (Supplemental Fig. 1). Cell counts are provided in Supplemental Figs. 2-3.
Tregs
As with the inflammatory cells, the lymphocytes were selected using FSC and SSC gating, and doublets were then eliminated based on SSC-A/H and FSC-A/H characteristics. Then CD4+ cells were selected by expression of CD4. Once CD4+ Cells were identified, the percentage of double positive CD25+Foxp3+ cells within the CD4+ population was determined (Supplemental Fig. 4). Cell counts are provided in Supplemental Fig. 5.
MDSCs
Lymphocytes were selected using FSC and SSC gating, and doublets were then eliminated based on SSC-A/H and FSC-A/H characteristics. MDSC were then identified based on their expression of CD11b, Ly6C, and Ly6G extracellular markers. Monocytic MDSCs and granulocytic MDSCs were differentiated based on their Ly6C and Ly6G profiles. Granulocytic MDSC are identified as CD11b+Ly6CloLy6G+ cells and monocytic MDSC are identified as CD11b+Ly6C+Ly6G− (Supplemental Fig. 4)(Crook and Liu 2014). Cell counts are provided in Supplemental Fig. 5.
Spinal Cord Infiltrates
As before, dead cells were excluded from analysis by selecting cells that were not stained by NIR FVD, doublets were excluded by their SSC-A/H and FSC-A/H characteristics, and lymphocytes were selected based on FSC and SSC gating. CD45+CD3+ cells were then selected and subdivided base on their expression of CD4+ and CD8+. Cell counts are provided in Supplemental Fig. 6. Further gating of CD3+CD4+ T cells was performed to determine the number of Tregs within the spinal cords based on their expression of CD25 and FoxP3.
Elisa
Plates were coated with purified mouse IL-17A or IFN-γ antibodies by filling each well with 100 μl of coating buffer containing the desired antibody at a 1:500 dilution, and incubating the plate overnight at 4 °C. The next day the plates were washed three times with PBS containing 0.05% Tween 20 (PBST), and then washed three times with distilled water. PBS containing 3% BSA (3% BSA-PBS) was placed in the plate for at least 1 h at RT to block non-specific binding, and then the plate was washed three times with PBST. 100 μl of recombinant IL-17A and IFN-γ standards were then pipetted into their respective wells at increasing concentrations (from 7.8–4000 pg/ml), and 50 μl of supernatants obtained from the 48 h cultures were pipetted into each plate to measure the concentration of IL-17A and IFN-γ in each supernatant. 1:10 dilutions of all supernatants were prepared in the same manner to detect higher ranges of these cytokines. 100 μl of 3% BSA-PBS was used as a blank control. The plates containing the standards and supernatants were then incubated for 1 h at RT, and subsequently washed three times with PBST. 100 μl of biotinylated IL-17A or biotinylated IFN-γ antibodies in 3% BSA-PBS at a 1:500 dilution were added to their respective plates and incubated for 1 h at RT. The plates were next washed three times with PBST. 100 μl of HRP-Avidin in 3% BSA-PBS at a dilution of 1:500 was added to each well and incubated for 1 h at RT, and then the plates were rinsed three times with PBST. 100 μl of TMB substrate was then added to each well and incubated in the dark until color development was detected in the lowest standard, then the reaction was stopped with 100 μl of 2 N H2SO4, and optical density (OD) was measured at 450 nm.
Processing of Brains and Spinal Cords for Histology
After the mice were euthanized, to make manipulation of the cranium and spinal column easier, the spinal column was severed at the base of the skull just caudal to the first cervical vertebra (C1), and the brain and spinal column were processed separately.
Brain
To remove the brain from the cranial cavity, the calvarium was removed, the brain was elevated out of the cranial cavity, and sharp dissection of the cranial nerves was performed to disconnect the brain from the skull. The brain was then cut along the sagittal plane using a razor blade and the right half of the brain was placed in 10% neutral buffered formalin (NBF) for fixation. Once fixation was complete, the brains were embedded in paraffin wax blocks so that sections of the tissue could be cut for histologic analysis.
Spinal Column
After the spinal column was separated from the skull, the excess tissue and ribs were trimmed, and the caudal end of the spinal cord was cut at the level of the L6 vertebra. Next the entire spinal column was placed in 10% NBF for fixation. The fixed spinal columns were rinsed in PBS to remove the 10% NBF, and were placed in a 10% EDTA solution at pH 7.4 for decalcification. 10% EDTA was made by diluting 0.5 M EDTA, pH 8 (ThermoFisher) with distilled water, and adjusting the solution to pH 7.4 with acetic acid. The spinal columns were left in this solution for 15 days and the solution was refreshed every 5 days. Once decalcification was complete, the spinal column was cut at the T7 vertebra to isolate the lumbar intumescence. This method of dissection left the spinal column between T7 and L6. The T7-L6 region was then cut at intervals of approximately 0.3 cm in length, which provided between 7 and 8 separate segments depending on the original length of the mouse. The segments were then placed in paraffin wax blocks so the tissue could be cut for histologic analysis.
Histologic Analysis
5-μm sections of brains and spinal cords from each time point were initially stained with hematoxylin and eosin (H&E) to determine the level of cellular invasion. Based on the results seen from this initial analysis, it was determined that brains from the day 18 mice in the SAL/CO, EAE/CO and EAE/CBD groups would be stained for CD3 to determine which regions contained T cells, and spinal cords would be used for quantification of T cell numbers and lesion size.
