
Abstract
Adult-onset neurodegenerative disorders, like Parkinson’s disease (PD) and dementia with Lewy bodies (DLB), that share the accumulation of aggregated α-synuclein (αSynagg) as their hallmark molecular pathology are collectively known as α-synucleinopathies. Diagnosing α-synucleinopathies requires the post-mortem detection of αSynagg in various brain regions. Recent efforts to measure αSynagg in living patients include quantifying αSynagg in different biofluids as a biomarker for PD. We adopted the real-time quaking-induced conversion (RT-QuIC) assay to detect very low levels of αSynagg. We first optimized RT-QuIC for sensitivity, specificity, and reproducibility by using monomeric recombinant human wild-type αSyn as a substrate and αSynagg as the seed. Next, we exposed mouse microglia to αSyn pre-formed fibrils (αSynPFF) for 24 h. RT-QuIC assay revealed that the αSynPFF is taken up rapidly by mouse microglia, within 30 min, and cleared within 24 h. We then evaluated the αSyn RT-QuIC assay for detecting αSynagg in human PD, DLB, and Alzheimer's disease (AD) post-mortem brain homogenates (BH) along with PD and progressive supranuclear palsy (PSP) cerebrospinal fluid (CSF) samples and then determined protein aggregation rate (PAR) for αSynagg. The PD and DLB BH samples not only showed significantly higher αSynagg PAR compared to age-matched healthy controls and AD, but RT-QuIC was also highly reproducible with 94% sensitivity and 100% specificity. Similarly, PD CSF samples demonstrated significantly higher αSynagg PAR compared to age-matched healthy controls, with 100% sensitivity and specificity. Overall, the RT-QuIC assay accurately detects αSynagg seeding activity, offering a potential tool for antemortem diagnosis of α-synucleinopathies and other protein-misfolding disorders.

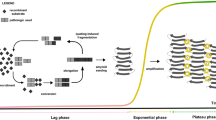
A schematic representation of αSyn RT-QuIC assay.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brain pathologies collectively known as α-synucleinopathies are characterized by the abnormal accumulation of α-synuclein aggregates (αSynagg). Some of these disorders include Parkinson’s disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA). Lewy bodies are a pathological hallmark in PD and DLB patients whose neurons are rich in deposits of αSynagg (Spillantini et al. 1998). In contrast, MSA patients have αSynagg deposits in oligodendrocytes as glial cytoplasmic inclusions (Tu et al. 1998). Although it is rare, the accumulation of Lewy bodies also occurs in progressive supranuclear palsy (PSP) cases, which is normally characterized by the accumulation of neurofibrillary tangles (NFTs) composed of phosphorylated tau protein (Mori et al. 2002). The highly charged, intrinsically disordered acidic protein αSyn is encoded by the SNCA gene and plays a key role in presynaptic vesicle trafficking (Spillantini et al. 1995; Allen Reish and Standaert 2015). This protein has the propensity to oligomerize and further trigger the host’s innate and adaptive immune responses that may contribute to PD pathology (Stone et al. 2009). Conversely, neuroinflammation has been implicated in promoting the prion-like oligomerization of αSyn (Lema Tomé et al. 2013). Epidemiological evidence has linked the use of non-aspirin anti-inflammatory drugs to a 15% reduction in PD incidence, suggesting that drugs that lower neuroinflammation might be an effective treatment against PD. Furthermore, targeting microglia-mediated neuroinflammation remains an attractive target for treating PD progression (Gupta et al. 2018; Williams et al. 2018).
A definitive diagnosis of α-synucleinopathy can be made only by identification of αSynagg in post-mortem brain tissues (Marti et al. 2003). Indeed, the clinical presentation of these movement disorders overlaps, thus making a conclusive diagnosis difficult and dependent primarily on a thorough history and an assessment of motor deficits during a comprehensive physical exam. Despite an urgent need, currently no biomarker is available to diagnose or monitor clinical outcomes in Parkinsonian disorders. Thus, the development of biomarkers for these disorders would be a major advance in the field as it would improve the accuracy of clinical diagnoses, treatment plans, and patient/family counseling; aid in monitoring the efficacy of therapeutic agents on disease processes; and facilitate the accurate recruitment of patients for clinical trials and research. Although biomarkers for the differential diagnosis of α-synucleinopathies currently are not available, several groups are actively pursuing them using a variety of approaches including the assessment of biofluids, motor performance tests, and imaging technologies. Developing a diagnostic test for these disorders is very challenging due to individual variation in the progression and severity of the diseases. Several teams have focused on the biomarker potential of αSyn and its various forms (total, oligomeric, and phosphorylated) in biofluids, employing distinct quantitative assays like ELISA, time-resolved Förster resonance energy transfer (TR-FRET), and immunoassays (Bidinosti et al. 2012; Wang et al. 2012; Compta et al. 2015; Hall et al. 2015). Levels of αSyn in the CSF differ widely between patient cohorts (Ohrfelt et al. 2009; Tokuda et al. 2010) and the labor-intensive preparation requirements of immunoassay techniques have limited the high-throughput, clinical application of these diagnostic tests.
