
Abstract
Purpose
Arsenic, especially inorganic trivalent species, is one of the most important poisons in the field of forensic toxicology. There are many reports on the speciation analysis of arsenic species in biological specimens by liquid chromatography–inductively coupled plasma-mass spectrometry (LC–ICP-MS). The aim of this study was to develop a rapid and robust analytical method for speciation/quantitative analysis of arsenic species in serum by LC–ICP-tandem mass spectrometry (MS/MS).
Methods
An analytical method for arsenous acid and its metabolites, dimethylarsinic acid, monomethylarsonic acid, and arsenic acid in serum was tested through the analysis of serum samples by an LC–ICP-MS/MS system consisting of different anion exchange columns and different mobile phases. Rapid pretreatment of serum samples by ultrafiltration was also tested.
Results
Robust speciation/quantitative analysis of arsenic in serum samples with LC–ICP-MS/MS was achieved by using a mildly acidic mobile phase. The limits of detection for the four arsenic species were in the range 0.19–0.68 ngAs/mL, and the well-known interference by argon chloride ion was removed by the MS/MS apparatus. This method was precise enough for quantitative analysis of four arsenic compounds in serum (0.24–3.68% precision; 97.0–104% accuracy; and 101–112% recovery for all analytes at 5 and 50 ngAs/mL). The total analytical time was 30 min (20-min pretreatment and 10-min analysis), and multiple serum samples could be pretreated simultaneously.
Conclusions
A rapid, sensitive, interference-free and robust speciation/quantitative analysis of toxic arsenous acid and related metabolites in serum by LC–ICP-MS/MS was developed. To our knowledge, this is the first report to use LC–ICP-MS/MS for analysis of arsenic species in human blood/serum samples.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Arsenic, especially arsenous acid (As(III)), is one of the most important poisons in the field of forensic toxicology. The toxicity of arsenic compounds largely depends on the chemical structure; thus, there have been many recent reports on speciation analyses of arsenic in biological specimens, mainly by using liquid chromatography–inductively coupled plasma-mass spectrometry (LC–ICP-MS) [1,2,3,4,5,6,7,8,9,10,11,12,13,14], and there are some reports focusing on blood/plasma/serum samples [8, 13,14,15], which is one of the most important biological specimens in forensic toxicology. In most reports on the speciation analysis of arsenic by LC–ICP-MS, LC separations are based on ion chromatography (IC) using anion exchange columns [4,5,6,7,8,9,10,11,12,13] or ion-pair chromatography (IPC) using ion-pair reagents and reversed-phase columns [14, 16]. IC is a powerful tool for protein purification [17]; however, in IC-based separations, proteins, peptides, fats or lipids, which are major components of blood, may strongly bind to the IC columns [18, 19] and may damage them; hence there are no reports of the robust and fully validated quantitative blood/plasma/serum analysis by IC-based LC–ICP-MS. In IPC-based separation, the LC system is robust for blood analysis; however, in the IPC-based separation of arsenic compounds, alkylsulfonic acids are used as IPC reagents [14, 16], which interfere with 32S/34S analysis by the same analytical system [20,21,22]. Recently, another solution for speciation analysis of arsenics in blood samples, LC–hydride generation-atomic fluorescence spectrometry (LC–HG-AFS) method, is also being utilized [23,24,25]; however, in principle, applications of these methods are limited for hydride-generating elements [26], and protein precipitation process using mineral acids is still needed [25]. LC–ICP-MS/MS analysis is still advantageous for simultaneous determination of hydride-generating and non-hydride-generating elements [2, 3, 8]. In this paper, we report a rapid and robust speciation and sensitive quantitative analysis method for As(III) and its metabolites, dimethylarsinic acid (DMAA), monomethylarsonic acid (MMAA), and arsenic acid (As(V)) (Fig. 1) in human serum by IC-based LC–ICP-MS/MS with minimal and easy pretreatment, although a few reports on analysis of arsenic species in seafood at ppm–ppb levels by LC–ICP-MS/MS have appeared very recently [27,28,29].