CD3 Staining
5-μm sections from brains and spinal cords of the day 18 mice from SAL/CO, EAE/CO and EAE/CBD groups were used for CD3 staining. Sections were first deparafinized and rehydrated by placing them in the following solutions for 2 min each: xylene, 100% ethanol, 95% ethanol, DI water. Antigen retrieval was performed in a steamer by treating the slides for 20 min with Target Retrieval Solution which is a modified citrate buffer with a pH 6.1. After retrieval slides were air dried for 10 mins, rinsed with phosphate buffered saline (PBS), and covered with 3% H2O2 for 30 min to block endogenous peroxidases. H2O2 was removed by washing the slides three times in PBS. A PBS solution containing 1% bovine serum albumin (BSA) and 4% goat serum was then added to each slide and incubated for 1 h to block non-specific binding of the primary antibodies. The blocking solution was removed, and polyclonal rabbit anti-human CD3 antibody in PBS/1% BSA/0.1% Triton X-100 (Tx) (1:2500) was added to each slide and incubated overnight at 4 °C. The following day, slides were rinsed three times with PBS and treated with a biotinylated goat anti-rabbit secondary antibody diluted in PBS/1% BSA/0.1% Tx (1:100) for 2 h at RT. Slides were then rinsed with distilled water three times, and incubated in a solution containing avidin D and biotinylated horseradish peroxidase H (Vectastain Elite ABC Kit) in PBS for 2 h. Sections were then rinsed three times with distilled water, and incubated with DAB for approximately 5 min. The reaction with DAB was stopped by dipping the slides in distilled water, and the slides were dipped in hematoxylin to make visualization of histologic structures easier. Each slide was then dipped in 0.3% ammonia water to lighten the hematoxylin stain. For dehydration, slides were dipped in each of the following solutions for 1 min: distilled water, 75% ethanol, 100% ethanol, and xylene. A cover slip was placed on the slide with permount and allowed to dry.
CD4 and CD8 Double Stain
Due to the extremely dim staining that resulted from CD4 immunofluorescent staining, it was decided that the best route to achieve a double stain would be to stain the CD4 first as an immunoperoxidase stain, and then follow up with a CD8 immunofluorescent stain. Much like the CD3 stain, slides for the double stain underwent deparafinization, antigen retrieval and blocking with 3% H2O2; however it is important to note that 1% Tx was added to the antigen retrieval solution to help uncover the antigens. Following the blocking step a biotinylated monoclonal rat CD4 antibody in PBS/1% BSA/1% Tx (1:40) was added to each slide and allowed to incubate overnight at 4 °C. The next day, the slides were rinsed with distilled water and incubated in a solution containing avidin D and biotinylated horseradish peroxidase H and an unconjugated monoclonal rabbit CD8 antibody in PBS (1:50) for 2 h at RT. Sections were then rinsed three times with distilled water, and incubated with DAB for approximately 20 min. The reaction with DAB was stopped by dipping the slides in distilled water, and the slides were dipped in hematoxylin to make visualization of histologic structures easier. Each slide was then dipped in 0.3% ammonia water to lighten the hematoxylin stain. Next an Alexa Fluor 488 conjugated goat anti-rabbit IgG antibody in PBS/1% BSA/0.1% Tx (1:200) was added to each slide and allowed to incubate for 2 h. To protect the Alexa Fluor 488 from degradation during visualization slides were coverslipped with Vectashield.
Glial Fibrillary Acidic Protein (GFAP)
Deparafinization, antigen retrieval and blocking with 4% goat serum was performed as it was for the CD3 stain. After blocking, slides were incubated with PBS/1% BSA/0.1% Tx containing unconjugated polyclonal rabbit GFAP antibody (1:200; Millipore, Burlington, MA) overnight at 4 °C in the dark. The next day, the slides were rinsed in distilled water and incubated for 2 h with PBS/1% BSA/0.1% Tx containing Alexa Fluor 488 conjugated goat anti-rabbit IgG antibody (1:200; ThermoFisher) and 0.5 μg/ml of 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI; ThermoFisher) as a nuclear counterstain. Slides were then rinsed and coverslipped with Vectashield.
Imaging
Images were captured using a Lumenera Digital Camera equipped with Infinity Analyze Software, and analyzed using the ImageJ software. To analyze the number of CD3+, CD4+, and CD8+ T cells present within the spinal cord a total of four lesions from two sections of lumbosacral intumescence of each mouse were analyzed at 400x magnification. The number of each cell type was averaged across the four lesions to get the average number of cells for lesions within the lumbosacral region of each mouse. To determine lesion size for each mouse, 40x magnification images of 2 sections from the same region were analyzed using the GFAP/DAPI stain, which allowed us to determine the size of the lesion within the parenchyma of the spinal cord and exclude the portion of the lesions within the meninges.
Statistical Analysis
Statistical analysis was done with GraphPad Prism 7 software. For clinical scoring the Mann-Whitney U test was use for comparisons between groups. All other comparisons were made between groups of the same day using ANOVA or t-test with the level of significance defined as p < 0.05. Transformations were performed on all percentages obtained by flow cytometry before ANOVA was performed. Graphical representations of data show averages within each group and the error bars represent the standard deviation of each group.
Results
Clinical Scores
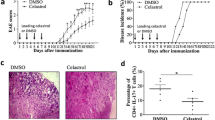
Average clinical scores recorded during the 18 day time course showed that clinical disease begins on approximately day 14 for all groups except for the SAL/CO group (n = 5). On day 18, the EAE/CO group (n = 11) had the highest average clinical score out of all the groups, with the EAE/CBD group (n = 11) having a significantly lower average clinical score than the EAE/CO group. The Mild EAE/CO group (n = 5) also had lower clinical disease than the EAE/CO group. There was no clinical difference seen between the Mild EAE/CO and Mild EAE/CBD (n = 5) groups. The SAL/CO group showed no signs of clinical disease (Fig. 1).

Clinical scores. Average clinical scores recorded for each group of mice during the 18 day time course. Mice from 3 and 10 day time points were included in each group until they were euthanized. Disease incidence in day 18 groups was as follows: SAL/CO = 0%, EAE/CO = 82%, EAE/CBD = 64%, Mild EAE/CO = 60%, Mild EAE/CBD = 40%. * p < 0.05 differences between SAL/CO and EAE/CO; # p < 0.05 difference between EAE/CO and EAE/CBD; @ p < 0.05 difference between EAE/CO and Mild EAE/CO.There were 5 mice in all groups except EAE/CO and EAE/CBD, which had 11 mice per group
Effects of CBD on Suppressor Cell Populations
Same day analysis by flow cytometry of suppressor cells isolated from the secondary lymphoid tissue showed that treatment with CBD did not significantly change the percentage of Tregs isolated from the lymph nodes and spleen, or MDSCs from the spleen (Fig. 2). However, significant increases were seen in the percentage of MDSCs recovered from the spleens of EAE and Mild EAE mice as compared to the SAL/CO group at several time points with the most pronounced differences being seen on day 10 (Fig. 2c-d). Significant decreases were also seen in the percentage of Tregs recovered from the spleens and lymph nodes of the EAE/CO and Mild EAE/CO groups. On day 10, a lower percentage of Tregs was recovered from the lymph nodes of EAE/CO and Mild EAE/CO groups as compared to the SAL/CO group, and the percentage of Tregs recovered from the spleens of the EAE/CO mice was significantly decreased as compared to the SAL/CO group and the Mild EAE/CO group (Fig. 2a-b). Lastly, a significant decrease was found in percentage of monocytic MDSCs present in splenocytes from Mild EAE/CBD as compared to the EAE/CBD group on day 10 (Fig. 2d).