Since αSynagg deposits appear as an early pathological event that occurs prior to the development of clinical signs (Braak et al. 2006), we explored its diagnostic utility as a biomarker. Prior to this work, we had demonstrated the viability of applying the real-time quaking-induced conversion (RT-QuIC) assay for the antemortem detection of chronic wasting disease-causing scrapie prions in rectal biopsies (Manne et al. 2017). Based on the observation that αSynagg shares prion-like properties in the templated conversion of monomeric αSyn, we similarly developed and optimized the RT-QuIC assay for the ultrasensitive detection of αSynagg in biological samples. In α-synucleinopathies, evidence suggests that activated microglia are the most efficient scavengers of extracellular αSynagg (Lee et al. 2008). We thus compared the uptake and clearance of αSyn pre-formed fibrils (αSynPFF) by microglia and detected the seeding activity from activated microglia using the αSyn RT-QuIC assay. After determining microglial seeding activity, we used the αSyn RT-QuIC assay for diagnosing α-synucleinopathies. Meanwhile, three other groups independently developed a similar αSyn RT-QuIC assay for diagnosing α-synucleinopathies with comparably high sensitivities and specificities (Fairfoul et al. 2016; Groveman et al. 2018; Sano et al. 2018). In the present study, we tested a total of 31 CSF samples and 45 brain homogenates (BHs) from two different patient cohorts using our αSyn RT-QuIC assay. Collectively, our results demonstrate that misfolded αSyn seeding activity can be detected in activated microglia, CSF and BHs using the αSyn RT-QuIC assay.
Results
Expression and Purification of Monomeric Human Wild-Type αSyn from E. coli
Since the purity of the monomeric αSyn substrate is a critical determinant of the RT-QuIC assay, we generated a highly suitable recombinant human wild-type (WT) αSyn using a bacterial expression system followed by fast-protein liquid chromatography for purification. As described in the Methods, E. coli expressing αSyn were pelleted and lysed. Overnight-dialyzed lysates were fractionated using size-exclusion (Fig. 1a) and anion-exchange chromatography (Fig. 1b). Coomassie staining of the fractions from size-exclusion (Fig. 1c) and subsequent anion-exchange (Fig. 1d) chromatography revealed the achievement of a pure monomer of recombinant WT αSyn protein. Only recombinant monomers of αSyn protein that passed our quality control criteria were used for the preparation of αSynagg and αSynPFF as well as a substrate in the αSyn RT-QuIC assay.

Chromatogram showing the purification of recombinant human wild-type (WT) αSyn and Coomassie staining of collected fractions. Here we purified the full length of recombinant human WT αSyn. a Absorbance spectra for purification of WT rαSyn using a Sephacryl 200 column. b Absorbance spectra of the HiPrep Q FF 16/10 anion-exchange column. Dashed box denotes the peak fraction of the eluted protein. Coomassie blue staining of total protein fractions collected from c size-exclusion and d anion-exchange columns
Generation and Characterization of αSynagg and αSynPFF from Recombinant Human αSyn Monomers
Like infectious prion proteins, endogenous αSynagg is required for conversion of monomeric αSyn into more pathogenic αSynagg. We generated and characterized αSynagg and αSynPFF from monomeric αSyn according to the protocol set forth by the Michael J. Fox Foundation (Polinski et al. 2018). We generated αSynagg from pure monomeric human αSyn by continuous shaking for seven days over a thermal cycler (described in Methods). The αSynagg was sonicated using a probe sonicator to further generate αSynPFF. Both the αSynagg and αSynPFF were visualized for morphology and size by transmission electron microscopy (TEM) performed before and after sonication. TEM pictures revealed significantly less αSynagg after sonication due to the presence of more αSynPFF (Fig. 2a). Next, we confirmed the presence of αSynagg using a sedimentation assay after subjecting the αSynagg to ultracentrifugation. The supernatant and sediment fractions of αSynagg were separated using polyacrylamide gel electrophoresis. The successful formation of protein aggregates was indicated by the presence of substantially higher amounts of protein in the pellet fraction than in the supernatant (Fig. 2b). To further confirm this result, we tested the seeding activity of our newly generated αSynagg using the αSyn RT-QuIC assay. Various concentrations of αSynagg were tested for their ability to convert monomeric recombinant αSyn into the aggregated form. We detected a dose-dependent amplification in fluorescence kinetics with various doses of αSynagg (Fig. 2c) and an increase in protein aggregation rates (PAR) (Fig. 2d). The enhanced thioflavin T (ThT) fluorescence of the αSynagg we generated exceeded that of monomeric αSyn by more than 80-fold (Fig. 2e), and a well-area scan image showed the deposition of intense amyloids in the αSynagg (red color) compared to the monomeric αSyn (grey color) (Fig. 2f).

Validation of generated αSynagg and αSynPFF prior to use. a Scanning transmission electron microscopy (STEM) images of αSynagg before (scale bar = 100 nm) and after (scale bar = 500 nm) sonication, which converted long αSynagg into short αSynPFF while retaining beta-sheet conformation. b Sedimentation assay of ultracentrifuged αSynagg showing more αSynagg in the pellet fraction than in the supernatant (Sup). c Dose-dependent seeding activity of αSynagg at various concentrations using the αSyn RT-QuIC assay. d PAR of αSynagg tested in the αSyn RT-QuIC assay increased from lower to higher αSynagg concentrations. e Thioflavin T (ThT) assay confirmed the presence of αSynagg with higher ThT fluorescence for αSynagg than for recombinant αSyn. f Well-scan image of αSynagg (red color) and recombinant αSyn (grey color) showing increased intensity of amyloid deposits for αSynagg
Determination of Misfolded αSyn Seeding Activity in Glial Cells
Studies have shown that αSynagg is taken up and cleared by microglia (Lee et al. 2008), inducing a neuroimmune response, but the clearance rate of αSynagg is not well characterized. To determine the αSynagg uptake and clearance rate in microglia, we exposed a mouse microglial cell line to 1 μM αSynPFF, which was generated by sonicating αSynagg as described in Methods. After αSynPFF exposure, cells were harvested in ice-cold PBS at the post-treatment endpoints 0, 0.5, 2, 6, 12 and 24 h. From each lysate, 5 pg/well (Fig. 3a & c) and 50 pg/well (Fig. 3b & d) were loaded into a 96-well plate that used monomeric αSyn as a substrate in the RT-QuIC assay. Steady-state αSyn protein seeding activity (Fig. 3a & b) and aggregation (Fig. 3c & d), reported as PAR, were detected starting at 0.5 h and maintained at steady-state up to 12 h post-αSynPFF treatment, and then both declined gradually over time before returning to basal levels at 24 h, suggesting that αSynPFF is taken up rapidly by microglia, within 30 min, and reduced within 24 h. Collectively, our data demonstrate the degradation efficiency rate at which microglial cells scavenge αSynPFF present in the extracellular milieu.

