
Abstract
Nicotine, the active ingredient in tobacco smoke, suppresses antiviral responses. Interferon regulatory factors (IRFs) regulate transcription of type I interferons (IFNs) and IFN-stimulated genes (ISGs) in this response. IRF7 is a key member of the IRF family. Expression of Irf7 is elevated in the brains of virus-infected animals, including human immunodeficiency virus-1 transgenic (HIV-1Tg) rats. We hypothesized that IRF7 affects nicotine’s modulation of antiviral responses. Using CRISPR/Cas9 system, IRF7-mutant cell lines were created from human embryonic kidney 293FT cells in which 16 nicotinic acetylcholine receptors (nAChRs) were detected. Decreased expression of IRF7 was confirmed at both the mRNA and protein levels, as was IRF7-regulated cell growth in two IRF7-mutant cell lines, designated IRF7-Δ7 and IRF7-Δ11. In IRF7-Δ7 cells, expression of two nAChR genes, CHRNA3 and CHRNA9, changed modestly. After stimulation with polyinosinic–polycytidylic acid (poly I:C) (0.25 μg/ml) for 4 h to mimic viral infection, 293FT wild-type (WT) and IRF7-Δ7 cells were treated with 0, 1, or 100 μM nicotine for 24 h, which increased IFN-β expression in both types of cells but elevation was higher in WT cells (p < 0.001). Expression was significantly suppressed in WT cells (p < 0.001) but not in IRF7-Δ7 cells by 24-h nicotine exposure. Poly I:C stimulation increased mRNA expression of retinoic-acid-inducible protein I (RIG-I), melanoma-differentiation-associated gene 5 (MDA5), IFN-stimulated gene factor 3 (ISG3) complex, and IFN-stimulated genes (IRF7, ISG15, IFIT1, OAS1); nicotine attenuated mRNA expression only in WT cells. Overall, IRF7 is critical to nicotine’s effect on the antiviral immune response.

Involvement of IRF7 in nicotine’s suppression of poly I:C-induced antiviral immune responses. PAMPs, such as a synthetic viral analogue of dsRNA poly I:C attack cells, will be recognized by PRRs, and the host innate immunity against viral infection will be activated. PRRs signaling trigger phosphorylation of IRF7 and IRF3 to induce their translocation to the nucleus and result in the production of type I IFNs. Then IFNs bind to IFNAR to activate the transcription factor ISGF3, a complex consisting of STAT1, STAT2, and IRF9. Further, it induces the expression of ISGs, including IFIT1, OAS1, IRF7, ISG15, etc. Nicotine suppresses the immune responses stimulated by poly I:C. In the IRF7-mutant cells, nicotine’s suppressive effects on poly I:C-stimulated immune responses were restrained.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The innate immune system is the first line of host defense against viral pathogens and is regulated by pattern-recognition receptors (PRRs) that identify viral pathogen-associated molecular patterns (PAMPs) (Akira et al. 2006). There are two classes of PRRs recognizing viral components that induce antiviral innate immunity. One is endosomal Toll-like receptors (TLRs), in which TLRs 3, 7, 8, and 9 are involved in antiviral innate immune responses, and the other is cytoplasmic helicase proteins, such as retinoic-acid-inducible protein I (RIG-I) and melanoma-differentiation-associated gene 5 (MDA5) (Medzhitov 2001; Kato et al. 2006; Seth et al. 2006). Both TLR3 and RIG-I/MDA5 are involved in the recognition of viral double-stranded RNA (dsRNA) and synthetic intracellular and cytoplasmic dsRNA, (Medzhitov 2001; Kato et al. 2006; Seth et al. 2006). Polyinosinic-polycytidylic acid (poly I:C) is a synthetic analogue of dsRNA (Alexopoulou et al. 2001; Kato et al. 2006) that mimics viral infection and triggers antiviral immune responses, producing both type I interferons (IFNs) and inflammatory cytokines that prevent virus infection (Kawai and Akira 2010).
Cigarette smoking is a world-wide health issue. According to a 2013 report from the World Health Organization (WHO), smoking causes about 6 million deaths every year, with more than 5 million related directly to cigarette smoking (WHO 2013). The mortality rate from any cause is two to three times higher among current smokers than in people who do not smoke (Carter et al. 2015). Smoking causes various adverse effects on different systems (Sopori 2002; Stampfli and Anderson 2009; National Center for Chronic Disease Prevention et al. 2014), including increases the risk of infection by bacteria and viruses (Arcavi and Benowitz 2004). The U.S. Surgeon General’s Report demonstrates that smoking causes inflammation and impairs immune function and may accelerate progression of many infectious diseases (National Center for Chronic Disease Prevention et al. 2014).
Nicotine is one of the key active and addictive ingredients in cigarette smoke (Li 2018). However, numerous studies have reported nicotine’s beneficial effects in several diseases including the neurologic effects of human immunodeficiency virus infection (neuroHIV), depression, skin disease, and Parkinson’s disease (Tizabi et al. 1999; McClernon et al. 2006; Picciotto and Zoli 2008; Ingram 2009; Quik et al. 2012; Hedstrom et al. 2013; Li-Sha et al. 2015; Han et al. 2018). On the other hand, nicotine has deleterious effects on health by activating multiple biological signaling pathways and increasing the risk of many diseases (National Center for Chronic Disease Prevention et al. 2014). Nicotine exerts its action mainly on nicotinic acetylcholine receptors (nAChRs), which include 16 subunits of human α1, α2, α3, α4, α5, α6, α7, α9, α10, β1, β2, β3. β4, γ, δ and ε (Xu et al. 1999; Elgoyhen et al. 2001; Graham et al. 2002; Lee et al. 2010; Dash et al. 2014; Jiang et al. 2014; Chatzidaki et al. 2015; Qian et al. 2016; Ren et al. 2018). Expression of nAChRs α5 and α7 was reported in human embryonic kidney 293 (HEK293) cells (Thomas and Smart 2005). Unless there is action on a target gene, constitutive expression usually remains after genetic engineering (Nowakowski et al. 2013) such as transmission from HEK293 (293) to HEK293FT (293FT). In our study, expression of nAChRs was confirmed in 293FT cells using RT-qPCR. Taking advantage of the fast growth of 293FT cells, we chose HEK293FT cells to study the expression of nAChRs and the mechanisms underlying nicotine suppression of antiviral immunity.
The interferon regulatory factor (IRF) members of the transcription factor family are primarily involved in the induction of genes encoding type I IFNs and the regulation of innate and adaptive immune responses (Honda and Taniguchi 2006; Ikushima et al. 2013). In addition, some IRFs play important roles in the regulation of cell growth, apoptosis, survival, differentiation, and oncogenesis (Taniguchi et al. 2001; Tamura et al. 2008). Some PRRs, especially endolysosomal TLRs such as endocytic TLR3, TLR7, TLR8, and TLR9, activate IRFs to produce type I IFNs (Barton and Kagan 2009; Ning et al. 2011). RIG-I has also been recognized as a cytosolic receptor for intracellular dsRNA to induce IFNs (Yoneyama et al. 2004), which bind to the type I IFN receptor (IFNAR) and activate the Jak-STAT pathway to induce expression of interferon-stimulated genes (ISGs) (Honda et al. 2006). The ISGs create a robust antiviral state and prevent the spread of infection.