Regulatory cells from secondary lymphoid organs. Lymph nodes and spleens were taken from each mouse on the day of necropsy and made into single cell suspensions by passing them through a 70 μm filter. Cellular isolates from the lymph nodes were then stained for the classic Treg markers (CD4+CD25+FoxP3+) (a), and cellular isolates from the spleens were stained for Treg markers (b), Granulocytic MDSC markers (CD11b+Ly6CloLy6G+) (c) or Monocytic MDSC markers (CD11b+Ly6C+Ly6G−) (d). * p < 0.05 differences between SAL/CO and EAE/CO; ‡ p < 0.05 difference between SAL/CO and Mild EAE/CO; @p < 0.05 difference between EAE/CO and Mild EAE/CO; € p < 0.05 difference between EAE/CBD and Mild EAE/CBD
Effects of CBD on IFN-γ and IL-17 Producing T Cells
Intracellular Cytokine Staining
Analysis of IFN-γ+ and IL-17A+ T cells after a 48 h restimulation with MOG35–55 peptide showed significant differences in the percentage of MOG35–55 specific IFN-γ producing CD8+ T cells isolated from the spleens of day 10 mice (Fig. 3c). On this day, EAE/CO had the highest percentage of IFN-γ producing CD8+ T cells as compared to all other groups, with the SAL/CO, EAE/CBD, and Mild EAE/CO groups having a significantly lower percentage than the EAE/CO group. Interestingly, this pattern was also seen in the CD4+ T cells from the spleen, but only the difference between EAE/CO and SAL/CO groups was significant. Lastly, the number IL-17A+ CD8+ T cells isolated from the spleen of EAE/CO mice on day 10 was significantly increased as compared to the SAL/CO group. There were no effects of CBD on IL-17-producing T cells in the spleen.

Cytokine production in T cells isolated from spleens after ex vivo restimulation. Splenocytes were restimulated ex vivo with MOG peptide for 48 h, and then treated with Brefeldin A to block the secretion of cytokines. Flow cytometry was then used to measure the percentage of IFN-γ and IL-17A producing cells within the CD4 and CD8 population. * p < 0.05 differences between SAL/CO and EAE/CO; ‡ p < 0.05 difference between SAL/CO and Mild EAE/CO; # p < 0.05 difference between EAE/CO and EAE/CBD; @ p < 0.05 difference between EAE/CO and Mild EAE/CO.
Analysis of IFN-γ+ and IL-17A+ T cells from the lymph nodes after a 48 h restimulation with MOG35–55 peptide revealed very low cytokine production at all time points tested (Fig. 4a-d). There were no significant changes noted in response to disease or CBD.

Cytokine production in T cells isolated from lymph nodes after ex vivo restimulation. Cells isolated from lymph nodes were restimulated ex vivo with MOG peptide for 48 h, and then treated with Brefeldin A to block the secretion of cytokines. Flow cytometry was then used to measure the percentage of IFN-γ and IL-17A producing cells within the CD4 and CD8 population
Cytokine Production in Supernatants
ELISAs were performed on the supernatants following 48 h restimulation with MOG35–55 peptide to determine the cumulative production of IFN-γ and IL-17A during culture. In general, the cultured cells from EAE mice produced more IFN-γ and IL-17A than Mild EAE groups, with this difference reaching significant levels within the cultured splenocytes, and cytokine production is increased earlier in the lymph nodes than in the spleen (Fig. 5a-d). There was also a significant increase in IFN-γ production in EAE/CBD as compared to EAE/CO in splenocytes at day 18.

ELISA of Cytokines after ex vivo Restimulation. After 48 h restimulation of lymphocytes from secondary lymphoid tissues with MOG35–55 peptide, the supernatants from the cultures were analyzed by ELISA for the total amount of IL-17A and IFN- γ. * p < 0.05 differences between SAL/CO and EAE/CO;; # p < 0.05 difference between EAE/CO and EAE/CBD; @p < 0.05 difference between EAE/CO and Mild EAE/CO € p < 0.05 difference between EAE/CBD and Mild EAE/CBD
Analysis of Brain and Spinal Cord
Brain
H&E stains were performed on all mice to examine the level of cellular infiltration, and it was determined that cellular infiltrates were primarily observable at day 18. H&E stains of the brain from day 18 EAE/CO mice revealed cellular infiltrates throughout the white matter tracts of the brain, with the highest levels of infiltration being consistently seen in the white matter of the cerebellum. The infiltrating cells in the cerebellum of EAE/CO mice are diffusely spread throughout the white matter tracts, and there is significant perivascular cuffing evident in these mice (Fig. 6b). Immunoperoxidase staining for CD3 also revealed that a significant number of the infiltrating cells within these sections were T cells (Fig. 6e and h). In contrast to this, the EAE/CBD group had fewer cellular infiltrates and T cells within the white matter tracts of the cerebellum as compared to the EAE/CO group, and the cells are localized primarily in the perivascular cuffs with CBD treatment (Fig. 6c, f and i). No cellular infiltrates or T cells were observed in the SAL/CO group (Fig. 6a, d and g).