Detection of misfolded αSyn seeding activity in microglia. a, b ThT kinetics of microglia tested in the αSyn RT-QuIC assay after being incubated with αSynPFF for various time intervals, showing the clearance of αSynPFF by activated microglia within 24 h. c, d Comparison of PARs among various time intervals of αSynPFF-incubated microglia. Each trace and symbol represents the average of 4 replicates. We used 5 pg/well (a, c) and 50 pg/well (b, d) of microglial lysate as seed in the αSyn RT-QuIC assay
Determination of Misfolded αSyn Seeding Activity from CSF Samples
We next performed a blinded analysis using the αSyn RT-QuIC assay on a subset (Table 1) of CSF samples obtained from PD (n = 15) and control (n = 11) subjects. All 15 PD CSF samples tested positive within 20–40 h, as compared with control cases, further highlighting the diagnostic sensitivity of the proposed assay (Fig. 4a). The calculated αSyn PAR of CSF differed significantly between controls and PD samples (Fig. 4b). A small set of non-synucleinopathy (NS) cases consisting of PSP (n = 5) and control (n = 5) subjects were also tested in the αSyn RT-QuIC assay. Two of the five PSP cases were amplified in the αSyn RT-QuIC assay, indicating the presence of Lewy body pathology in PSP cases (Fig. 4c). However, the PAR values of amplified PSP cases did not significantly differ from control cases (Fig. 4d). None of the controls met criteria to be considered positive RT-QuIC responses, further supporting the specificity of the assay. Our results indicate that the CSF αSyn RT-QuIC assay can be applied to a larger PD cohort to further validate the diagnostic accuracy of the assay.

Blinded analysis of CSF samples for misfolded αSyn seeding activity. a ThT fluorescence showing more αSynagg in PD CSF than in controls. b Analysis of PAR showing a higher load of αSynagg detected in PD CSF samples [n = 15 PD (red) and 11 control (green) CSF samples]. c Enhanced ThT fluorescence among two PSP samples indicative of αSynagg in CSF of PSP samples. d Comparison of PAR among PSP and control CSF samples [n = 5 PSP (orange) and 5 control (green) CSF samples]. All samples were tested in quadruplicates and expressed as the mean of 4 technical replicates. ***p < 0.001
Determination of Misfolded αSyn Seeding Activity from BHs
To further validate the αSyn RT-QuIC assay, we analyzed BH samples from two independent brain banks: the UC Davis Alzheimer’s Disease Center provided DLB (n = 5), Alzheimer’s disease (AD; n = 10), and control (n = 8) samples, while the University of Miami’s Brain Endowment Bank provided PD and control samples (n = 11 each) (Table 2). Among these, PD and DLB represent α-synucleinopathies, whereas AD and controls represent NS samples. NS cases did not show ThT fluorescence above the threshold level over the entire 70-h RT-QuIC reaction period. To determine the optimal working concentration of BH, various BH dilutions (1:1000 to 1:1,000,000) from a small set of samples (3 PD and 2 controls) were tested in the αSyn RT-QuIC assay (Fig. 5). Of all the dilutions tested within the specified range, only the 1:10,000 dilution amplified all the biological replicates at a similarly early time (around 10 h) (Fig. 5b vs a, c, d); therefore, we used 1:10,000 dilution for further testing of a larger set of samples. Next, all BHs of 10−4 dilutions were tested for misfolded αSyn seeding activity in the αSyn RT-QuIC assay (Fig. 6). All DLB (5/5) and most of the PD (10/11) samples were amplified using the RT-QuIC assay for a period of 20 or 30 h, respectively, whereas the control BHs failed to demonstrate any amplification (Fig. 6a & c), indicating high sensitivity and specificity of the αSyn RT-QuIC assay. Similarly, the PAR for controls was significantly lower than that for the DLB and PD groups (Fig. 6b & d). Interestingly, two AD samples showed enhanced seeding activity and PAR (Fig. 6a-b), suggesting the presence of some αSyn pathology in these AD BHs, which is consistent with previous findings (Hamilton 2000; Waxman and Giasson 2011).

Choosing the optimal BH dilution required for the αSyn RT-QuIC assay. Enhanced ThT fluorescence of a 10−3, b 10−4, c 10−5, and d 10−6 dilutions in the αSyn RT-QuIC assay of PD (red, n = 3) and control (green, n = 2) BHs. Each trace represents the average of 4 technical replicates

Testing of misfolded αSyn seeding activity in BH samples from α-synucleinopathy and NS cases using the RT-QuIC assay. a Enhanced ThT fluorescence in DLB (red, n = 5), AD (purple, n = 10), and control (green, n = 8) brain samples, showing more αSynagg in DLB samples. b Comparison of PAR between the same control, AD and DLB samples showing higher αSynagg load in DLB brain samples. c Enhanced ThT fluorescence in PD (red, n = 11) brain samples compared to controls (green, n = 11). d Comparison of PAR between the same samples showing higher PAR in PD relative to controls. Each trace and symbol represents the average of 4 technical replicates. **p < 0.01, and ***p < 0.001
Immunoreactivity Using a αSyn Filament Conformation-Specific Antibody
To further confirm the presence of misfolded αSyn in human BHs, we performed a dot blot analysis using an αSyn filament conformation-specific antibody in BHs of PD, DLB, AD and controls obtained from the two brain banks. Immunoblotting revealed the presence of misfolded αSyn in DLB (Fig. 7a, top panel; b) and PD (Fig. 7d, top panel; e) BHs compared to control and AD cases. Interestingly, the same two AD BH samples that amplified in the αSyn RT-QuIC assay also showed positive results using the αSyn filament-specific antibody, indicating the possible presence of αSyn pathology in these two AD cases (Hamilton 2000) (Fig. 7a, top panel). One control exhibited higher staining intensity for the αSyn filament form (Fig. 7a). This may be due to a non-PD-related increase in the staining intensity. This control subject had a metastatic tumor, and the role of its underlying pathology on αSyn aggregation is not known. Furthermore, our study used only one brain region (SN) obtained from the autopsy brains. Considering the history of this control subject, and its lack of apparent seeding activity in RT-QuIC reactions, we removed this control from statistical analysis in Fig. 7b using the robust regression and outlier removal method (ROUT in Prism 7.0, GraphPad). The immunoblots also were stained for total αSyn to reveal the presence of similar αSyn levels among these groups (Fig. 7a & d, bottom panels; c & f). Collectively, these results suggest that significantly higher levels of misfolded αSyn can be observed among α-synucleinopathy patients as compared to NS cases, but total αSyn levels remained unchanged across all groups.