IRF7 is a key member of the interferon regulatory factor family. We previously reported that the expression of Irf7 was significantly increased in several brain areas, including the striatum, prefrontal cortex, and hippocampus, in human immunodeficiency virus-1 (HIV-1) transgenic (HIV-1Tg) rats compared with control F344 rats (Li et al. 2013; Yang et al. 2016). These regions are important in regulating learning, memory, and motivation, suggesting that Irf7 plays a key role in the neurologic abnormalities in HIV-infected patients (Eichenbaum et al. 1996; Wise 2000; Packard and Knowlton 2002). Further, IRF7 is involved in various other infections, such as by influenza A virus (IAV) (Ciancanelli et al. 2015; Hatesuer et al. 2017), human rhinoviruses (HRV) (Bosco et al. 2016), and Epstein-Barr virus (EBV) (Xu et al. 2015), although the particular function of IRF7 differs depending on the cell type and the virus (Daffis et al. 2009). IRF7 is the master regulator of type I IFN-dependent innate antiviral immunity responses (Honda et al. 2005) and has been reported to be involved in cell growth and proliferation (Honda et al. 2005; Li et al. 2017; Yang et al. 2017; Zhao et al. 2017). Cigarette smoking has been shown to attenuate innate antiviral responses (Bauer et al. 2008; Mian et al. 2009; Eddleston et al. 2011; Wu et al. 2014), however, whether IRF7 plays any role in the inhibition of antiviral responses by smoking remains unknown.
The CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9) technique is a genome-editing method. Three types of CRISPR/Cas9 systems have been described. Type II CRISPR from Streptoccus pyogenes is the simplest and most used. It consists of Cas9 nuclease, as well as two noncoding CRISPR RNAs (crRNAs): trans-activating crRNA (tracrRNA) and a precursor crRNA (pre-crRNA) array. Both tracrRNA and crRNA can be replaced by an engineered sgRNA, and the Cas9 endonuclease directed by the sgRNA can induce double-strand DNA breaks (DSB) at specific loci. In the Streptoccus pyogenes CRISPR/Cas9 system, the 5′-NGG protospacer adjacent motif (PAM) sequence must precede the target DNA (Cong et al. 2013; Doudna and Charpentier 2014). This system has been widely used in biomedical research, as it facilitates a variety of targeted genome engineering applications (Hsu et al. 2014). This technique enables much more efficient and faster generation of stable edited cell lines.
Using the CRISPR/Cas9 system, we edited the human embryonic kidney (HEK) 293FT cell line to generate stable IRF7-mutant lines with partial knockdown. Knockdown of IRF7 was confirmed at the DNA, RNA, protein, and functional levels. Poly I:C was used to mimic various virus infections to examine involvement of IRF7 in nicotine’s modulation of innate antiviral immune responses. Treatment with nicotine suppressed poly I:C-mediated expression elevation of PRRs, IFN-stimulated gene factor 3 (ISG3) complex (STAT1, STAT2, IRF9), and ISGs (IRF7, ISG15, IFIT1, OAS1) in wild-type (WT) 293FT cells. Nicotine suppression of these genes that had been elevated via treatment with poly I:C was not evident in the IRF7 mutant cells. CRISPR/Cas9 editing of IRF7 confirmed the involvement of IRF7 in nicotine’s suppression of antiviral immune responses.
Materials and Methods
Single Guide RNA (sgRNA) Design and sgRNA-Cas9 Co-expression Vector Construction
The sgRNA of IRF7 was designed using the CRISPR Guide Design Resources (http://crispr.mit.edu). The 20-nucleotide sequences were selected to precede a 5′-NGG PAM sequence located on exon 3 in the coding sequence (CDS) region (Fig. 1a). Oligo nucleotide-containing ligation adapters were synthesized (Eurofins Genomics LLC, Louisville, KY). The sgRNA sequences of IRF7 are: forward: 5′-CACCGACTCTCCGAACAGCACGCGT-3′ and reverse: 5′-AAACACGCGTGCTGTTCGGAGAGTC-3′.

Schematic diagram of sgRNA and Cas9 co-expressed vector construction. a Design of sgRNA of IRF7. The underlined letters in blue indicate sgRNA. The letters in red are the PAM sequence. b The sequence of vector and the insertion site cut with the BbsI restriction enzyme. The triangle marks the sgRNA insertion site. c Sequence of sgRNA and Cas9 co-expressed vector validated by Sanger sequencing. The words in blue are the sequence of sgRNA inserted into the vector. d The sequence of selected positive single-colony cell lines. The dots around the PAM sequence show the deletion sites of the nucleotides in IRF7-Δ7 (7 nucleotides deleted) and IRF7-Δ11 (11 nucleotides deleted). CDS: coding sequence
Using the procedure reported by Ran et al. (2013), the sense and antisense sgRNA oligos were annealed and cloned into the BbsI (Thermo Fisher Scientific, Hanover Park, IL) site downstream from the human U6 promoter in the pSpCas9(BB)-2A-GFP (PX458) and pSpCas9(BB)-2A-Puro (PX459) plasmids, which were gifts from Dr. Feng Zhang (Addgene plasmids #48138 and #48139, Cambridge, MA) (Ran et al. 2013). The cloned plasmid DNA was then transformed into Invitrogen™ One Shot Stbl3 chemically competent E. coli (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. The positive colonies were cultured at 37 °C overnight for amplification. The plasmid DNA was isolated with a QIAprep Spin Miniprep Kit (Qiagen, Germantown, MD) according to the manufacturer’s instructions. The sequence of the construction was validated by DNA Sanger sequencing (Genewiz, South Plainfield, NJ), and the sequence was analyzed with Chromas (v. 2.6.2). The primers used for Sanger sequencing were: forward: 5′-GAGGGCCTATTTCCCATGATTCC-3′ and reverse: 5′-TGTCTGCAGAATTGGCGCAC-3′.
Cell Culture, Transfection, and Stable Cell-Line Generation
The HEK293FT cell line was purchased from Invitrogen and maintained in standard DMEM culture medium supplemented with 10% GIBCO fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA) and 1% penicillin–streptomycin (Thermo Fisher Scientific, Waltham, MA) at 37 °C in a humidified atmosphere with 5% CO2 and 95% air.
A total of 2 × 105 cells were plated in each well of a 24-well plate. When the cells had grown to 70%–90% confluence, transfection was performed using Invitrogen™ Lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA) with 1 μg of PX458/459-IRF7 co-expressed plasmid DNA according to manufacturer’s instructions. Twenty-four hours after transfection, stably transfected cells were selected using puromycin (InvivoGen, San Diego, CA) with a final concentration of 3 μg/ml for 2–3 days. The cells were harvested, and a serial dilution in a 96-well plate was applied to obtain single-cell colonies. The genomic DNA was isolated using a QIAamp DNA Mini Kit (Qiagen, Germantown, MD) according to the manufacturer’s instructions. The polymerase chain reaction (PCR) was performed with forward (5′-CTGAAGAGGGGGACAGAACAC-3′) and reverse (5′-AGCCCTTACCTCCCCTGTTA-3′) primers. The PCR products were sequenced by the Sanger procedure (Genewiz, South Plainfield, NJ) to confirm that there were insertions and deletions (Indels).
Cell Stimulation and Nicotine Treatment
Approximately 1 × 106 wild-type (WT) and IRF7-mutant HEK293FT cells were seeded in each well of a 6-well plate. Cells were transfected with poly I:C (InvivoGen, San Diego, CA) at a concentration of 0.25 μg/ml using Lipofectamine 3000 following the manufacturer’s protocol. After transfection for 4 h at 37 °C, fresh complete medium was added to each well with free-base nicotine pH 7.0 at a final concentration of 1 μM or 100 μM. After treatment with nicotine for 24 h, cells were harvested for RNA isolation.