Structures of a toxic arsenous acid (As(III)) and its metabolites, b dimethylarsinic acid (DMAA), c monomethylarsonic acid (MMAA), and d arsenic acid (As(V))
Materials and methods
Chemicals and reagents
Ultrapure water was prepared with Milli-Q Gradient A10 system (Merck-Millipore Japan, Tokyo, Japan). Stock solutions of 100 μgAs/mL for As(III), DMAA, MMAA, As(V) and arsenobetaine (AB) were prepared by dissolving sodium metaarsenite (special grade; Wako Pure Chemical Industries, Osaka, Japan), DMAA, and MMAA (Tri Chemical Laboratories Inc., Yamanashi, Japan), disodium hydrogen arsenate heptahydrate and arsenobetaine (Wako Pure Chemical Industries) in ultrapure water, respectively. Human serum was purchased from Sigma-Aldrich (St. Louis, MO, USA). All other chemicals were of analytical grade commercially available.
Instrumentation
The LC–ICP-MS/MS system consisted of an Agilent 1260 infinity II LC system and an Agilent 8800 ICP-tandem mass spectrometer (Agilent Technologies Japan, Ltd., Tokyo, Japan). Two LC columns based on the hydrophilic methacrylate strong anion exchange resin were employed: an Agilent Excelpak G3288-80000 strong anion exchange column for direct urine analysis (250 × 4.6 mm i.d.) in combination with an Agilent Excelpak G3154-65002 guard column (10 × 4.6 mm i.d.) (column 1 system), and an Agilent Excelpak G1836A/101 strong anion exchange column for environmental water analysis (150 × 4.6 mm i.d.) in combination with an Agilent Excelpak G1836A/102 guard column (10 × 4.6 mm i.d.) (column 2 system). The mobile phases for both columns were fixed to the manufacturer-recommended composition without any modifications to evaluate the influence of multiple serum sample injections onto the columns. Both mobile phases are shown in Table 1. The MS/MS conditions are also shown in Table 1. Amongst the MS/MS modes, including helium collision mode, hydrogen reaction mode and oxygen reaction mode (O2 mode), O2 mode showed the highest sensitivity for all the arsenic compounds. The optimized O2 reaction conditions are shown in Table 1.
Serum sample preparation
Serum samples (500 µL) were placed into Amicon Ultra 0.5 3 K ultrafiltration cartridges (0.5 mL, nominal molecular weight limit (NMWL) 3000 Da, Merck-Millipore Japan) without dilution, and centrifuged at 14,000 × g for 20 min. Ten microliters of each filtrate (300–400 μL) was injected into LC–ICP-MS/MS.
Quantitative analysis of the arsenic compounds in serum samples
Mixed standard solutions (10 μgAs/mL) of As(III), DMAA, MMAA, and As(V) were prepared by mixing 0.1-mL volume of each stock solution (100 μgAs/mL), followed by appropriate dilution with water, and were used in the following experiments. Calibrators for the calibration curves for four arsenic compounds were prepared at concentrations of 2, 5, 10, 20, 50, 100, 200 and 500 ngAs/mL by adding 50 μL of mixed standard solutions with appropriate concentrations prepared from the 10-μgAs/mL mixed standard solution to 950 μL of blank serum. Absolute calibration curves were plotted using the peak area of analytes (y-axis) and the concentration of the calibration standard (x-axis), and fitted with a linear equation by least-squares approximation. The limit of detection (LOD) and the limit of quantification (LOQ) of arsenic species were calculated according to the definition stipulated by Japanese Industrial Standards [30]. Extraction recoveries for four analytes in serum were determined at concentrations of 5 ngAs/mL (asymptomatic level [13, 14]) and 50 ng/mL (poisoning level [8]). For each concentration, five replicate aliquots (1.0 mL) of the serum sample were fortified with the appropriate amounts of analytes (pre-spiked samples). An additional five replicate aliquots (1.0 mL) had no analytes added (post-spiked samples). After ultrafiltration of the post-spiked samples, the filtrates were fortified with the same numbers of analytes as the pre-spiked samples. Then, 10 μL of the filtrates of the pre- and post-spiked samples were injected into the LC–ICP-MS/MS. Recoveries were calculated by comparing the peak area ratios of analytes of the pre-spiked samples to those of the post-spiked samples. The means were obtained for the results of each set of five replicates. Quality control samples were also independently prepared at 5 ngAs/mL and 50 ngAs/mL. Quantification of the samples was performed using the above calibration curves. The accuracy was expressed as the difference between the concentration at which the sample was spiked and the measured concentration. Intraday precision was evaluated using the relative standard deviation (RSD) of the calculated concentrations for five replicates spiked at two levels and analyzed on the same day. Interday precision was assessed using the RSD of the mean concentrations measured on three different days at two levels (three replicates on each day).