Dot blot analysis of α-synucleinopathy and NS BHs. a Dot blot analysis of AD (purple, n = 10), DLB (red, n = 5) and control (green, n = 8) BH with αSyn filament conformation-specific (top panel) and total αSyn (bottom panel) antibodies. b, c Densitometric analyses of (b) αSyn filament conformation-specific and (c) total αSyn levels showing that DLB BH has significantly increased levels of αSyn filaments compared to AD and control BH, while total αSyn levels did not differ among the 3 groups. One outlier was identified and excluded from the control group in (b) using the robust regression and outlier (ROUT) detection method with a ROUT coefficient of 1%. d Dot blot analysis of PD (red, n = 5) and control (green, n = 5) BH with αSyn filament conformation-specific (top panel) and total αSyn (bottom panel) antibodies. e, f Densitometric analyses of (e) αSyn filament conformation-specific and (f) total αSyn levels showing significantly increased levels of αSyn filaments for PD compared to control BH, while total αSyn levels did not differ between groups. **p < 0.01, and ***p < 0.001
Discussion
The accumulation of αSynagg is the main pathological event in neurodegenerative α-synucleinopathies like PD, DLB, and MSA. Early and accurate diagnosis of these disorders has been hampered by the lack of a well-characterized, sensitive diagnostic assay, thereby resulting in ineffective treatment strategies and delayed counseling of patients and their families. Diagnosis of PD is made after ~50–70% of dopaminergic neurons are lost and is based on patient history and presence of the cardinal motor symptoms, such as bradykinesia, rigidity, and tremor (Hughes et al. 1992). A recent study indicates that the accuracy of clinical diagnoses of PD is only 60% (Beach and Adler 2018) due to PD symptoms overlapping with other Parkinsonian syndromes such as PSP and MSA. A definitive diagnosis has been possible only by post-mortem evaluation using immunohistochemistry (Braak and Braak 2000; Holdorff 2006). Currently, no in vivo biomarkers are approved to differentiate between these clinically similar syndromes and to capture their distinctive pathological patterns and molecular characteristics. Detection of αSynagg as a diagnostic biomarker offers a promising approach. However, recent reports of using the αSyn RT-QuIC assay to detect αSynagg in CSF and BH samples of α-synucleinopathy (Fairfoul et al. 2016; Groveman et al. 2018; Sano et al. 2018) stopped short of assessing reproducibility and sensitivity of the assay. Herein, we assessed the reproducibility, sensitivity, and specificity of the αSyn RT-QuIC assay using CSF and BH samples from two different brain banks. To achieve an accurate diagnosis, having pure recombinant αSyn is a key determinant in the RT-QuIC assay, and hence we generated a recombinant human WT αSyn and optimized the RT-QuIC assay conditions by generating αSynagg from recombinant αSyn. Other causes of neurodegeneration in α-synucleinopathy include neuronal damage caused by activated microglia. Anti-inflammatory therapies that target activated microglia have been shown to slow-down disease progression (Block et al. 2007). We adopted the αSyn RT-QuIC assay to detect αSynPFF internalized in microglia by analyzing the seeding activity within microglia incubated with αSynPFF for varying time periods. Our results demonstrated that αSynPFF stays at steady state for 12 h before degrading within 24 h, suggesting the anti-inflammatory therapy should be administered at early stages of protein aggregation.
Next, we exploited the ability of the αSyn RT-QuIC assay to measure αSynagg levels in human CSF samples. We achieved 100% sensitivity and specificity with a blinded analysis of a subset of CSF samples. Among the PSP cases tested in the αSyn RT-QuIC assay, two cases showed enhanced ThT fluorescence, indicating the presence of Lewy body pathology consistent with previous findings (Mori et al. 2002; Uchikado et al. 2006). Later, we tested a set of BH samples received from two independent brain banks for determining misfolded αSyn seeding activity. Based on results using BHs, we were able to differentiate α-synucleinopathy (DLB and PD patients) from controls with 94% sensitivity and 100% specificity. Along with α-synucleinopathy samples, two AD samples also amplified in the RT-QuIC assay and this might be due to the presence of αSyn pathology (Hamilton 2000). Collectively, these results demonstrate the power of the αSyn RT-QuIC assay in distinguishing α-synucleinopathy cases from controls. Furthermore, dot blot quantification of αSynagg levels in BHs using an αSyn filament conformation-specific antibody also revealed significant differences among α-synucleinopathy cases and controls. Furthermore, dot blot intensities for αSyn filament conformation-specific antibody in BH samples from DLB and PD patients were positively correlated with their respective PARs from the αSyn RT-QuIC assay (supplement Fig. 1).