RNA Isolation, Reverse Transcription, and Real-Time Quantitative PCR (RT-qPCR)
After the various treatments, total RNA was isolated from the WT and IRF7-mutant HEK293FT cells using the RNeasy Mini Kit (Qiagen, Germantown, MD). RNA (1 μg) was converted to cDNA using an RT2 First-Strand Kit (Qiagen, Germantown, MD) following the manufacturer’s manual. The RT-qPCR was conducted in a volume of 10 μl containing 5 μl 2× Power SYBR™ Green PCR Master Mix (Applied Biosystems, Hanover Park, IL) and combined forward and reverse primers (1.5 μl; final concentration 500 nM) in a 384-well plate using the 7900 HT Fast Real-Time PCR System (Applied Biosystems, Foster, CA) with the following thermal cycling conditions: 1 cycle at 50 °C for 2 min, initial denaturation at 95 °C for 10 min, and 40 cycles of denaturation at 95 °C for 15 s and annealing/extension at 60 °C for 1 min. Primers were synthesized by Eurofins (Eurofins Genomics LLC, Louisville, KY). The primer sequences are listed in Supplementary Table 1. The relative mRNA expression of the genes of interest was normalized to the expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and calculated using a 2-∆∆Ct method.
Western Blotting
Total protein was extracted from the WT and IRF7-mutant HEK293FT cells using cell lysis buffer (Cell Signaling Technology, Danvers, MA) with protease inhibitor cocktail tablets (Roche, Basel, Switzerland). The protein concentration was measured using a Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford, IL). A 15-μl aliquot consisting of protein sample, Invitrogen™ 4× LDS sample buffer, and 10× reducing agent) (Thermo Fisher Scientific, Waltham, MA) was incubated at 70 °C for 10 min and then loaded on a gradient (4%–12%) of Bis-Tris gel (Thermo Fisher Scientific, Waltham, MA). Electrophoresis was carried out in 1× MES SDS running buffer (Thermo Fisher Scientific, Waltham, MA) at 200 V for 35 min at room temperature. After electrophoresis, the gel was transferred to a nitrocellulose membrane supplied in iBlot2 nitrocellulose mini transfer packs (Thermo Fisher Scientific, Waltham, MA) and blotted using iBlot2 dry blotting system (Thermo Fisher Scientific, Waltham, MA). The membrane was blocked with 5% non-fat dry milk for 1 h and incubated with anti-IRF7 primary antibody (Abcam, Cambridge, MA) overnight at 4 °C. The anti-rabbit horseradish peroxidase-linked secondary antibody (Cell Signaling Technology, Danvers, MA) was incubated for 1 h at room temperature. Signals were detected using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Rockford, IL).
Flow Cytometry
A total of 1 × 106 cells was transferred to each well of a 96-well plate and centrifuged, and the medium was discarded. The cells were blocked in phosphate-buffered saline (PBS) with 1% bovine serum albumin (BSA) for 60 min at room temperature. After centrifugation at 400×g for 5 min, the supernatant liquid was removed by gently flicking the plates over. Then the cells were fixed and permeabilized using a fixation/permeabilization solution kit (BD Biosciences, San Jose, CA) according to the manufacturer’s protocol. The cells were re-suspended in 100 μl of Cytofix/Cytoperm™ solution and incubated for 20 min at 4 °C. They then were washed twice in 1× Perm/Wash™ solution (200 μl/wash) and pelleted, and the supernatant liquid was removed. The fixed/permeabilized cells were thoroughly resuspended in 100 μl of Perm/Wash™ solution containing primary anti-IRF7 antibody (1:200; Abcam, Cambridge, MA) or anti-IgG isotype (1:150; Abcam, Cambridge, MA). After incubation for 45 min at 4 °C in the dark, cells were pelleted by centrifugation and washed twice with 1× Perm/Wash™ solution. Then they were resuspended in a fluorochrome-conjugated secondary anti-IgG Alexa Fluor 488 antibody (1:3000; Abcam, Cambridge, MA) and incubated at 4 °C for 60 min in the dark. The cells then were pelleted and washed twice with 1× Perm/Wash™ solution. All samples were resuspended in PBS with 1% BSA and subjected to flow cytometry (Miltenyi Biotec, San Diego, CA). Data were analyzed with FlowJo software (Flowjo LLC, Ashland, OR).
Cell Proliferation Assay
Cell proliferation was measured using a Cell Counting Kit-8 (CCK-8, Dojindo, Rockville, MD). According to the manufacturer’s instructions, a total of 2 × 104 cells (100 μl/well) were seeded in a 96-well plate and incubated in a 5% CO2 incubator at 37 °C for 24, 48, or 72 h. Then 10 μl of the CCK-8 solution was added to each well. Four hours later, the absorbance was measured at 450 nm using a SpectraMax® 190 Microplate Reader (Molecular Devices, San Jose, CA).
Cell Cycle Analysis
Invitrogen™ FxCycle PI/RNase Staining Solution (Thermo Fisher Scientific, Waltham, MA) was used to analyze the cell cycle according to the manufacturer’s instructions. Briefly, cells were harvested, and a single-cell suspension was prepared in ice-cold PBS buffer. Cells were washed and spun at 1000×g for 5 min. About 2 × 106 cells were transferred in a 5-ml polypropylene tube and fixed in cold 70% ethanol overnight at 4 °C. On the following day, cells were centrifuged at 1000×g for 5 min, resuspended in cold PBS, and washed twice. Finally, 0.5 ml of PI/RNase staining solution was mixed well with the cell pellet. After incubation for 30 min at room temperature in the dark, the distribution in the cell cycle was analyzed using MACS Quant flow cytometer (Miltenyi Biotec, San Diego, CA) for detection of the propidium iodide signal in the PI/RNase staining solution. Data were analyzed with FlowJo software (Flowjo LLC, Ashland, OR).
Enzyme-Linked Immunosorbent Assay
Secreted IFN-α and IFN-β from the WT and IRF7-mutant HEK293FT cells were measured using an IFN-α and IFN-β ELISA kit after both poly I:C stimulation and nicotine treatment (LumiKine Xpress hIFN-α and hIFN-β, InvivoGen) according to the manufacturer’s protocol. One hundred microliters of supernatant liquid was used to measure the concentration of IFN-α and IFN-β. The reading was obtained immediately using a luminometer (GloMax® Multi Detection System; Promega, Madison, WI) after QUANTI-Luc™ solution was added to each well.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA). Data are presented as mean ± standard error of the mean (SEM) and analyzed by one-way or two-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test or Bonferroni post-test correction, respectively. Expression of nAChRs was analyzed using Student’s t test. A value of p < 0.05 was considered significant.
Results
Construction of CRISPR/Cas9 Plasmids Targeting the IRF7 Gene
To construct plasmids containing IRF7-targeted sgRNA, 20-nucleotide sequences were selected followed by the PAM sequence 5′-NGG in the IRF7 gene (Fig. 1a). As described above, the IRF7-targeted sequence was cloned into the PX458/459 plasmid at the BbsI restriction enzyme digestion site (Fig. 1b and c). The sequences of sgRNA and the Cas9 co-expressed vector were validated by Sanger sequencing.
Generation and Validation of IRF7-Mutant HEK293FT Cells by CRISPR/Cas9
HEK293FT cells were transfected with PX458/459-sgRNA co-expressed plasmids, and positive clones were selected as described above. Two positive cell lines were obtained. Figure 1d shows Sanger sequencing results. One line had a 7-nucleotide deletion, and the other had an 11-nucleotide deletion compared with the WT HEK293FT cells. These two cell lines were designated IRF7-Δ7 and IRF7-Δ11, respectively. The “Δ7” and “Δ11” stand for deletion of 7 nucleotides and 11 nucleotides of the IRF7-Δ7 cell line and the IRF7-Δ11 cell line, respectively.