Results and discussion
Chromatographic separation of arsenic compounds in serum
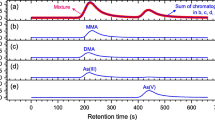
First, the column 1 system with the strong alkaline mobile phase (pH 11; Table 1) was tested for the separation of the arsenic species in water and serum samples containing 5 ngAs/mL of As(III), DMAA, MMAA, and As(V) for each, and good separation was found for each sample (Fig. 2a, b), as with our past work with a single-stage MS [31]. The retention time of AB, the minor interference compound in serum, is also shown in Fig. 2.

Chromatograms of As(III), DMAA, MMAA, and As(V) in a aqueous and b serum (10 ngAs/mL each) samples obtained by liquid chromatography–inductively coupled plasma-mass spectrometry (LC–ICP-MS) analysis using the column 1 system with a strong alkaline mobile phase (pH 11). The retention time of arsenobetaine (AB), the minor interference compound in serum, is also shown in a and b
However, the column 1 system was severely and irreversibly damaged after 12 injections of the serum samples (data not shown). In this study, the serum samples were analyzed after minimal pretreatment only and could have still contained peptides with NMWLs of less than 3000 Da, phospholipids, fatty acids, and so on. They would be negatively charged at pH 11 and would strongly bind to the anion exchange resin, thus resulting in the irreversible damage to the column. If the mobile phase were a mild alkaline, the damage of the column would be reduced, but it would not be an ideal solution.
In contrast, the column 2 system with a mild acidic mobile phase (pH 6, Table 1) showed robustness against the serum samples. A water sample containing the four arsenic compounds As(III), DMAA, MMAA, and As(V) (10 ngAs/mL each), which is the original target sample of this column, was separated with good peak shapes (Fig. 3a). Following on the figure, a serum sample containing 50 ngAs/mL of As(III), DMAA, MMAA, and As(V) was analyzed, which showed broadening of the peaks (Fig. 3b), and quantitative analysis was possible. After 100 injections of the 50-ngAs/mL serum samples, the peak shapes of the arsenic compounds were retained (Fig. 3c). After 100 injections of serum samples, a 10-ngAs/mL water sample was injected again and the peak shapes mostly recovered (Fig. 3d) to the initial state (Fig. 3a). Thus, the peak broadening of the serum samples was thought to be a reversible phenomenon caused by coexisting components in the serum, such as peptides (< 3000 Da), phospholipids, fatty acids, and so on, which would not bind to the anion exchange resin, but was coeluted with the analytes under acidic conditions. Therefore, the column 2 system with pH 6 mobile phase was used for the following experiments.

Chromatograms of As(III), DMAA, MMAA, and As(V) in water and serum samples obtained by LC–ICP-MS/MS analysis using the column 2 system with a mild acidic mobile phase (pH 6). a Aqueous sample (5 ngAs/mL each), b serum sample (50 ngAs/mL each) injected just after a, c 100th injection of serum sample (10 ngAs/mL each) after b, and d an aqueous sample (5 ngAs/mL each) injected just after c. The retention time of AB is also shown in a–d
Validation of the analytical method
The chromatograms of blank serum (Fig. 4a), and 0.5-ngAs/mL-fortified (Fig. 4b) and 50-ngAs/mL-fortified (Fig. 4c) serum samples are shown in Fig. 4.