Overall, our results highlight the high level of sensitivity and reproducibility of the αSyn RT-QuIC assay using a recombinant human αSyn as a substrate as well as the detection of αSynagg sequestered within microglia, CSF, and BHs. Clearly, further validation using larger sample sizes is needed to demonstrate the utility of the αSyn RT-QuIC assay in discriminating among α-synucleinopathies by further establishing the efficacy of αSynagg as a biomarker for those neurodegenerative disorders having the propensity to accumulate aggregate-prone proteins. In terms of clinical relevance to disease progression in Parkinsonism, monitoring disease progression and/or response to therapy continues to be a valuable objective of this technique. Using larger cohorts consisting of samples at various stages of disease progression, the kinetics associated with the αSyn RT-QuIC assay could be validated for indirectly assessing the concentration of pathological αSyn aggregates in samples. We propose that integrating αSyn and tau RT-QuIC assays with multi-model brain imaging modalities would have important clinical value in monitoring disease progression, differential diagnosis and clinical monitoring of disease-modifying pharmacotherapies.
Materials and Methods
Lumbar Puncture and Cerebrospinal Fluid (CSF) Sample Storage
CSF samples were collected as part of the NINDS Parkinson’s Disease Biomarker Program (PDBP) according to guidelines (Rosenthal et al. 2016) for the lumbar puncture. The protocol was reviewed and approved by the Penn State Hershey Internal Review Board and informed written consent was obtained from all subjects. After CSF collection, samples were centrifuged for 5 min at 4 °C and 1300 x g to remove cellular debris. The supernatant was aliquoted into 1-mL samples and stored long-term in cryovials at −80 °C. Individual 1-mL samples from control and PD subjects (n = 5 each) were thawed, and then 300 μL was transferred to a cryovial with a generic label to blind investigators at Iowa State. These samples then were frozen and shipped overnight on dry ice to Iowa State University. Samples were stored at −80 °C upon arrival and prior to analysis (Table 2).
Autopsy Brain Samples
Brain tissues were provided by the Brain Endowment Bank from the University of Miami and UC Davis Alzheimer’s Disease Center. All tissues were frozen at −80 °C upon arrival and stored at −80 °C prior to analysis. From the University of Miami, substantia nigra tissue from 11 PD and 11 control patients was obtained. The UC Davis Alzheimer’s Disease Center provided slices of basal ganglia from 5 DLB, 10 AD, and 8 control patients. All tissues had been examined histologically for accurate diagnoses using accepted criteria (Montine 2012) prior to testing in the RT-QuIC assay.
Preparation of BHs
From the frozen brain sections, 10% w/v BHs were prepared in sterile PBS using a Bullet Blender (Next Advance) with 0.5-mm zirconium oxide beads for 2 min at the maximum speed. The 10% homogenates were stored at −80 °C until use. Various dilutions of BHs, ranging from 10−2 to 10−6, were made using PBS, and the 10−4 dilution was used for testing all the samples in the RT-QuIC assay.
Recombinant αSyn Protein Purification
A plasmid expressing human WT αSyn was a generous gift from Dr. Julien Roche (Iowa State University). Recombinant human WT αSyn was purified as described previously (Maltsev et al. 2012; Volpicelli-Daley et al. 2014) with a few modifications. In brief, a loop-full inoculum of human WT αSyn-expressing Rosetta cells in glycerol stock was added to 5 mL of LB media supplemented with 50 μg/mL kanamycin and grown overnight at 225 rpm and 37 °C. Next, mini-cultures were grown in 1 L of kanamycin-LB media, and when the OD600 reached 0.7, 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added into the bacteria. Cells were harvested 4.5 h later by pelleting at 4200 x g for 20 min at 4 °C. Bacterial pellets were lysed by dissolving them in 10 mL of 50 mM Tris and 500 mM NaCl at pH 7.4 using an Omni tissue homogenizer. Following lysis, the cell suspension was sonicated for 2 min using a high setting on a probe sonicator that was paused every 15 s. Samples then were heat-precipitated at 85 °C for 15 min. Later, the precipitates were removed by centrifugation at 15,000 x g for 10 min at 4 °C and DNA was precipitated by adding streptomycin (10 mg/mL) to the supernatants. The solution then was centrifuged at 23,000 x g for 30 min, and the supernatant was 10-fold diluted and dialyzed using a 3-kDa molecular weight cut-off snakeskin dialysis tubing in a 20 mM Tris HCl buffer at pH 8 overnight at 4 °C. The next morning, the dialyzed supernatant was concentrated and filtered with a 0.2-μM filter before loading onto a Sephacryl 200 column (GE Healthcare Life Sciences) for size-exclusion chromatography using a 20 mM Tris-HCl buffer of pH 8 at 4 °C. Fractions having recombinant αSyn were pooled, concentrated and filtered using a 3-kD cut-off filter and analyzed with gel electrophoresis. Later, Coomassie staining was performed and fractions having recombinant αSyn were combined, concentrated, and filtered using a 0.2-μm filter before loading onto a HiPrep Q FF 16/10 anion-exchange column. A linear gradient was run up to 1 M NaCl in 20 mM Tris, pH 8, and αSyn protein was recovered between 300 and 350 mM NaCl. Fractions were collected and analyzed through polyacrylamide gel electrophoresis followed by Coomassie staining to visualize the protein fractions. Fractions having protein were pooled and dialyzed in 20 mM Tris, pH 8, at 4 °C overnight. On the next day, the buffer was replaced with a new buffer for one hour and the protein was filtered using a 0.2-μm filter. Protein concentrations were estimated using a NanoDrop spectrophotometer with an extinction coefficient of 0.5960 M-1cm-1. The protein was lyophilized in aliquots with a final concentration of 1 mg/mL and stored at −80 °C until use.