Figure 2a shows the mRNA expression of IRF7 in both WT and mutant cells. By using RT-qPCR (Fig. 2a) and Western blotting (Fig. 2b), we found that the mRNA and protein expression of IRF7 were significantly decreased in the two mutant cell lines compared with the WT cells. Using flow cytometry, we found that there was a peak shift of IRF7 in IRF7-Δ7 (Fig. 2c, blue) and IRF7-Δ11 (Fig. 2d, pink) mutant cells compared with WT cells.

Validation of IRF7 expression in the twoIRF7-mutant HEK293FT cells created by CRISPR/Cas9. a, b The mRNA and protein expression of IRF7 in WT, IRF7-Δ7, and IRF7-Δ11 HEK293FT cells determined by real-time qPCR and Western blotting, respectively. GAPDH was used as housekeeping gene. c, d Using flow cytometry to determine the expression of IRF7 in WT and two IRF7-mutant HEK293FT cell lines. Solid lines show WT cells; dashed lines indicate IRF7-mutant cells; shaded histogram marks isotype control; open histogram illustrates IRF7 staining with FITC. * p < 0.05 and *** p < 0.001 compared with WT cells
Cell Growth Changes in the IRF7-Mutant HEK293FT Cells
IRF7 has been shown to be involved in cell growth and proliferation (Honda et al. 2005; Li et al. 2017; Yang et al. 2017; Zhao et al. 2017). To confirm the functional knockdown of IRF7 in the two IRF7-mutant HEK293FT cell lines, cell proliferation was measured with the CCK-8 assay at 24, 48, and 72 h. As shown in Fig. 3a, the proliferation of mutant cells was dramatically decreased compared with that of the WT cells. Cell cycle was examined by staining with propidium iodide and analysis using flow cytometry (Fig. 3b and c). The cell number in the G2 phase rose in IRF7-Δ7 cells but not in IRF7-Δ11 cells. We also measured some markers related to the cell cycle and apoptosis (Fig. 3d). Cyclin B1, a marker of the G2 phase (Maity et al. 1995), was significantly increased in IRF7-Δ7 cells (p < 0.001). The marker of the G1 phase, cyclin D1 (Baldin et al. 1993), did not show any significant difference in either mutant line.

Cell proliferation and cell-cycle changes in the twoIRF7-mutant HEK293FT cells created by CRISPR/Cas9. a Cell proliferation was measured with CCK-8 assay at indicated time points. b, c Cell cycle measured by propidium iodide staining and analyzed by flow cytometry. IRF7-Δ7 and IRF7-Δ11 cells are overlaid with WT cells; G1, S, and G2 phases are noted on the chart. The arrow indicates arrest in G2 phase. d Expression of genes related to cell cycle (PCNA, cyclin B1, and cyclin D1) and apoptosis (Bcl-2, Bax, and caspase 3) was measured by RT-qPCR. Data are presented as mean ± SEM (n = 3). * p < 0.05, ** p < 0.01, *** p < 0.001 compared with WT cells
Proliferating cell nuclear antigen (PCNA) functions in cell-cycle regulation (Kurki et al. 1986). However, it did not show significant changes in the mutant cells, although there was a trend to an increase in IRF7-Δ7 cells. Bcl-2-associated X protein (Bax) is an apoptotic activator that was the first-identified pro-apoptotic member of the Bcl-2 protein family (Oltvai et al. 1993). The mRNA expression of Bax was significantly increased in both IRF7-Δ7 (p < 0.01) and IRF7-Δ11 (p < 0.001) cells. The expression of anti-apoptosis protein, Bcl-2, showed a significant increase only in IRF7-Δ11 (p < 0.05) cells. There was no significant change in caspase 3 expression in either cell line. The IRF7-mutant HEK293FT cells showed attenuated growth, especially the IRF7-Δ7 cells, which showed significant changes in proliferation, the marker of the G2 phase, and the pro-apoptotic marker. Therefore, in the later studies, IRF7-Δ7 cells were used to determine whether IRF7 is involved in nicotine’s effects on the immune response.
Expression of nAChR Genes in HEK 293FT WT Cells and IRF7- Δ7 HEK293FT Cells
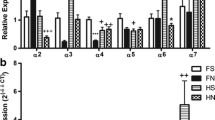
Nicotine exerts its action mainly on nicotinic acetylcholine receptors (nAChRs). Expression of nAChR subunits was assayed in WT and IRF7-Δ7 cells using RT-qPCR. As shown in Fig. 4, all 16 nAChRs, α1, α2, α3, α4, α5, α6, α7, α9, α10, β1, β2, β3, β4, δ, ε, and γ (encoded by the genes CHRNA1, CHRNA2, CHRNA3, CHRNA4, CHRNA5, CHRNA6, CHRNA7, CHRNA9, CHRNA10, CHRNB1, CHRNB2, CHRNB3, CHRNAB4, CHRND, CHRNE, CHRNAG, respectively), were detected in both WT and IRF7-Δ7 cells. nAChR subunit transcripts α3, α5, α7, and β1 were detected at high levels. The nAChR subunit transcripts α1, α2, α4, α5, α6, α7, α10, β1, β2, β3, β4, δ, ε, and γ showed no significant change between the two cells. The nAChR subunit transcript α9 had low expression, but it was upregulated 2.5 fold in IRF7-Δ7 cells compared with WT cells, whereas α3 exhibited high expression in these cells but modest downregulation in IRF7-Δ7 cells (0.2 fold decrease).

Expression of nAChR genes in HEK 293FT WT cells and IRF7-mutant HEK293FT cells. The mRNA expression of CHRNA1, CHRNA2, CHRNA3, CHRNA4, CHRNA5, CHRNA6, CHRNA7, CHRNA9, CHRNA10, CHRNB1, CHRNB2, CHRNB3, CHRNAB4, CHRND, CHRNE, CHRNAG) in WT, and IRF7-Δ7 cells determined by real-time qPCR. Data are presented as mean ± SEM (n = 3). * p < 0.05
Effects of Nicotine on Expression of Genes Related to the Cell Cycle and Apoptosis in IRF7-Δ7 HEK293FT Cells Stimulated with Poly I:C
The IRF7- Δ7 and WT HEK293FT cells were stimulated with poly I:C to determine whether IRF7 is involved in nicotine’s effects on the regulation of cell growth under immune stimulation. As shown in Fig. 5a and b, there was no significant difference in either the WT or mutant cells after nicotine treatment and poly I:C stimulation compared with the control group.