Chromatograms of blank serum and As(III), DMAA, MMAA, and As(V) in serum samples. a Blank serum, b serum sample (0.5 ngAs/mL each), and c serum sample (50 ngAs/mL each). The retention time of AB is also shown in a–c. *Unknown As compounds in blank serum different from AB
In the chromatogram of the blank serum (Fig. 4a), peaks of very small amounts of unknown arsenic compounds, probably originating from foods, were observed around 1.4–1.7 min. Thus, the peak area of As(III) up to 1.7 min was cut off in the quantitative calculations. No other major interference peaks were observed in Fig. 4a. In the analysis of arsenic compounds in urine samples by monitoring of 75As+ (m/z 75) with single-stage LC–ICP-MS, a large peak of argon chloride ion (m/z 75) was generated from a large amount of urinary chloride ions in the analysis [32]; hence, there are many reports on the reduction of argon chloride ion by a collision cell with helium or hydrogen gases [7, 18] or dynamic reaction cells [6, 20]. Serum samples also contain chloride ions, but in this study, the interference by argon chloride ion was completely removed by the MS/MS apparatus, and reliable argon chloride-free analysis was fully implemented. In the chromatogram of the blank serum used in this experiment (Fig. 4a), AB, a non-toxic As species mainly originating from seafood, was not detected. However, some ethnic groups, e.g., Japanese, have higher daily dietary intake of AB [33,34,35] from seafood. AB is rapidly removed from blood [36] and mainly excreted into urine [13, 33,34,35], and there is no report of the AB levels of real blood/plasma/serum samples. About a decade ago, we looked for real serum samples containing AB obtained from healthy volunteers in Japan, but we were unsuccessful (unpublished observations). Just in case, we studied the influence of AB in serum on the detection of other As species. Thus, we used AB-spiked blank serum for the experiment. In the LC condition using the column 2 system, AB spiked in blank serum sample was eluted at 1.5 min (Fig. 5). The peak of AB did not show baseline separation from As(III), but did not seriously interfere with the detection of As(III), and as described above, we cut off the peak area of As(III) up to 1.7 min for the quantitative calculation. Therefore, in this study, non-toxic AB was omitted in the quantification and validation experiments, and only the retention time of AB was shown in Fig. 4a–c.

Chromatogram of AB, As(III), DMAA, MMAA, and As(V) in a serum sample (50 ngAs/mL each)
The calibration equation, coefficient of determination, LOD, and LOQ for each arsenic compound tested are shown in Table 2. The LOD values for all the arsenic compounds were in ppt-order, and the LOQ value of the most toxic As(III) was also in ppt-order, showing the very high sensitivity and selectivity of LC–ICP-MS/MS. The calibration range covered from 2 ngAs/mL (As(III), DMAA, and MMAA) and 5 ngAs/mL (As(V)), which are asymptomatic levels [13, 14], to 500 ngAs/mL, which is the poisoning level. The linearities of all the calibration curves were over 0.999, demonstrating good quantitative performance of LC–ICP-MS/MS. The chromatogram of 0.5-ngAs/mL-fortified serum sample (Fig. 4b), which was nearly LOD level, showed enough ion intensity for detection of the most toxic As(III). The satisfactory recoveries of the arsenic compounds are shown in Table 3. They were around 100% for all the arsenic compounds, and the RSDs were not greater than 4.68% for all analytes and concentrations. Intraday (n = 5) and interday (n = 3) precisions for each arsenic compound are shown in Table 4. For each compound, this method was reproducible enough (0.24–3.68% intra- and interday precisions, and 97.0–104% accuracies at the two fortifying levels) showing the outstanding performance of this method for quantitative analysis.
Conclusions
Many poisoning cases from toxic inorganic arsenic occur worldwide every year; in such cases, speciation and quantitative analysis of arsenic compounds in biological specimens are conducted by LC–ICP-MS to clarify the cause of poisoning. However, to our knowledge, there have been no reports on the toxicological analysis of arsenic species in blood/plasma/serum samples by LC–ICP-MS/MS with detailed validation, even though blood/plasma/serum samples are quite important in forensic toxicology. In addition, we have found that serum samples may collapse anion exchange columns when using a strong alkaline mobile phase; but the use of a mild acidic mobile phase enables robust analysis of arsenic compounds in serum samples. Using our LC conditions, the pretreatment of serum could be simplified to 20-min ultrafiltration. This method also enabled high-precision quantitative analysis, and will be useful in the field of forensic toxicology.