CBD reduces neuroinflammation in the cerebellum of EAE mice. Images of H&E stains show increased cellular infiltration into the white matter tracts of the cerebellum in EAE/CO mice as compared to EAE/CO and EAE/CBD mice (a-c). Similarly, CD3 stains revealed a higher level of T cell infiltrates in the EAE/CO group as compared to the SAL/CO and EAE/CBD groups. CD3 stains are shown at 40x (d-f) and 200x (g-i) magnifications
Spinal Cord
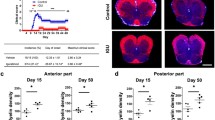
H&E stains were performed on all mice to examine cellular infiltration, and it was determined that cellular infiltrates were present primarily at the day 18 time point. The most prominent feature observed in the H&E stains of lumbosacral spinal cord from day 18 EAE/CO mice was the presence of subpial and perivascular cellular infiltrates that were comprised largely of CD3+ T cells (Fig. 7b, e and h). Subpial and perivascular infiltration was also seen in the spinal cords of EAE/CBD mice, but to a lower extent (Fig. 7c, f and i). Double staining of the spinal cord for CD4+ and CD8+ T cells (Fig. 7p-r) from each group showed similar patterns of infiltration compared to those seen with the CD3 stain. To maximize our ability to accurately analyze the T cell compartment, quantifications of CD3+ cells were performed on the same lesions as the quantifications for CD4+ and CD8+ cells in the next consecutive slice taken from the paraffin blocks containing the spinal cords. No cellular infiltrates or T cells were observed in the SAL/CO group (Fig. 7a, d, g, j, m and p). Spinal cord sections were also analyzed for lesion size using a GFAP/DAPI stain, which allowed for a more accurate measurement of lesion size within the parenchyma by eliminating the cells that were located in the meninges (Fig. 8). As with the T cell stains, the GFAP/DAPI stain showed a large amount of subpial and perivascular infiltration, but this stain also highlights the boundary between meningitis and myelitis in these sections. An observation that is of particular note in all of the stains that were performed is a surprising lack of cellular infiltrates in the white matter of the nerve roots surrounding the lumbosacral spinal cord. Quantification and statistical analysis of the T cells and lesions of the spinal cords revealed a modest decrease in the number of T cells and size of the lesions in the EAE/CBD group as compared to EAE/CO group at the day 18 timepoint (Fig. 9a-c).

CBD reduces neuroinflammation in the spinal cord of EAE mice. Images of H&E stains show increased cellular infiltration around large blood vessels and in peripheral white matter tracts of the spinal cords of EAE/CO mice as compared to SAL/CO and EAE/CBD mice (a-c). Similarly, CD3 stains revealed a higher number of T cell in these regions in the EAE/CO group as compared to the SAL/CO and EAE/CBD groups. CD3 stains are shown at 100x (d-f) and 400x (g-i) magnifications. CD4 and CD8 double staining was performed on the next slice taken from the paraffin blocks containing the spinal cords, thus allowing for intralesional comparisons to be made between the number of CD3, CD4 and CD8 positive cells. CD4 staining (j-l) revealed lower numbers of CD4+ T cells present within the lesions of the spinal cords, while CD8 staining (m-o) revealed no substantial difference between the number of CD8+ T cells when comparing the EAE/CO and EAE/CBD groups. To overcome the dark background of the CD8 immunofluorescent stain in the combined CD4/CD8 images (p-r) the contrast of the original CD4 stains (j-l) was enhanced by 0.2% using ImageJ software

Measuring lesion size within the parenchyma of the spinal cord. By using an immunofluorescent GFAP Stain (a-c & j) to define the boundary of the spinal cord parenchyma created by the glial limitans, and a DAPI nuclear stain (d-f & j) to identify lesions within the spinal cord, we were able to accurately determine the total lesion area within the parenchyma of spinal cord sections by overlaying images of the two stains and removing cells within the meninges from consideration. Images were taken at 40x magnification (a-i) and 200x magnification (j-l). Images j-l show the accumulation of cells outside of the glial limitans around major vessels, and help to illustrate the difference between meningitis and myelitis

Quantification of T cells and lesion area within the spinal cord. A total of 4 lesions from the lumbosacral intumescence were analyzed for the number and type of T cells present, as well as lesion size. CD3 (a) and CD4/CD8 double staining (c) were done on consecutive slices from the paraffin blocks, which allowed for intralesional comparisons to be made for the T cell counts. Quantification of T cells was performed by counting the number of cells present within an image captured at 400x magnification for each of the four lesions, and averaging the counts for each mouse. Lesion area within the parenchyma of the spinal cord (b) was measured first outlining the area of the lesion seen in the DAPI image, then overlaying the GFAP image and adjusting the outline so that only cells within the parenchyma were considered (See Fig. 8). Analysis of spinal cord infiltrates by flow cytometry also revealed a moderate increase in the percentage of CD3+CD4+ T cells within the spinal cord (d)
Analysis of Spinal Cord Infiltrates by Flow Cytometry
Flow cytometry performed on cells isolated from the spinal cord of EAE/CO and EAE/CBD mice showed a modest reduction in the percentage of CD3+CD4+ T cells (Fig. 9d), which closely resembles the results found by histologic analysis of the spinal cord (Fig. 9c). No difference was seen in the percentage of CD3+CD8+ or CD3+CD4+CD8+ T cells. Further analysis of CD25 and FoxP3 expression by CD4+ cells also revealed very small numbers of Tregs present within the spinal cords of both groups, and no significant difference was seen between groups (data not shown).
Discussion
Currently a main method of treatment for MS involves a two step approach that first uses immunosuppressive drugs, such as corticosteroids, to reduce the severity of disease, and then uses immunomodulatory drugs, such as glatiramer acetate and IFN-β, to reduce the recurrence of relapses. While this method does seem to help patients that are in the relapsing and remitting stage of disease, it does very little to stop the long term progressive stages of MS (Burstein 2015). For this reason the search for more viable therapies is still under way, and the use of cannabinoids as immunomodulatory compounds is one of the routes that are being examined. Recently, CBD has shown promise as a treatment for MS through its use in the EAE model and the use of Sativex, which is an oral mixture of THC and CBD, in MS patients; however the number of studies looking at CBD in the EAE model is limited, and none have examined CBD alone in MS. One study using the MOG35–55 peptide to induce EAE has shown that when CBD was administered intraperitoneally (i.p.) for 3 days after the first signs of disease it was capable of reducing microglial activation and T cell infiltration in the CNS (Kozela et al. 2011). Similarly, a study looking at the i.p. administration of CBD in the Theiler’s murine encephalomyelitis virus (TMEV) model of MS highlighted CBD’s ability to reduce the expression of vascular cell-adhesion molecule-1 (VCAM-1) on vascular endothelial cells, suppress the production of chemokines and cytokines, and reduce the activation of microglia (Mecha et al. 2013). While these studies provide vital information about the effects of CBD on neuroinflammatory components of the MS models, they did not evaluate the systemic immune response associated with the CBD treatment. Furthermore, the i.p. route of administration is used less often in a clinical setting as compared to oral administration or inhalation. For these reasons, in the present study we sought to examine the response of EAE to oral CBD by exploring the systemic immune system in addition to the neuroinflammatory component of the model. By administering CBD for 5 days after the initiation of disease, we provide evidence that early treatment of EAE with CBD reduced clinical disease in EAE mice, which was accompanied by a modest reduction in neuroinflammation.