Effects of nicotine on expression of genes related to cell cycle and apoptosis after stimulation by poly I:C (0.25 μg/ml) in the WT andIRF7-mutant HEK293FT cells created by CRISPR/Cas9. a The mRNA expression of cell-cycle markers (PCNA, cyclin D1, and cyclin B1) in WT and IRF7-Δ7 HEK293FT cells by RT-qPCR. b Expression of cell-cycle marker mRNAs (Bcl-2, Bax, and caspase 3) in WT and IRF7-Δ7 HEK293FT cells by RT-qPCR. GAPDH was used as housekeeping gene. WT: wild type, P: poly I:C, N1: 1 μM nicotine, N100: 100 μM nicotine, PN1: poly I:C + 1 μM nicotine, PN100: poly I:C + 100 μM nicotine. Statistical analysis was performed with two-way ANOVA followed by Bonferroni post-test correction. Data are presented as mean ± SEM (n = 3). #p < 0.05, ##p < 0.01, ###p < 0.001 compared with WT cells subjected to same treatment
Effects of Nicotine on Type I IFNs in IRF7-Δ7 HEK293FT Cells Stimulated with Poly I:C
Because IRF7 is the master regulator of type I IFN-dependent immune responses (Honda et al. 2005), we determined the amounts of type I IFNs in both WT and IRF7- Δ7 cells stimulated with poly I:C and nicotine. The poly I:C stimulus enhanced IFN-β mRNA expression compared with unstimulated control cells of both WT and IRF7- Δ7, the response being lower in IRF7- Δ7 cells. Further, nicotine exposure attenuated the expression of IFN-β mRNA stimulated by poly I:C (Fig. 6a). Similarly, as shown in Fig. 6b, the IFN-β concentration in the supernatant liquid was increased after poly I:C stimulation but was significantly decreased in IRF7-Δ7 cells (p < 0.001). Compared with the group transfected with poly I:C, nicotine significantly attenuated IFN-β production in WT cells (p < 0.01), but no significant difference was found in IRF7-Δ7 cells. In addition, we measured both the mRNA and protein of IFN-α in the supernatant liquid and found the expression too low to be detected (data not shown). This result suggests that the IRF7 mutation suppressed the poly I:C-induced type I IFN increase, mainly of IFN-β, and nicotine further inhibited its production.

Effects of nicotine on type 1 IFN gene expression in WT andIRF7-mutant HEK293FT cells by CRISPR/Cas9 after stimulation by poly I:C (0.25 μg/ml). a IFN-β mRNA expression in WT and IRF7-Δ7 HEK293FT cells was measured by RT-qPCR, and 1% agarose gel electrophoresis was run to measure the expression normalized to GAPDH. b Concentration of IFN-β in supernatant liquid of WT and IRF7-Δ7 HEK293FT cells quantified by ELISA. Statistical analysis was performed with two-way ANOVA followed by Bonferroni post-test correction. Values represent mean ± SEM (n = 3). * p < 0.05, ** p < 0.01, *** p < 0.001 compared with poly I:C treatment group in same cell line. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with WT group having same treatment. WT: wild type, P: poly I:C, N1: 1 μM nicotine, N100: 100 μM nicotine, PN1: poly I:C + 1 μM nicotine, PN100: poly I:C + 100 μM nicotine, ns: not significant; ND: not detected
Effects of Nicotine on the Expression of Genes Related to IFN Production in IRF7-Δ7HEK293FT Cells Stimulated with Poly I:C
As mentioned above, poly I:C is an immunostimulant that induces the production of type I IFN. To identify nicotine’s effects on innate antiviral immune responses and determine whether IRF7 mutation affects this process, we stimulated the WT and mutant HEK293FT cells with poly I:C prior to exposure to nicotine. Expression of PRRs and genes related to IFN production via RIG-I/MDA5 pathway were determined. As shown in Fig. 7, poly I:C stimulation enhanced mRNA expression of RIG-I, MDA5, and IRF7 compared with unstimulated cells in both the WT and IRF7-Δ7 lines, whereas the increase was lower in IRF7- Δ7 cells (p < 0.001). Nicotine exposure significantly suppressed RIG-I, MDA5, and IRF7 increase after stimulation by poly I:C in WT cells (p < 0.05) but not in IRF7-Δ7 cells. The interferon-stimulated gene factor 3 (ISGF3) complex, including signal transducer and activator of transcription 1 (STAT1), STAT2, and IRF9, showed similar changes. The mRNA expression of ISGs, including IFN-induced protein with tetratricopeptide repeats 1 (IFIT1), 2′-5′-oligoadenylate synthetase 1 (OAS1), and ISG15, also showed increased expression after poly I:C stimulation, and exposure to nicotine significantly inhibited mRNA expression in WT cells (p < 0.05) but not in IRF7-Δ7 cells. However, we did not find any significant changes of IRF3 in either cells (p > 0.05). Collectively, these results suggest that IRF7 knockdown suppressed poly I:C-induced type I IFN production and genes downstream, and nicotine’s attenuation effects on the expression of genes was not significant in IRF7-Δ7 HEK293FT cells.

Effects of nicotine on expression of genes related to IFN production after stimulation by poly I:C (0.25 μg/ml) in WT andIRF7-mutant HEK293FT cells created by CRISPR/Cas9. a Amounts of RIG-I, MDA5, IRF7, STAT1, STAT2, IRF9, ISG15, IFIT1, and OAS1 mRNA in WT and IRF7-Δ7 HEK293FT cells determined by RT-qPCR. Statistical analysis was performed with two-way ANOVA followed by Bonferroni post-test correction. Data are presented as mean ± SEM (n = 3). * p < 0.05, ** p < 0.01, *** p < 0.001 compared with the poly I:C treatment group in the same cell line. #p < 0.05, ##p < 0.01, ###p < 0.001 comparing WT group with the same treatment. WT: wild type, P: poly I:C, N1: 1 μM nicotine, N100: 100 μM nicotine, PN1: poly I:C + 1 μM nicotine, PN100: poly I:C + 100 μM nicotine. ND: not detected, ns: not significant
Discussion
By using the CRISPR/Cas9 system, we established two IRF7-mutant cell lines of HEK293FT with 7- and 11- nucleotide deletions. IRF7 is the master regulator of the type I IFN-dependent antiviral innate immune response, and treatment with poly I:C was used to mimic viral infection. We found that poly I:C-induced expression elevation of the genes related to the antiviral response was decreased in the IRF7-mutant cells in comparison with the WT cells. Treatment with nicotine attenuated the expression elevation of these genes in the WT cells. Nicotine suppression of the poly I:C-induced effects at both the protein and mRNA levels was not observed in the IRF7-Δ7 cells. Our data suggest that IRF7 plays a vital role in nicotine suppression of innate antiviral immune responses.
The CRISPR/Cas9 system has been used to knock down some target genes (Cui et al. 2017; Yu et al. 2017). Cui et al. constructed an erythropoietin-producing hepatocellular A1 (EPHA1) knockdown ovarian cancer cell line using the CRISPR/Cas9 system (Cui et al. 2017). In another in vivo study, Yu et al. generated neural retina leucine zipper (Nrl) knockdown mice by adeno-associated virus-delivered CRISPR/Cas9 (Yu et al. 2017). Using a similar strategy, we employed the CRISPR/Cas9 editing system with sgRNA targeting IRF7 gene, to generate two stable IRF7 knockdown cell lines with deletion of 7 or 11 nucleotides, designated IRF7-Δ7 and IRF7-Δ11 cell lines, respectively. Knockdown of IRF7 expression at DNA, RNA, and protein levels was confirmed by Sanger sequencing (Fig. 1), RT-qPCR, Western blotting, and flow cytometry (Fig. 2). These IRF7-mutant cells are the models to study molecular and cellular functions and signaling pathways in which IRF7 might be involved.