References
Benramdane L, Bressolle F, Vallon JJ (1999) Arsenic speciation in humans and food products: a review. J Chromatogr Sci 37:330–344
Delafiori J, Ring G, Furey A (2016) Clinical applications of HPLC-ICP-MS element speciation: a review. Talanta 153:306–331
Marcinkowska M, Barałkiewicz D (2016) Multielemental speciation analysis by advanced hyphenated technique—HPLC/ICP-MS: a review. Talanta 161:177–204
Okina M, Yoshida K, Kuroda K, Wanibuchi H, Fukushima S, Endo G (2004) Determination of trivalent methylated arsenicals in rat urine by liquid chromatography-inductively coupled plasma mass spectrometry after solvent extraction. J Chromatogr B 799:209–215
Heitland P, Köster HD (2009) Comparison of different medical cases in urinary arsenic speciation by fast HPLC-ICP-MS. Int J Hyg Environ Health 212:432–438
Verdon CP, Caldwell KL, Fresquez MR, Jones RL (2009) Determination of seven arsenic compounds in urine by HPLC-ICP-DRC-MS: a CDC population biomonitoring method. Anal Bioanal Chem 393:939–947
Serrano IN, Ballesteros MTL, Pacheco SSF, Álvarez SI, Colón JLL (2016) Total and speciated urinary arsenic levels in the Spanish population. Sci Total Environ 571:164–171
Heitland H, Blohm M, Breuer C, Brinkert F, Achilles EG, Pukite I, Köster HD (2017) Application of ICP-MS and HPLC-ICP-MS for diagnosis and therapy of a severe intoxication with hexavalent chromium and inorganic arsenic. J Trace Elem Med Biol 41:36–40
Wang D, Shimoda Y, Wang S, Wang Z, Liu J, Liu X, Jin H, Gao F, Tong J, Yamanaka K, Zhang J, An Y (2017) Total arsenic and speciation analysis of saliva and urine samples from individuals living in a chronic arsenicosis area in China. Environ Health Prev Med 22:45. https://doi.org/10.1186/s12199-017-0652-5
Carioni VMO, McElroy JA, Guthrie JM, Ngwenyama RA, Brockman JD (2017) Fast and reliable method for As speciation in urine samples containing low levels of As by LC-ICP-MS: focus on epidemiological studies. Talanta 165:76–83
Yuan C, Lu X, Oro N, Wang Z, Xia Y, Wade TJ, Mumford J, Le XC (2008) Arsenic speciation analysis in human saliva. Clin Chem 54:163–171
Kintz P, Ginet M, Marques N, Cirimele V (2007) Arsenic speciation of two specimens of Napoleon’s hair. Forensic Sci Int 170:204–206
Contreras-Acuña M, García-Barrera T, García-Sevillano MA, Gómez-Ariza JL (2014) Arsenic metabolites in human serum and urine after seafood (Anemonia sulcata) consumption and bioaccessibility assessment using liquid chromatography coupled to inorganic and organic mass spectrometry. Microchem J 112:56–64
Fukai Y, Hirata M, Ueno M, Ichikawa N, Kobayashi H, Saito H, Sakurai T, Kinoshita K, Kaise T, Ohta S (2006) Clinical pharmacokinetic study of arsenic trioxide in an acute promyelocytic leukemia (APL) patient: speciation of arsenic metabolites in serum and urine. Biol Pharm Bull 29:1022–1027
Ito K, Goessler W, Gürleyük H, Wels B, Palmer CD, Verostek MF, Parsons PJ (2011) An interlaboratory study of arsenic speciation analysis of whole blood. J Anal At Spectrom 26:1740–1745
Araujo-Barbosa U, Peña-Vazquez E, Barciela-Alonso MC, Costa Ferreira SL, Pinto Dos Santos AM, Bermejo-Barrera P (2017) Simultaneous determination and speciation analysis of arsenic and chromium in iron supplements used for iron-deficiency anemia treatment by HPLC-ICP-MS. Talanta 170:523–529
Yigzaw Y, Hinckley P, Hewig A, Vedantham G (2009) Ion exchange chromatography of proteins and clearance of aggregates. Current Pharm Biotechnol 10:421–426
Goheen SC, Gibbins BM (2000) Protein losses in ion-exchange and hydrophobic interaction high-performance liquid chromatography. J Chromatogr A 890:73–80
Muca R, Marek W, Żurawski M, Piątkowski W, Antos D (2017) Effect of mass overloading on binding and elution of unstable proteins in hydrophobic interaction chromatography. J Chromatogr A 1492:79–88
Bandura DR, Baranov VI, Tanner SD (2002) Detection of ultratrace phosphorus and sulfur by quadrupole ICPMS with dynamic reaction cell. Anal Chem 74:1497–1502