As part of our exploration into the effects of oral CBD on EAE we chose to examine several suppressor cell types that could potentially contribute to CBD’s immunosuppressive properties. Tregs and MDSCs represent two distinct linages of suppressor cells that may have a role in the pathogenesis of MS. Tregs are of particular interest because several studies have suggested that Treg function is decreased in MS patients, leading some to speculate that dysregulation of Tregs may lead to the CNS based autoimmunity seen in MS (Haas et al. 2005; Vos et al. 2005; Feger et al. 2007). On the other hand, MDSCs have been reported to be increased in the peripheral blood of MS patients and both the secondary lymphoid tissues and CNS lesions of EAE mice, and are capable of suppressing activated T cells (Moline-Velazquez et al. 2011; Ioannou et al. 2012; Crook and Liu 2014). In previous studies, CBD has been shown to be capable of inducing both Tregs and MDSCs (Hegde et al. 2011; Hegde et al. 2015; Dhital et al. 2017; Elliott et al. 2018); however, in the present study analysis by flow cytometry showed CBD had no effect on the percentage of either suppressor cell population in the secondary lymphoid organs, or on the percentage of Tregs present in the spinal cord. One possible explanation for the discrepancy between previous MDSC studies and the results reported here is that the previous studies used i.p. injection for CBD, while we used oral gavage to administer CBD. This is supported by results from Hegde et al. which suggests that the induction of MDSCs seen after the i.p. injection of CBD is dependent on the production of G-CSF by peritoneal mast cells (Hegde et al. 2015). Additionally, i.p. injection limits CBD modification by the metabolic enzymes of the liver, which could cause differential effects as compared to oral administration of CBD. In an earlier paper, Hegde et al. also showed that i.p. administration of CBD was capable of modest induction of Tregs in conjunction with increased MDSC in an autoimmune hepatitis model (Hegde et al. 2011). These results were more recently supported by our own in vitro study showing CBD’s ability to induce Tregs at low level T cell stimulation (Dhital et al. 2017). In the EAE model, i.p. injections of CBD have been shown to decrease severity, which correlated to an increase number of MDSCs in the peritoneal cavity, and when these MDSCs were placed in culture with MOG restimulated splenocytes or adoptively transfered into EAE mice the MDSCs were capable of suppressing T cell proliferation and disease respectively (Elliott et al. 2018). These results suggest that induction of suppressor cells by CBD may be an important mechanism in CBD’s ability to suppress neuroinflammation. However, despite the lack of significant enhancement of either of the suppressor cell populations in the spleen in the current study, CBD was still an effective treatment in the EAE model, which suggests that CBD may have differential effects on the immune response depending on the route of administration.
In addition to examining the suppressor cell populations, ex vivo restimulation of cellular isolates from the spleen and lymph nodes of each mouse with MOG35–55 peptide allowed us to examine the effects of oral CBD on four separate MOG-specific inflammatory T cell populations at three separate time points: CD4+IFN-γ+ (Th1 cells), CD4+IL-17A+ (Th17 cells), CD8+IFN-γ+ (Tc1 cells), and CD8+IL-17A+ (Tc17 cells). Analysis of these T cell populations by flow cytometry after restimulation revealed a significant decrease in the percentage of MOG-specific Tc1 cells by CBD in EAE on day 10. This decrease in the percentage of Tc1 cells within the spleen corresponded to a reduction in clinical disease and neuroinflammation at the day 18 time point. In previous studies IFN-γ production was shown to be reduced in vitro and in vivo with CBD treatment, and very recently this relationship has been shown in the CNS of EAE mice treated with i.p. CBD, although no work was done to identify the source of the IFN-γ within the CNS (Malfait et al. 2000; Kaplan et al. 2008; Giacoppo et al. 2017). Here we have shown that CBD suppresses the peripheral Tc1 population and is capable of reducing clinical disease in the EAE model even when administered before the clinical signs or neuroinflammation are present. This correlation suggests that CBD could be effective in the pathogenesis of MS, since CD8+ T cells have been shown to be one of the predominant cell types in the CNS lesions of MS patients (Babbe et al. 2000). Furthermore, CD8+ T cells have been shown to be capable of initiating EAE, which might explain why inhibition of the Tc1 cells has such a profound effect on EAE pathogenesis (Huseby et al. 2001). Interestingly, when ELISAs were performed on the supernatants from the cultures to determine total cytokine production there was an unexpected spike in IFN-γ production by CBD in EAE mice on day 18. This increase in IFN-γ production could represent a delay in the immune response in this group of mice due to the CBD treatment, and suggests that given enough time these mice would eventually begin to develop more severe disease; however, further work must be done to determine the population of cells that are responsible for this increase in IFN-γ production. In the study by Elliott, et. al. a method similar to the one here was used to restimulated splenocytes from EAE mice, and they also found that supernatants from splenocyte cultures of CBD treated mice had lower levels of IFN-γ and IL-17A, and higher levels of IL-10 as compared to EAE mice that did not receive CBD. Together our results and theirs suggest that CBD administration may affect the long term suppression of the IFN-γ response (Elliott et al. 2018). Unlike the effects seen on the Tc1 population, no correlation was seen in the present study between CBD treatment and IL-17A producing T cells from either secondary lymphoid tissue, despite previous reports of CBD’s effects on IL-17 production (Kozela et al. 2013; Giacoppo et al. 2017; Elliott et al. 2018). In particular, a study by Kozela et al. showed that in vitro treatment of T cells with CBD reduced IL-17 production, but did not affect IFN-γ production (Kozela et al. 2013). The contrast in cytokine production seen between their study and ours might hint at a temporal component to CBD’s effects, since their study examined the in vitro effects of CBD only 24 h after the MOG35–55 specific T cells were stimulated, and our study examined the in vivo effects of CBD after 48 h of restimulation. Another explanation for the lack of effect seen with IFN-γ in the Kozela et al. paper could be that the concentration of CBD used was not high enough to affect IFN-γ levels. In a study by Kaplan et al. a range of concentrations were examined, and the results shown by this study suggest that higher concentrations provide more IFN-γ suppression (Kaplan et al. 2008). Based on these inconsistencies, it is apparent that more work needs to be done to explore the temporal and dose dependent effects of CBD on cytokine production, with specific attention on IL-17 and IFN-γ, so that a more conclusive connection can be made between CBD and its effects on these two cytokines.