The HEK293FT cell line is a highly transfectable clonal isolate derived from HEK293 cells (Cong et al. 2013; Ran et al. 2013), It is fast growing and has been widely used as a cell model. Because of its fast growth property, we chose HEK293FT for our studies. HEK293 cells have no tissue-specific gene expression signatures. The cells express the biomarker proteins of many cells including renal progenitor, neuronal, and adrenal. For example, HEK293 was used as a model for cancer because these cells express cancer-associated genes (Stepanenko and Dmitrenko 2015). Some of the genes specifically expressed in neurons are detectable in HEK293 cells (Thomas and Smart 2005). The HEK293 cells express nicotinic receptors such as CHRNA5 and CHRNA7 (Thomas and Smart 2005). Unless there is action on a target gene, constitutive expression usually remains after genetic engineering (Nowakowski et al. 2013) such as transmission from HEK293 (293) to HEK293FT (293FT). We showed differential expression of 16 nAChRs in both WT and IRF7–Δ7 cells using RT-qPCR (Fig. 4). These 16 nAChRs are α1, α2, α3, α4, α5, α6, α7, α9, α10, β1, β2, β3, β4, δ, ε, and γ in human (encoded by the genes CHRNA1, CHRNA2, CHRNA3, CHRNA4, CHRNA5, CHRNA6, CHRNA7, CHRNA9, CHRNA10, CHRNB1, CHRNB2, CHRNB3, CHRNAB4, CHRND, CHRNE, CHRNAG, respectively).
The IRF family members have multiple functions (Taniguchi et al. 2001; Tamura et al. 2008). Many regulate oncogenesis and apoptosis in addition to immune responses (Ning et al. 2011). For example, IRF1 induces cell-cycle arrest and apoptosis; IRF2 and IRF4 have oncogenic potential; IRF5 is involved in several autoimmune diseases; IRF3 mediates virus-induced apoptosis; IRF7 also has oncogenic properties, but in recent studies, it has been reported to have biological functions other than antiviral actions (Li et al. 2017; Yang et al. 2017; Zhao et al. 2017). In addition, IRF7 has been reported to be involved in cell growth and proliferation (Honda et al. 2005; Li et al. 2017; Yang et al. 2017; Zhao et al. 2017). Thus in our study, growth and viability of WT and IRF7-mutant cells (both IRF7-Δ7 and IRF7-Δ11) were assessed to judge the functional knockdown of IRF7 (Fig. 3a). Significant attenuation in cell growth was observed in both cell lines. This attenuation was characterized by: (1) the decrease in cell viability (Fig. 3a); (2) the percentage increase of G2 phase in IRF7- Δ7 cells (Fig. 3b and c): and (3) expression of a G2 phase marker, cyclin B1, increased in IRF7-Δ7 cells (Fig. 3d). In addition, expression of the genes of the proteins involved in apoptosis were also examined. The pro-apoptotic gene Bax increased in both IRF7-Δ7 and IRF7-Δ11 cells, while the anti-apoptotic gene, Bcl-2, showed a significant increase only in IRF7-Δ11 cells (Fig. 3d). We believe that these differential changes in the two IRF7-mutant cell lines reflect mechanisms underlying the significant attenuation in the growth of these cell lines while the changes in IRF7-Δ7 cells were more obvious. Interestingly, neither treatment with nicotine nor infection with poly I:C caused changes in expression of genes related to apoptosis or cell proliferation (Fig. 5). These data suggest that involvement of IRF7 in cell growth did not engage in nicotine’s actions in either the WT or the IRF7-Δ7 cell line.
IRF7 plays a key role in inducing production of type I IFN via virus-mediated and TLR-dependent signaling pathways (Honda et al. 2005; Honda and Taniguchi 2006). IRF3 is highly homologous with IRF7 (Servant et al. 2002), and it also participates in some of the same classical pathways as IRF7 (Sato et al. 2000; Yoneyama et al. 2004). However, the contribution of IRF3 alone is limited without IRF7, probably because IRF3 and IRF7 interact to cause full function (Honda et al. 2005). Honda et al. reported that the production of IFN-β mRNA is reduced in Irf7−/− mice (Honda et al. 2005). Our studies showed that partial knockdown of IRF7 in IRF7-Δ7 cells did not change gene expression of IRF3 (data not shown). Following transfection with poly I:C, expression elevation of IFN-β was lower in the IRF7-Δ7 cells in comparison with the WT cells. Treatment with nicotine suppressed expression of both IRF7 and IFN-β mRNA.
The induced type I IFNs bind to the IFNAR, leading to activation of transcriptional activator ISGF3, including STAT1, STAT2, and IRF9 (Darnell et al. 1994). Then ISGF3 translocates to the nucleus and induces a large set of ISGs, which display combinatorial antiviral properties, inhibiting infection and viral spread (Sato et al. 1998; Schmid et al. 2010). In our studies, we determined expression of ISGF3 and some ISGs, including ISG15, IFIT1, and OAS1. ISG15 is an IFN-induced protein playing a central role in the host antiviral response (Perng and Lenschow 2018). IFIT1, also known as ISG56, is normally silent in most cells but is strongly induced by virus infection or molecular compounds such as dsRNA (Fensterl and Sen 2011). OAS1 is another ISG, which also is expressed to only a small degree in normal cells but is upregulated by dsRNA or IFNs (Sadler and Williams 2008). In this study, poly I:C-mediated stimulation of IFN-beta protein was decreased by IRF7 knockdown. The effect of poly I:C was inhibited by treatment with nicotine in the WT cells but was not observable in IRF7-Δ7 cells (Fig. 6). These data are in line with the expression of genes related to IFN-beta production, shown in Fig. 7, that expression of RIG-I and MDA5, STAT1, STAT2, IRF9, IRF7, ISG15, IFIT1, OAS1 was increased by poly I:C treatment, and exposure to nicotine inhibited that increase in WT cells. In IRF7-Δ7 cells, nicotine-induced suppression of poly I:C-mediated increase in expression of these genes was not observed. Taking these data together with a similar trend in IFN-β protein expression, as shown in Fig. 6, the global effects of IRF7 knockdown have been clearly confirmed at various nodes of the signaling pathway responsible for IFN-beta production. These findings suggest that, in the absence of IRF7, the antiviral immune response was restrained, and nicotine’s attenuated effects were no longer expressed.
As noted previously, nicotine exerts its action mainly on nAChRs. Both the WT and IRF7-Δ7 cell lines differentially expressed 16 nAChR subunits (α1, α2, α3, α4, α5, α6, α7, α8, α9, α10, β1, β2, β3. β4, γ, δ and ε; Fig. 4). The nAChR subunit transcripts α3, α5, α7, and β1 were detected at high levels. The other nAChRs also were detected in both WT and IRF7-Δ7 cells but at much lower levels. Thus, these nAChR subunits may collectively mediate the nicotine actions we have studied using both cell lines. The nAChR subunit transcripts α9 has low expression, but it was upregulated 2.5 fold in IRF7-Δ7 cells compared with WT cells, while α3 showed high expression but was modestly downregulated in IRF7-Δ7 cells (0.2 fold decrease). The similar expression profiles of these nAChR subunits in both the WT and IRF7-Δ7 cells, as shown in Fig. 4, do not support the contention that the modest change in the expression of CHRNA3 and CHRNA9 could contribute to different biological effects between the WT and IRF7-Δ7 cell lines, including nicotine’s suppression of antiviral immune responses. In future studies outside our current research scope, our expression data could be helpful to choose specific agonists and antagonists to study involvement of these 16 nAChR subunits in nicotine’s actions.