Polyacrylamide Gel Electrophoresis
Protein fractions from size-exclusion or anion-exchange chromatography were mixed with an equal volume of 2X sample-loading buffer and 2-mercaptoethanol (2X Laemmli sample buffer, BIO-RAD), and boiled for 5 min. Proteins were separated by polyacrylamide gel electrophoresis using 4–20% gradient gels (BIO-RAD) followed by staining and destaining with Coomassie blue.
Generation of αSynagg and αSynPFF from Recombinant WT αSyn
We generated αSynagg and αSynPFF from the recombinant αSyn according to published methods (Polinski et al. 2018). In brief, 100 μL solution of 5 mg/mL recombinant WT αSyn in PBS with 100 mM NaCl was subjected to continuous 1000 rpm shaking at 37 °C for 7 days in a 1.5-mL tube. Later, the products were verified for the presence of ThT-positive αSyn aggregates. Western blot analysis of ultra-centrifuged aggregates revealed that pellets have more αSynagg than supernatant fractions. To generate αSynPFF, the αSynagg was probe-sonicated at power level 2 for a total of 60 pulses, pausing every 10–12 pulses.
Sedimentation Assay
The αSynagg was diluted 10-fold using sterile PBS. Diluted αSynagg later was ultracentrifuged at 100,000 x g for 30 min at 25 °C. Supernatants and pellets were diluted with 5X Laemmli buffer and boiled for 5 min. Equal volumes of supernatants and pellets then were separated using 4–20% gradient gels (BIO-RAD).
ThT Assay
A 1-mM stock of ThT was diluted in PBS to a final concentration of 25 μM. This was used to add 95 μL of ThT to each well of a 384-well plate. A 2.5-μL sample of αSynagg, monomeric recombinant αSyn or PBS alone was added to the 95 μL of ThT in the wells and incubated at RT for 2 min. Fluorescence readings were then taken on a plate reader at excitation and emission wavelengths of 450 and 480 nm, respectively.
Scanning Transmission Electron Microscopy (STEM)
For STEM characterization, αSynagg and αSynPFF were resuspended in sterile PBS. Next, 20 μL of resuspended sample was mixed with 2% uranyl acetate and incubated for 5 min. Then, 5 μL of sample was applied to carbon-coated copper grids and images were taken with a JEOL 2100 200-kV STEM operated at 80 kV. The analysis was done using a Thermo Fisher Noran System 6 elemental analysis system.
Cell Culture and Treatments
A mouse microglial cell line was a gift from Douglas T Golenbock (U Mass) and cultured as described previously (Halle et al. 2008). Cells were maintained in DMEM F12 media supplemented with 9% FBS, penicillin/streptomycin, glutamine, and sodium pyruvate. Microglia were treated with 1 μM of αSyn PFFs prepared in serum-free media. After treatments, cells were collected at 0, 0.5, 2, 6, 12 and 24 h using ice-cold PBS. After estimating the protein concentrations, 5 and 50 pg of microglial lysate was used as a seed in the RT-QuIC assay.
RT-QuIC Method
The αSyn RT-QuIC assay was done as published previously (West Greenlee et al. 2016; Moore et al. 2017) using a 96-well clear bottom plate (Nalgene Nunc International). Briefly, samples were loaded into each well of a 96-well plate preloaded with 6 silica beads 0.8 mm in diameter (OPS Diagnostics). For all the αSyn RT-QuIC assays, the reaction mixture consists of 40 mM phosphate buffer (pH 8.0), 170 mM NaCl, 10 μM ThT, and 0.1 mg/mL of recombinant αSyn. But for testing microglia and CSF samples, along with the above components, we also added final concentrations of 0.0015% sodium dodecyl sulfate (SDS) to the reaction mixture. For microglia samples, we loaded 5 μL of microglial lysate along with 95 μL of the reaction mixture in each well of a 96-well plate. For CSF samples, we loaded 15 μL of CSF sample along with 85 μL of the reaction mixture in each well of a 96-well plate. For BH samples, we loaded 2 μL of diluted BH along with 98 μL of the reaction mixture in each well of a 96-well plate. After loading the samples, plates were sealed with a plate sealer (Nalgene Nunc International) and ThT fluorescence readings were taken at excitation and emission wavelengths of 450 and 480 nm, respectively, every 30 min using a CLARIOstar (BMG) plate reader with alternating 1-min shake and rest cycles (double orbital, 400 rpm) at 42 °C. Samples were run in quadruplicates and considered positive when at least two of the wells crossed the threshold fluorescence. Threshold fluorescence was calculated by taking the average fluorescence of the first 10 cycles for all samples plus 10 standard deviations. PAR was calculated by taking the inverse of the time required to cross the threshold fluorescence.
Immunostaining
Immunoreactivity of αSyn filaments from BHs of α-synucleinopathy and NS cases was determined by dot blot using a Bio-Dot Microfiltration System per manufacturer’s protocol as described previously (Kondru et al. 2017). In brief, after determining the protein concentrations of BHs using the Bradford assay, 1 μg of homogenate was mixed with 200 μL of Tris-buffered saline (1X TBS) with 0.1% Tween-20 and allowed to adsorb onto a nitrocellulose membrane for 1 h. The membrane was blocked with 1X LI-COR blocking buffer (LBB) for 30 min after washing twice with 200 μL of 1X TBS using a gentle vacuum. The membrane then was incubated with a rabbit monoclonal (MJFR-14-6-4-2) αSyn filament conformation-specific antibody (dilution 1:2000) and a mouse monoclonal (BD Biosciences, #610787) total αSyn antibody for 1 h at RT, and triple-washed with 1X TBS. Membranes then were incubated with their respective secondary antibody made in LBB (1:10,000) for 30 min followed by 3 washes with 1X TBS. Immunoreactive proteins were detected using the Odyssey IR Imaging system along with the densitometric quantification of dots.