Despite the differences in CBD’s effect on cytokine production between our studies and others, the reduction by CBD on neuroinflammation and T cells present in the white matter tracts of the brain and spinal cord of EAE mice was consistent with other studies. Specifically, our results mirror those seen by Kozela et al. who noted reduced levels of T cell infiltrates in the spinal cord with i.p. CBD treatment, and observations seen by Mecha et al. who noted reduced leukocyte infiltrates within the brain of EAE mice with i.p. CBD treatment (Kozela et al. 2011; Mecha et al. 2013). However, in this study the reduction in neuroinflammation, as measured by histological analysis and flow cytometry at the day 18 timepoint, did not reach the level of statistical significance expected, suggesting that reduced leukocyte infiltration is only one of the neuroprotective mechanisms by which early CBD treatment reduces clinical disease in EAE. It is also important to note that the functional capabilities of the cells within the CNS were not examined, so the possibility exists that the cells present within the CNS of CBD treated mice have more of an immunosuppressive phenotype which would contribute to the reduced clinical disease seen in EAE/CBD mice. In consideration of possible functional changes within the T cell compartment, the number of colocalization events was determined between the CD4+ and CD8+ T cells within each lesion to estimate possible changes in communication between the two cell types. It was determined that low numbers of colocalization existed within each lesion with no increases or decreases noted between treatment groups (results not shown). When examining the longitudinal sections of brain tissue from day 18 mice the highest levels of infiltrates were noted in the cerebellum of EAE/CO mice, where the T cells can be seen diffusely and specifically spread throughout the white matter tracts with notable amounts of perivascular cuffing, while the grey matter of the cerebellum remains relatively untouched. Inflammation is also notably seen in the white matter of the cerebellum of EAE/CBD mice, but to a lesser extent than in the EAE/CO mice. The choroid plexus of each mouse was also examined for buildup of inflammatory cells with CBD treatment that might indicate a reduction in the ability of inflammatory cells to migrate into the CNS; however, there was no evidence of leukocyte buildup within the choroid plexus of any group at any time point (results not shown). In the spinal cord CBD showed a similar effect where meningeal, subpial, and perivascular infiltrates are increased in the EAE/CO mice, and reduced by administration of CBD. Notably, the spinal cord sections seen in this study show an almost complete lack of infiltration in the white matter of the nerve roots even in the EAE/CO group. This observation was unexpected, since one would presume all myelinated tracts to be targeted equally when the mouse was inoculated with MOG35–55 peptide. One possible explanation may be that leukocytes infiltrate primarily in the perivascular spaces through the glial limitans and not through the CSF, which would limit their ability to attack the myelin of the nerve roots that run through the subarachnoid space. This is supported by the two step pathogenesis put forward by Sallutso et al. in which the initial step of pathogenesis involves the migration of CCR6+ T cells across the choroid plexus which facilitate the flow of other leukocytes through the blood brain barrier at various points in the CNS (Sallusto et al. 2012). Another possible explanation for this observation is that the myelin sheaths in the nerve roots are fundamentally different from the myelin sheaths of the spinal cord and brain, and do not contain high levels of MOG. This latter explanation is supported by observations made by Pagany et al. who found that although MOG can be expressed by Schwann cells intracellularly in vitro and MOG mRNA can be identified by PCR analysis of the sciatic nerve, expression of MOG by Schwann cells could not be detected in vivo using IHC (Pagany et al. 2003). Either way, this observation raises some important questions about the pathogenesis of EAE, and provides insight into how peripheral nerves might be immunologically distinct from the CNS even before they leave the vertebral column.
Conclusion
The exploration of CBD as an immunosuppressive therapy has shown promise in models of rheumatoid arthritis (RA), inflammatory bowel disease (IBD), and MS. However, when exploring the effects CBD on the EAE model the peripheral immune responses are often overlooked even though they likely play a large role in the pathogenesis of EAE and MS, and observations in the RA and IBD models would suggest that CBD can have significant effects on the peripheral immune system (Kozela et al. 2011; Mecha et al. 2013; Burstein 2015). In this study we examined both peripheral and CNS based immune responses in an attempt to track the immune response from initiation of disease to the point of peak neuroinflammation and clinical pathology. By using this unique approach we were able to determine that early treatment with oral CBD significantly decreased the Tc1 responses in the spleen of CBD-treated EAE mice on day 10, which correlated with modest reduction in neuroinflammation of the CBD-treated EAE mice on day 18. These data are significant since suppression of immune function endpoints were observed following oral CBD and the reduction of clinical and neurological disease seen at later time points after early treatment with CBD suggest that residual effects of CBD treatment are still present for over a week after discontinuing the treatment.