Conclusions
Figure 8 schematically summarizes the involvement of IRF7 in nicotine’s suppression of poly I:C-induced antiviral immune responses. On virus infection or PAMP stimulation, PRRs such as TLR3 and RIG-I/MDA5 bind to their respective viral ligands, activating host innate immunity against viral infection. The phosphorylation of IRF7 and IRF3 by virus-activated kinases is induced to activate and translate to the nucleus, resulting in transcriptional activation of type I IFNs. The secreted IFNs bind to their cognate receptors (IFNAR1/2), activating the transcription factor ISGF3, which is a complex consisting of STAT1, STAT2, and IRF9 that induces the expression of ISGs, including IFIT1, OAS1, IRF7, ISG15, etc. Nicotine attenuates poly I:C-stimulated immune responses. The mutation of IRF7 inhibits nicotine’s suppression of poly I:C-stimulated immune responses. Our studies demonstrate that IRF7 is involved in nicotine’s attenuation of antiviral innate immune responses after poly I:C stimulation. Nicotine is the key ingredient of cigarette smoke Cigarette smoking may enhance risk of virus infection because of nicotine’s actions.

Involvement of IRF7 in nicotine’s suppression of poly I:C-induced antiviral immune responses. PAMPs, such as a synthetic viral analogue of dsRNA poly I:C attack cells, will be recognized by PRRs, and the host innate immunity against viral infection will be activated. PRRs signaling trigger phosphorylation of IRF7 and IRF3 to induce their translocation to the nucleus and result in the production of type I IFNs. Then IFNs bind to IFNAR to activate the transcription factor ISGF3, a complex consisting of STAT1, STAT2, and IRF9. Further, it induces the expression of ISGs, including IFIT1, OAS1, IRF7, ISG15, etc. Nicotine suppresses the immune responses stimulated by poly I:C. In the IRF7-mutant cells, nicotine’s suppressive effects on poly I:C-stimulated immune responses were restrained
References
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA (2001) Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature 413:732–738
Arcavi L, Benowitz NL (2004) Cigarette smoking and infection. Arch Intern Med 164:2206–2216
Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G (1993) Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev 7:812–821
Barton GM, Kagan JC (2009) A cell biological view of toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol 9:535–542
Bauer CM, Dewitte-Orr SJ, Hornby KR, Zavitz CC, Lichty BD, Stampfli MR, Mossman KL (2008) Cigarette smoke suppresses type I interferon-mediated antiviral immunity in lung fibroblast and epithelial cells. J Interferon Cytokine Res 28:167–179
Bosco A, Wiehler S, Proud D (2016) Interferon regulatory factor 7 regulates airway epithelial cell responses to human rhinovirus infection. BMC Genomics 17:76
Carter BD, Abnet CC, Feskanich D, Freedman ND, Hartge P, Lewis CE, Ockene JK, Prentice RL, Speizer FE, Thun MJ, Jacobs EJ (2015) Smoking and mortality--beyond established causes. N Engl J Med 372:631–640
Chatzidaki A, Fouillet A, Li J, Dage J, Millar NS, Sher E, Ursu D (2015) Pharmacological characterisation of nicotinic acetylcholine receptors expressed in human iPSC-derived neurons. PLoS One 10:e0125116
Ciancanelli MJ, Huang SXL, Luthra P, Garner H, Itan Y, Volpi S, Lafaille FG, Trouillet C, Schmolke M, Albrecht RA, Israelsson E, Lim HK, Casadio M, Hermesh T, Lorenzo L, Leung LW, Pedergnana V, Boisson B, Okada S, Picard C, Ringuier B, Troussier F, Chaussabel D, Abel L, Pellier I, Notarangelo LD, Garcia-Sastre A, Basler CF, Geissmann F, Zhang SY, Snoeck HW, Casanova JL (2015) Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 348:448–453
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Cui Y, Wu BO, Flamini V, Evans BAJ, Zhou D, Jiang WG (2017) Knockdown of EPHA1 using CRISPR/CAS9 suppresses aggressive properties of ovarian cancer cells. Anticancer Res 37:4415–4424
Daffis S, Suthar MS, Szretter KJ, Gale M Jr, Diamond MS (2009) Induction of IFN-beta and the innate antiviral response in myeloid cells occurs through an IPS-1-dependent signal that does not require IRF-3 and IRF-7. PLoS Pathog 5:e1000607
Darnell JE Jr, Kerr IM, Stark GR (1994) Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421
Dash B, Lukas RJ, Li MD (2014) A signal peptide missense mutation associated with nicotine dependence alters alpha2*-nicotinic acetylcholine receptor function. Neuropharmacology 79:715–725
Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096
Eddleston J, Lee RU, Doerner AM, Herschbach J, Zuraw BL (2011) Cigarette smoke decreases innate responses of epithelial cells to rhinovirus infection. Am J Respir Cell Mol Biol 44:118–126
Eichenbaum H, Schoenbaum G, Young B, Bunsey M (1996) Functional organization of the hippocampal memory system. Proc Natl Acad Sci U S A 93:13500–13507
Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann SF, Boulter J (2001) alpha10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci U S A 98:3501–3506
Fensterl V, Sen GC (2011) The ISG56/IFIT1 gene family. J Interferon Cytokine Res 31:71–78
Graham A, Court JA, Martin-Ruiz CM, Jaros E, Perry R, Volsen SG, Bose S, Evans N, Ince P, Kuryatov A, Lindstrom J, Gotti C, Perry EK (2002) Immunohistochemical localisation of nicotinic acetylcholine receptor subunits in human cerebellum. Neuroscience 113:493–507
Han H, Yang Z, Chang SL, Li MD (2018) Modulatory Effects of Nicotine on neuroHIV/neuroAIDS. J Neuroimmune Pharmacol 13:467–478
Hatesuer B, Hoang HT, Riese P, Trittel S, Gerhauser I, Elbahesh H, Geffers R, Wilk E, Schughart K (2017) Deletion of Irf3 and Irf7 genes in mice results in altered interferon pathway activation and granulocyte-dominated inflammatory responses to influenza a infection. J Innate Immun 9:145–161
Hedstrom AK, Hillert J, Olsson T, Alfredsson L (2013) Nicotine might have a protective effect in the etiology of multiple sclerosis. Mult Scler 19:1009–1013
Honda K, Taniguchi T (2006) IRFs: master regulators of signalling by toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 6:644–658
Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T (2005) IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777
Honda K, Takaoka A, Taniguchi T (2006) Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360
Hsu PD, Lander ES, Zhang F (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278
Ikushima H, Negishi H, Taniguchi T (2013) The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol 78:105–116
Ingram JR (2009) Nicotine: does it have a role in the treatment of skin disease? Postgrad Med J 85:196–201
Jiang Y, Dai A, Zhou Y, Peng G, Hu G, Li B, Sham JS, Ran P (2014) Nicotine elevated intracellular Ca(2)(+) in rat airway smooth muscle cells via activating and up-regulating alpha7-nicotinic acetylcholine receptor. Cell Physiol Biochem 33:389–401
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol 11:373–384
Kurki P, Vanderlaan M, Dolbeare F, Gray J, Tan EM (1986) Expression of proliferating cell nuclear antigen (PCNA)/cyclin during the cell cycle. Exp Cell Res 166:209–219
Lee CH, Huang CS, Chen CS, Tu SH, Wang YJ, Chang YJ, Tam KW, Wei PL, Cheng TC, Chu JS, Chen LC, Wu CH, Ho YS (2010) Overexpression and activation of the alpha9-nicotinic receptor during tumorigenesis in human breast epithelial cells. J Natl Cancer Inst 102:1322–1335