Statistical Analysis
GraphPad 7.0 was used for statistical analysis with p ≤ 0.05 considered statistically significant. Raw data were analyzed using Student’s t test for comparing two groups, one-way ANOVA for analyzing more than two groups, and then Tukey’s post-test was performed to compare different groups. Asterisks were assigned as follows: *p ≤ 0.05, **p < 0.01, and ***p < 0.001. The number of biological replicates is expressed as “n” unless otherwise mentioned.
References
Allen Reish HE, Standaert DG (2015) Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J Parkinsons Dis 5:1–19
Beach TG, Adler CH (2018) Importance of low diagnostic accuracy for early Parkinson's disease. Mov Disord 33:1551–1554
Bidinosti M, Shimshek DR, Mollenhauer B, Marcellin D, Schweizer T, Lotz GP, Schlossmacher MG, Weiss A (2012) Novel one-step immunoassays to quantify alpha-synuclein: applications for biomarker development and high-throughput screening. J Biol Chem 287:33691–33705
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69
Braak H, Braak E (2000) Pathoanatomy of Parkinson's disease. J Neurol 247(Suppl 2):Ii3–I10
Braak H, Bohl JR, Muller CM, Rub U, de Vos RA, Del Tredici K (2006) Stanley Fahn lecture 2005: the staging procedure for the inclusion body pathology associated with sporadic Parkinson's disease reconsidered. Mov Disord 21:2042–2051
Compta Y, Valente T, Saura J, Segura B, Iranzo A, Serradell M, Junque C, Tolosa E, Valldeoriola F, Munoz E, Santamaria J, Camara A, Fernandez M, Fortea J, Buongiorno M, Molinuevo JL, Bargallo N, Marti MJ (2015) Correlates of cerebrospinal fluid levels of oligomeric- and total-alpha-synuclein in premotor, motor and dementia stages of Parkinson's disease. J Neurol 262:294–306
Fairfoul G, McGuire LI, Pal S, Ironside JW, Neumann J, Christie S, Joachim C, Esiri M, Evetts SG, Rolinski M, Baig F, Ruffmann C, Wade-Martins R, Hu MT, Parkkinen L, Green AJ (2016) Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol 3:812–818
Groveman BR, Orrù CD, Hughson AG, Raymond LD, Zanusso G, Ghetti B, Campbell KJ, Safar J, Galasko D, Caughey B (2018) Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathologica Communications 6
Gupta N, Shyamasundar S, Patnala R, Karthikeyan A, Arumugam TV, Ling EA, Dheen ST (2018) Recent progress in therapeutic strategies for microglia-mediated neuroinflammation in neuropathologies. Expert Opin Ther Targets 22:765–781
Hall S, Surova Y, Ohrfelt A, Zetterberg H, Lindqvist D, Hansson O (2015) CSF biomarkers and clinical progression of Parkinson disease. Neurology 84:57–63
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9:857–865
Hamilton RL (2000) Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol (Zurich, Switzerland) 10:378–384
Holdorff B (2006) Fritz Heinrich Lewy (1885-1950). J Neurol 253:677–678
Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55:181–184
Kondru N, Manne S, Greenlee J, West Greenlee H, Anantharam V, Halbur P, Kanthasamy A, Kanthasamy A (2017) Integrated Organotypic slice cultures and RT-QuIC (OSCAR) assay: implications for translational discovery in protein Misfolding diseases. Sci Rep 7:43155
Lee HJ, Suk JE, Bae EJ, Lee SJ (2008) Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem Biophys Res Commun 372:423–428
Lema Tomé CM, Tyson T, Rey NL, Grathwohl S, Britschgi M, Brundin P (2013) Inflammation and α-Synuclein’s prion-like behavior in Parkinson's disease—is there a link? Mol Neurobiol 47:561–574
Maltsev AS, Ying J, Bax A (2012) Impact of N-terminal acetylation of alpha-synuclein on its random coil and lipid binding properties. Biochemistry 51:5004–5013
Manne S, Kondru N, Nichols T, Lehmkuhl A, Thomsen B, Main R, Halbur P, Dutta S, Kanthasamy AG (2017) Ante-mortem detection of chronic wasting disease in recto-anal mucosa-associated lymphoid tissues from elk (Cervus elaphus nelsoni) using real-time quaking-induced conversion (RT-QuIC) assay: a blinded collaborative study. Prion 11:415–430
Marti MJ, Tolosa E, Campdelacreu J (2003) Clinical overview of the synucleinopathies. Mov Disord 18(Suppl 6):S21–S27
Montine TJ (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach 123:1–11
Moore SJ, West Greenlee MH, Kondru N, Manne S, Smith JD, Kunkle RA, Kanthasamy A, Greenlee JJ (2017) Experimental transmission of the chronic wasting disease agent to swine after Oral or intracranial inoculation. J Virol 91
Mori H, Oda M, Komori T, Arai N, Takanashi M, Mizutani T, Hirai S, Mizuno Y (2002) Lewy bodies in progressive supranuclear palsy. Acta Neuropathol 104:273–278
Ohrfelt A, Grognet P, Andreasen N, Wallin A, Vanmechelen E, Blennow K, Zetterberg H (2009) Cerebrospinal fluid alpha-synuclein in neurodegenerative disorders-a marker of synapse loss? Neurosci Lett 450:332–335
Polinski NK, Volpicelli-Daley LA, Sortwell CE, Luk KC, Cremades N, Gottler LM, Froula J, Duffy MF, Lee VMY, Martinez TN, Dave KD (2018) Best practices for generating and using alpha-Synuclein pre-formed fibrils to model Parkinson's disease in rodents. J Parkinsons Dis 8:303–322
Rosenthal LS, Drake D, Alcalay RN, Babcock D, Bowman FDB, Chen-Plotkin A, Dawson TM, Dewey RB Jr, German DC, Huang X, Landin B, McAuliffe M, Petyuk VA, Scherzer CR, Hillaire-Clarke CS, Sieber BA, Sutherland M, Tarn C, West A, Vaillancourt D, Zhang J, Gwinn K, on behalf of the PDBP consortium (2016) The NINDS Parkinson's disease biomarkers program. Mov Disord 31:915–923