Abbreviations
- THC:
-
Δ9-tetrahydrocannabinol
- BSA:
-
Bovine serum albumin
- CBD:
-
Cannabidiol
- CB1/CB2:
-
Cannabinoid receptors
- CO:
-
Corn oil
- DAB:
-
Diaminobenzidine
- EAE:
-
Experimental Autoimmune Encephalomyelitis
- FVD:
-
Fixable Viability Dye
- FC buffer:
-
Flow cytometry buffer
- GFAP:
-
Glial Fibrillary Acidic Protein
- HKMT:
-
Heat-killed Mycobacterium tuberculosis H37Ra
- H&E:
-
Hematoxylin and eosin
- IBD:
-
Inflammatory bowel disease
- i.p.:
-
Intraperitoneal
- MS:
-
Multiple Sclerosis
- MBP:
-
Myelin basic protein
- MOG:
-
Myelin oligodendrocyte protein
- MDSC:
-
Myeloid derived suppressor cell
- NBF:
-
Neutral buffered formalin
- PBST:
-
PBS containing 0.05% Tween 20
- PTX:
-
Pertussis toxin
- PMA/Io:
-
Phorbol 12-myristate-13-acetate and ionomycin
- PBS:
-
Phosphate buffered saline
- PLP:
-
Proteolipid protein
- RA:
-
Rheumatoid arthritis
- TMEV:
-
Theiler’s murine encephalomyelitis virus
- Tx:
-
Triton X-100
- VCAM-1:
-
Vascular cell-adhesion molecule-1
References
Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, Giudici F, Romagnani P, Parronchi P, Tonelli F, Maggi E, Romagnani S (2007) Phenotypic and functional features of human Th17 cells. J Exp Med 204:1849–1861
Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schroder R, Deckert M, Schmidt S, Ravid R, Rajewsky K (2000) Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med 192:393–404
Bogie JF, Stinissen P, Hendriks JJ (2014) Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol 128:191–213
Bongioanni P, Lombardo F, Moscato G, Mosti S, Meucci G (1999) T-cell interferon gamma receptor binding in interferon beta-1b-treated patients with multiple sclerosis. Arch Neurol 56:217–222
Boven LA, Van Meurs M, Van Zwam M, Wierenga-Wolf A, Hintzen RQ, Boot RG, Aerts JM, Amor S, Nieuwenhuis EE, Laman JD (2006) Myelin-laden macrophages are anti-inflammatory, consistent with foam cells in multiple sclerosis. Brain 129:517–526
Brucklacher-Waldert V, Stuerner K, Kolster M, Wolthausen J, Tolosa E (2009) Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 132:3329–3341
Burstein S (2015) Cannabidiol (CBD) and its analogs: a review of their effects on inflammation. Bioorg Med Chem 23:1377–1385
Crook KR, Liu P (2014) Role of myeloid-derived suppressor cells in autoimmune disease. World J Immunol 4:26–33
Dhital S, Stokes JV, Park N, Seo KS, Kaplan BL (2017) Cannabidiol (CBD) induces functional Tregs in response to low-level T cell activation. Cell Immunol 312:25–34
Elliott DM, Singh N, Nagarkatti M, Nagarkatti PS (2018) Cannabidiol attenuates experimental autoimmune encephalomyelitis model of multiple sclerosis through induction of myeloid-derived suppressor cells. Front Immunol 9:1782
Feger U, Luther C, Poeschel S, Melms A, Tolosa E, Wiendl H (2007) Increased frequency of CD4+ CD25+ regulatory T cells in the cerebrospinal fluid but not in the blood of multiple sclerosis patients. Clin Exp Immunol 147:412–418
Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH (2010) T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol 162:1–11
Giacoppo S, Pollastro F, Grassi G, Bramanti P, Mazzon E (2017) Target regulation of PI3K/Akt/mTOR pathway by cannabidiol in treatment of experimental multiple sclerosis. Fitoterapia 116:77–84
Gravano DM, Hoyer KK (2013) Promotion and prevention of autoimmune disease by CD8+ T cells. J Autoimmun 45:68–79
Haas J, Hug A, Viehover A, Fritzsching B, Falk CS, Filser A, Vetter T, Milkova L, Korporal M, Fritz B, Storch-Hagenlocher B, Krammer PH, Suri-Payer E, Wildemann B (2005) Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol 35:3343–3352
Hegde VL, Nagarkatti PS, Nagarkatti M (2011) Role of myeloid-derived suppressor cells in amelioration of experimental autoimmune hepatitis following activation of TRPV1 receptors by cannabidiol. PLoS One 6:e18281
Hegde VL, Singh UP, Nagarkatti PS, Nagarkatti M (2015) Critical role of mast cells and peroxisome proliferator-activated receptor gamma in the induction of myeloid-derived suppressor cells by marijuana Cannabidiol in vivo. J Immunol 194:5211–5222
Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J (2001) A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med 194:669–676
Ioannou M, Alissafi T, Lazaridis I, Deraos G, Matsoukas J, Gravanis A, Mastorodemos V, Plaitakis A, Sharpe A, Boumpas D, Verginis P (2012) Crucial role of granulocytic myeloid-derived suppressor cells in the regulation of central nervous system autoimmune disease. J Immunol 188:1136–1146
Kaplan BL, Rockwell CE, Kaminski NE (2003) Evidence for cannabinoid receptor-dependent and -independent mechanisms of action in leukocytes. J Pharmacol Exp Ther 306:1077–1085
Kaplan BL, Springs AE, Kaminski NE (2008) The profile of immune modulation by cannabidiol (CBD) involves deregulation of nuclear factor of activated T cells (NFAT). Biochem Pharmacol 76:726–737
Karmaus PW, Wagner JG, Harkema JR, Kaminski NE, Kaplan BL (2013) Cannabidiol (CBD) enhances lipopolysaccharide (LPS)-induced pulmonary inflammation in C57BL/6 mice. J Immunotoxicol 10:321–328
Kozela E, Lev N, Kaushansky N, Eilam R, Rimmerman N, Levy R, Ben-Nun A, Juknat A, Vogel Z (2011) Cannabidiol inhibits pathogenic T cells, decreases spinal microglial activation and ameliorates multiple sclerosis-like disease in C57BL/6 mice. Br J Pharmacol 163:1507–1519
Kozela E, Juknat A, Kaushansky N, Rimmerman N, Ben-Nun A, Vogel Z (2013) Cannabinoids decrease the th17 inflammatory autoimmune phenotype. J NeuroImmune Pharmacol 8:1265–1276
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201:233–240
Li R, Rezk A, Healy LM, Muirhead G, Prat A, Gommerman JL, Bar-Or A, Team MCBciM (2015) Cytokine-defined B cell responses as therapeutic targets in multiple sclerosis. Front Immunol 6:626