Li MD (2018) Tobacco smoking addiction: epidemiology, Genetics, Mechanisms, and Treatment. Springer, Singapore
Li MD, Cao J, Wang S, Wang J, Sarkar S, Vigorito M, Ma JZ, Chang SL (2013) Transcriptome sequencing of gene expression in the brain of the HIV-1 transgenic rat. PLoS One 8:e59582
Li Z, Huang Q, Chen H, Lin Z, Zhao M, Jiang Z (2017) Interferon regulatory factor 7 promoted glioblastoma progression and stemness by modulating IL-6 expression in microglia. J Cancer 8:207–219
Li-Sha G, Jing-Lin Z, Guang-Yi C, Li L, De-Pu Z, Yue-Chun L (2015) Dose-dependent protective effect of nicotine in a murine model of viral myocarditis induced by coxsackievirus B3. Sci Rep 5:15895
Maity A, McKenna WG, Muschel RJ (1995) Evidence for post-transcriptional regulation of cyclin B1 mRNA in the cell cycle and following irradiation in HeLa cells. EMBO J 14:603–609
McClernon FJ, Hiott FB, Westman EC, Rose JE, Levin ED (2006) Transdermal nicotine attenuates depression symptoms in nonsmokers: a double-blind, placebo-controlled trial. Psychopharmacology 189:125–133
Medzhitov R (2001) Toll-like receptors and innate immunity. Nat Rev Immunol 1:135–145
Mian MF, Stampfli MR, Mossman KL, Ashkar AA (2009) Cigarette smoke attenuation of poly I:C-induced innate antiviral responses in human PBMC is mainly due to inhibition of IFN-beta production. Mol Immunol 46:821–829
National Center for Chronic Disease P, Health Promotion Office on S, Health (2014) Reports of the surgeon general. In: The health consequences of smoking-50 years of progress: a report of the surgeon general. Centers for Disease Control and Prevention (US), Atlanta
Ning S, Pagano JS, Barber GN (2011) IRF7: activation, regulation, modification and function. Genes Immun 12:399–414
Nowakowski A, Andrzejewska A, Janowski M, Walczak P, Lukomska B (2013) Genetic engineering of stem cells for enhanced therapy. Acta Neurobiol Exp 73:1–18
Oltvai ZN, Milliman CL, Korsmeyer SJ (1993) Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74:609–619
Packard MG, Knowlton BJ (2002) Learning and memory functions of the Basal Ganglia. Annu Rev Neurosci 25:563–593
Perng YC, Lenschow DJ (2018) ISG15 in antiviral immunity and beyond. Nat Rev Microbiol 16:423–439
Picciotto MR, Zoli M (2008) Neuroprotection via nAChRs: the role of nAChRs in neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease. Front Biosci 13:492–504
Qian J, Mummalaneni SK, Alkahtani RM, Mahavadi S, Murthy KS, Grider JR, Lyall V (2016) Nicotine-induced effects on nicotinic acetylcholine receptors (nAChRs), Ca2+ and brain-derived neurotrophic factor (BDNF) in STC-1 cells. PLoS One 11:e0166565
Quik M, Perez XA, Bordia T (2012) Nicotine as a potential neuroprotective agent for Parkinson’s disease. Mov Disord 27:947–957
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308
Ren A, Wu H, Liu L, Guo Z, Cao Q, Dai Q (2018) Nicotine promotes atherosclerosis development in apolipoprotein E-deficient mice through alpha1-nAChR. J Cell Physiol. https://doi.org/10.1002/jcp.27728
Sadler AJ, Williams BR (2008) Interferon-inducible antiviral effectors. Nat Rev Immunol 8:559–568
Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N (1998) Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett 441:106–110
Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T (2000) Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539–548
Schmid S, Mordstein M, Kochs G, Garcia-Sastre A, Tenoever BR (2010) Transcription factor redundancy ensures induction of the antiviral state. J Biol Chem 285:42013–42022
Servant MJ, Tenoever B, Lin R (2002) Overlapping and distinct mechanisms regulating IRF-3 and IRF-7 function. J Interferon Cytokine Res 22:49–58
Seth RB, Sun L, Chen ZJ (2006) Antiviral innate immunity pathways. Cell Res 16:141–147
Sopori M (2002) Effects of cigarette smoke on the immune system. Nat Rev Immunol 2:372–377
Stampfli MR, Anderson GP (2009) How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol 9:377–384
Stepanenko AA, Dmitrenko VV (2015) HEK293 in cell biology and cancer research: phenotype, karyotype, tumorigenicity, and stress-induced genome-phenotype evolution. Gene 569:182–190
Tamura T, Yanai H, Savitsky D, Taniguchi T (2008) The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol 26:535–584
Taniguchi T, Ogasawara K, Takaoka A, Tanaka N (2001) IRF family of transcription factors as regulators of host defense. Annu Rev Immunol 19:623–655
Thomas P, Smart TG (2005) HEK293 cell line: a vehicle for the expression of recombinant proteins. J Pharmacol Toxicol Methods 51:187–200
Tizabi Y, Overstreet DH, Rezvani AH, Louis VA, Clark E Jr, Janowsky DS, Kling MA (1999) Antidepressant effects of nicotine in an animal model of depression. Psychopharmacology 142:193–199
World Health Organization (WHO) (2013) WHO report on the global tobacco epidemic, 2013: enforcing bans on tobacco advertising, promotion and sponsorship. World Health Organization, Geneva
Wise RA (2000) Interactions between medial prefrontal cortex and meso-limbic components of brain reward circuitry. Prog Brain Res 126:255–262
Wu W, Zhang W, More S, Booth JL, Duggan ES, Liu L, Zhao YD, Metcalf JP (2014) Cigarette smoke attenuates the RIG-I-initiated innate antiviral response to influenza infection in two murine models. Am J Physiol Lung Cell Mol Physiol 307:L848–L858
Xu W, Gelber S, Orr-Urtreger A, Armstrong D, Lewis RA, Ou CN, Patrick J, Role L, De Biasi M, Beaudet AL (1999) Megacystis, mydriasis, and ion channel defect in mice lacking the alpha3 neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci U S A 96:5746–5751
Xu D, Zhang Y, Zhao L, Cao M, Lingel A, Zhang L (2015) Interferon regulatory factor 7 is involved in the growth of Epstein-Barr virus-transformed human B lymphocytes. Virus Res 195:112–118
Yang Z, Nesil T, Connaghan KP, Li MD, Chang SL (2016) Modulation effect of HIV-1 viral proteins and nicotine on expression of the immune-related genes in brain of the HIV-1 transgenic Rats. J Neuroimmune Pharmacol 11:562–571
Yang Q, Li X, Chen H, Cao Y, Xiao Q, He Y, Wei J, Zhou J (2017) IRF7 regulates the development of granulocytic myeloid-derived suppressor cells through S100A9 transrepression in cancer. Oncogene 36:2969–2980
Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737
Yu W, Mookherjee S, Chaitankar V, Hiriyanna S, Kim JW, Brooks M, Ataeijannati Y, Sun X, Dong L, Li T (2017) Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat Commun 8:14716
Zhao Y, Chen W, Zhu W, Meng H, Chen J, Zhang J (2017) Overexpression of interferon regulatory factor 7 (IRF7) reduces bone metastasis of prostate cancer cells in mice. Oncol Res 25:511–522
Acknowledgments
We thank Judith Gunn Bronson, M.S., F.S.T.C., for editing this manuscript.
Funding
The study was in part supported by the U.S. National Institutes of Health grants DA026356, and DA046258.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
All authors declare no conflict of interest on this paper.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOC 89 kb)
Rights and permissions
About this article

Cite this article
Han, H., Huang, W., Du, W. et al. Involvement of Interferon Regulatory Factor 7 in Nicotine’s Suppression of Antiviral Immune Responses. J Neuroimmune Pharmacol 14, 551–564 (2019). https://doi.org/10.1007/s11481-019-09845-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-019-09845-2