Schaumlöffel D, Giusti P, Preud’Homme H, Szpunar J, Łobiński R (2007) Precolumn isotope dilution analysis in nanoHPLC-ICPMS for absolute quantification of sulfur-containing peptides. Anal Chem 79:2859–2868
Zinn N, Krüger R, Leonhard P, Bettmer J (2008) μLC coupled to ICP–SFMS with post-column isotope dilution analysis of sulfur for absolute protein quantification. Anal Bioanal Chem 391:537–543
Šlejkovec Z, Podgornik H, Černelč P, Falnoga I (2016) Exceptions in patterns of arsenic compounds in urine of acute promyelocytic leukaemia patients treated with As2O3. Biometals 29:107–118
Šlejkovec Z, Falnoga I, Goessler W, van Elteren JT, Raml R, Podgornik H, Černelč P (2008) Analytical artefacts in the speciation of arsenic in clinical samples. Anal Chim Acta 607:83–91
Guo M, Wang W, Hai X, Zhou J (2017) HPLC-HG-AFS determination of arsenic species in acute promyelocytic leukemia (APL) plasma and blood cells. J Pharm Biomed Anal 145:356–363
Chen YW, Belzile N (2010) High performance liquid chromatography coupled to atomic fluorescence spectrometry for the speciation of the hydride and chemical vapour-forming elements As, Se, Sb and Hg: a critical review. Anal Chim Acta 671:9–26
Schmidt L, Landero JA, Santos RF, Mesko MF, Mello PA, Flores EMM, Caruso JA (2017) Arsenic speciation in seafood by LC-ICP-MS/MS: method development and influence of culinary treatment. J Anal At Spectrom 32:1490–1499
Guimarães D, Roberts AA, Tehrani MW, Huang R, Smieska L, Woll AR, Lin S, Parsons PJ (2018) Characterization of arsenic in dried baby shrimp (Acetes sp.) using synchrotron-based X-ray spectrometry and LC coupled to ICP-MS/MS. J Anal At Spectrom 33:1616–1630
Schmidt L, Landero JA, La Rosa Novo D, Duarte FA, Mesko MF, Caruso JA, Flores EMM (2018) A feasible method for As speciation in several types of seafood by LC-ICP-MS/MS. Food Chem 255:340–347
Japanese Standards Association (2006) High frequency plasma mass spectrometry general notice, K0133. In: JIS Handbook. Japanese Standards Association, Tokyo, pp 371–388 (in Japanese)
Higashikawa Y, Kazui Y, Suzuki S, Ohtsuru O (2008) Arsenic speciation of arsine-exposed blood samples by high-performance liquid chromatography-inductively coupled plasma mass spectrometry and As-adduct, a possible indicator of AsH3 exposure. J Anal Toxicol 32:344–348
Tan SH, Horlick G (1986) Background spectral features in inductively coupled plasma/mass spectrometry. Appl Spectrosc 40:445–460
Kaise T (2002) Speciation of arsenic compounds in biological samples by high performance liquid chromatography-inductively coupled plasma mass spectrometry system. J Plasma Fusion Res 78:646–652 (in Japanese with English abstract)
Ohwaki S, Matsuoka M, Sato T, Kikukawa K, Kobayashi M, Kaneko S (2018) Arsenic speciation analysis in seaweed and rice flour samples by ion chromatography combined with inductively coupled plasma-mass spectrometry. Studies Sci Technol 7:69–74 (in Japanese)
Yoshinaga J, Chatterjee A, Shibata Y, Morita M, Edmonds JS (2000) Human urine certified reference material for arsenic speciation. Clin Chem 46:1781–1786
Oliver LK (2009) Laboratory assessment of exposure to neurotoxic agents. In: Dobbs M (ed) Clinical neurotoxicology. Saunders, Philadelphia, pp 213–221
Acknowledgements
This work was supported by the research project “R&D of chemical examination method by the application of highly effective instruments for elemental analysis to the discrimination of evidential material” by National Research Institute of Police Science.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The coauthor Yasuo Seto is a Chief Editor of the Forensic Toxicology, but was not involved in the peer-reviewing of this article. Other authors declare no competing interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kazui, Y., Ohta, H., Watanabe, D. et al. Rapid and robust speciation/quantitative analysis of arsenous acid and related metabolites in serum by liquid chromatography–inductively coupled plasma-tandem mass spectrometry. Forensic Toxicol 37, 424–431 (2019). https://doi.org/10.1007/s11419-019-00479-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11419-019-00479-w