M. Mecha AF, F.J. Carrillo-Salinas, C. Guaza (2017) Chapter 93: Cannabidiol and Multiple Sclerosis. In: Handbook of Cannabis and Related Pathologies (Preedy VR, ed), p 1 online resource. S.l.: Academic Press
Malfait AM, Gallily R, Sumariwalla PF, Malik AS, Andreakos E, Mechoulam R, Feldmann M (2000) The nonpsychoactive cannabis constituent cannabidiol is an oral anti-arthritic therapeutic in murine collagen-induced arthritis. Proc Natl Acad Sci U S A 97:9561–9566
Mallucci G, Peruzzotti-Jametti L, Bernstock JD, Pluchino S (2015) The role of immune cells, glia and neurons in white and gray matter pathology in multiple sclerosis. Prog Neurobiol 127-128:1–22
Mangmool S, Kurose H (2011) G(i/o) protein-dependent and -independent actions of pertussis toxin (PTX). Toxins (Basel) 3:884–899
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564
Mecha M, Feliu A, Inigo PM, Mestre L, Carrillo-Salinas FJ, Guaza C (2013) Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: a role for A2A receptors. Neurobiol Dis 59:141–150
Miller SD, Karpus WJ (1994) The immunopathogenesis and regulation of T-cell-mediated demyelinating diseases. Immunol Today 15:356–361
Moline-Velazquez V, Cuervo H, Vila-Del Sol V, Ortega MC, Clemente D, de Castro F (2011) Myeloid-derived suppressor cells limit the inflammation by promoting T lymphocyte apoptosis in the spinal cord of a murine model of multiple sclerosis. Brain Pathol 21:678–691
Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65
Nichols JM, Kaplan BL (2020) Immune responses regulated by cannabidiol. Cannabis Cannabinoid Res 5:12–31
Pagany M, Jagodic M, Schubart A, Pham-Dinh D, Bachelin C, Baron van Evercooren A, Lachapelle F, Olsson T, Linington C (2003) Myelin oligodendrocyte glycoprotein is expressed in the peripheral nervous system of rodents and primates. Neurosci Lett 350:165–168
Rao P, Segal BM (2012) Experimental autoimmune encephalomyelitis. Methods Mol Biol 900:363–380
Sallusto F, Impellizzieri D, Basso C, Laroni A, Uccelli A, Lanzavecchia A, Engelhardt B (2012) T-cell trafficking in the central nervous system. Immunol Rev 248:216–227
Sinha S, Boyden AW, Itani FR, Crawford MP, Karandikar NJ (2015) CD8(+) T-cells as immune regulators of multiple sclerosis. Front Immunol 6:619
Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L (2008) Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 172:146–155
Vos CM, Geurts JJ, Montagne L, van Haastert ES, Bo L, van der Valk P, Barkhof F, de Vries HE (2005) Blood-brain barrier alterations in both focal and diffuse abnormalities on postmortem MRI in multiple sclerosis. Neurobiol Dis 20:953–960
Acknowledgments
The authors thank Dr. T. Graham Rosser for help with illustrations. Funding was provided by National Institutes of Health training grant T35OD010432, Center of Biomedical Research Excellence (COBRE) grant P20GM103646 and Institutional Development Award (IDeA) grant P20GM103476.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Experiments were approved by Mississippi State University Institutional Animal Care and Use Committee (IACUC).
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplemental Fig. 1
Gating strategy for identifying Tc1, Tc17, Th1, and Th17cells. CD8+IFN-γ+, CD8+IL17A+, CD4+IFN-γ+, CD4+IL-17A+ lymphocytes were identified in the live singlet population. (PNG 1007 kb)
Supplemental Fig. 2
Cell counts of inflammatory T cells from ex vivo restimulated splenocytes. These cell counts correspond to the percentages of inflammatory T cells given in Fig. 3 of the main body of the paper. * p < 0.05 differences between SAL/CO and EAE/CO; ‡ p < 0.05 difference between SAL/CO and Mild EAE/CO; # p < 0.05 difference between EAE/CO and EAE/CBD; @p < 0.05 difference between EAE/CO and Mild EAE/CO € p < 0.05 difference between EAE/CBD and Mild EAE/CBD. (PNG 305 kb)
Supplemental Fig. 3
Cell counts of inflammatory T cells from ex vivo restimulated cells isolated from lymph nodes. These cell counts correspond to the percentages of inflammatory T cells given in Fig. 4 of the main body of the paper. * p < 0.05 differences between SAL/CO and EAE/CO; ‡ p < 0.05 difference between SAL/CO and Mild EAE/CO; § p < 0.05 difference between Mild EAE/CO and Mild EAE/CBD. (PNG 375 kb)
Supplemental Fig. 4
Gating strategy for identifying MDSCs and Tregs. Tregs were identified as CD4+CD25+FoxP3+ Lymphocytes in the singlet population. Granulocytic MDSC are identified as CD11b+Ly6CloLy6G+ cells and monocytic MDSC are identified as CD11b+Ly6C+Ly6G− cells in the singlet population. (PNG 1288 kb)
Supplemental Fig. 5
Cell counts of regulatory cells from secondary lymphoid organs. These cell counts correspond to the percentages of regulator cells given in Fig. 2 of the main body of the paper. * p < 0.05 differences between SAL/CO and EAE/CO; ‡ p < 0.05 difference between SAL/CO and Mild EAE/CO; # p < 0.05 difference between EAE/CO and EAE/CBD; @p < 0.05 difference between EAE/CO and Mild EAE/CO € p < 0.05 difference between EAE/CBD and Mild EAE/CBD. (PNG 409 kb)
Supplemental Fig. 6
Cell counts of T cells isolated from the spinal cord of EAE mice. These cell counts correspond to the percentages of T cells given in Fig. 9D of the main body of the paper. (PNG 236 kb)
Rights and permissions
About this article

Cite this article
Nichols, J.M., Kummari, E., Sherman, J. et al. CBD Suppression of EAE Is Correlated with Early Inhibition of Splenic IFN-γ + CD8+ T Cells and Modest Inhibition of Neuroinflammation. J Neuroimmune Pharmacol 16, 346–362 (2021). https://doi.org/10.1007/s11481-020-09917-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-020-09917-8