Sano K, Atarashi R, Satoh K, Ishibashi D, Nakagaki T, Iwasaki Y, Yoshida M, Murayama S, Mishima K, Nishida N (2018) Prion-like seeding of misfolded alpha-Synuclein in the brains of dementia with Lewy body patients in RT-QUIC. Mol Neurobiol 55:3916–3930
Spillantini MG, Divane A, Goedert M (1995) Assignment of human alpha-synuclein (SNCA) and beta-synuclein (SNCB) genes to chromosomes 4q21 and 5q35. Genomics 27:379–381
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A 95:6469–6473
Stone DK, Reynolds AD, Mosley RL, Gendelman HE (2009) Innate and adaptive immunity for the pathobiology of Parkinson's disease. Antioxid Redox Signal 11:2151–2166
Tokuda T, Qureshi MM, Ardah MT, Varghese S, Shehab SA, Kasai T, Ishigami N, Tamaoka A, Nakagawa M, El-Agnaf OM (2010) Detection of elevated levels of alpha-synuclein oligomers in CSF from patients with Parkinson disease. Neurology 75:1766–1772
Tu PH, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, Iwatsubo T, Trojanowski JQ, Lee VM (1998) Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol 44:415–422
Uchikado H, DelleDonne A, Ahmed Z, Dickson DW (2006) Lewy bodies in progressive supranuclear palsy represent an independent disease process. J Neuropathol Exp Neurol 65:387–395
Volpicelli-Daley LA, Luk KC, Lee VMY (2014) Addition of exogenous α-Synuclein pre-formed fibrils to primary neuronal cultures to seed recruitment of endogenous α-Synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc 9:2135–2146
Wang Y et al (2012) Phosphorylated alpha-synuclein in Parkinson's disease. Sci Transl Med 4:121ra120
Waxman EA, Giasson BI (2011) Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci 31:7604–7618
West Greenlee MH, Lind M, Kokemuller R, Mammadova N, Kondru N, Manne S, Smith J, Kanthasamy A, Greenlee J (2016) Temporal resolution of misfolded prion protein transport, accumulation, glial activation, and neuronal death in the retinas of mice inoculated with scrapie. Am J Pathol 186:2302–2309
Williams GP, Schonhoff AM, Jurkuvenaite A, Thome AD, Standaert DG, Harms AS (2018) Targeting of the class II transactivator attenuates inflammation and neurodegeneration in an alpha-synuclein model of Parkinson's disease. J Neuroinflammation 15:244
Acknowledgments
We thank Gary Zenitsky for proofreading the manuscript and Griffin Clabaugh for technical assistance. We would like to thank Kayla Guthals for assistance with the figures. We also are indebted to Dr. Julien Roche, Iowa State University, for providing us with human WT αSyn-expressing plasmid, Dr. Douglas T. Golenbock, U Mass, for the mouse microglial cell line, Alzheimer’s disease Center at UC Davis for supplying AD, DLB and control samples (funded by NIH/NIA P30 AG10129), and the University of Miami Brain Endowment Bank of the NIH Neurobiobank for providing PD and control samples. The Lloyd and Armbrust endowments to AGK and Salisbury endowment to AK are also acknowledged. This study was supported in part by the following sources: National Institutes of Health grants ES026892 and NS100090 to AGK, and NS088206 to AK, as well as the Presidential Interdisciplinary Research Initiative for the Big Data Brain Research from Iowa State University. The financial support for human CSF and brain samples received by XH include NS060722 and NS082151 to the Hershey Medical Center Clinical Research Center (National Center for Research Resources, Grant UL1 RR033184 that is now at the National Center for Advancing Translational Sciences, Grant UL1 TR000127), National Center for Advancing Translational Sciences (TL1 TR002016), the PA Department of Health Tobacco CURE Funds (XH), the Translational Brain Research Center, the Michael J. Fox Foundation for Parkinson’s Research, Alzheimer’s Association, Alzheimer’s Research UK, and the Weston Brain Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
A.G.K. and V.A. have an equity interest in PK Biosciences Corporation located in Ames, IA. The terms of this arrangement have been reviewed and approved by Iowa State University in accordance with its conflict of interest policies. Other authors declare no actual or potential competing financial interests.
Ethics Approval
CSF samples were collected as part of the NINDS Parkinson’s Disease Biomarker Program (PDBP) according to guidelines (Rosenthal et al. 2016) for the lumbar puncture. The protocol was reviewed and approved by the Penn State Hershey Internal Review Board and informed written consent was obtained from all subjects. All brain tissues of human subjects were from the University of Miami Brain Endowment Bank and the UC Davis Alzheimer’s Disease Center.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author Contributions
Conceptualization, S.M., N.K., and A.G.K.; Experiment design and performance, S.M. and N.K.; microglial uptake of αSyn, M.H., N.K., and S.M.; H.J. and V.A. assisted in the preparation of the manuscript; Clinical specimen acquisition, X.H., and M.L.; Funding Acquisition, A.G.K., A.K.; Supervision, A.G.K.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 129 kb)
Rights and permissions
About this article

Cite this article
Manne, S., Kondru, N., Hepker, M. et al. Ultrasensitive Detection of Aggregated α-Synuclein in Glial Cells, Human Cerebrospinal Fluid, and Brain Tissue Using the RT-QuIC Assay: New High-Throughput Neuroimmune Biomarker Assay for Parkinsonian Disorders. J Neuroimmune Pharmacol 14, 423–435 (2019). https://doi.org/10.1007/s11481-019-09835-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-019-09835-4