
Abstract
In diabetic patients, diabetic cardiomyopathy (DCM) is one of the most common causes of death. The inflammatory response is essential in the pathogenesis of DCM. Rhein, an anthraquinone compound, is extracted from the herb rhubarb, demonstrating various biological activities. However, it is unclear whether rhein has an anti-inflammatory effect in treating DCM. In our research, we investigated the anti-inflammatory properties as well as its possible mechanism. According to the findings in vitro, rhein could to exert an anti-inflammatory effect by reducing the production of NO, TNF-α, PGE2, iNOS, and COX-2 in RAW264.7 cells that had been stimulated with advanced glycosylation end products (AGEs). In addition, rhein alleviated H9C2 cells inflammation injury stimulated by AGEs/macrophage conditioned medium (CM). In vivo have depicted that continuous gavage of rhein could improve cardiac function and pathological changes. Moreover, it could inhibit the accumulation of AGEs and infiltration of inflammatory factors inside the heart of rats having DCM. Mechanism study showed rhein could suppress IKKβ and IκB phosphorylation via down-regulating TRAF6 expression to inhibit NF-κB pathway in AGEs/CM-induced H9C2 cells. Moreover, the anti-inflammation effect of rhein was realized through down-regulation phosphorylation of JNK MAPK. Furthermore, we found JNK MAPK could crosstalk with NF-κB pathway by regulating IκB phosphorylation without affecting IKKβ activity. And hence, the protective mechanism of rhein may involve the inhibiting of the TRAF6-NF/κB pathway, the JNK MAPK pathway, and the crosstalk between the two pathways. These results suggested that rhein may be a promising drug candidate in anti-inflammation and inflammation-related DCM therapy.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus (DM), is a chronic disease threatening human health, with its prevalence rising annually from a younger age. As per the epidemiological surveys, DM affects 463 million people around the world, and expected to rise to 578 million by the year 2030 [1]. DM could entail various complications, such as cardiovascular disease being one of the leading causes of death and disability, with approximately 2/3rd of DM patients dying yearly [2, 3]. One of the many cardiovascular complications of DM is diabetic cardiomyopathy (DCM). It is independent of myocardial ischemia, coronary atherosclerotic heart disease, heart valve disease, hypertension, and other cardiomyopathies, leading to structural or/and functional abnormalities in heart. DCM is characterized by myocardial fibrosis, myocardial inflammation, apoptosis, cardiac dysfunction, etc. DCM induces changes in diastolic and/or systolic cardiac function, leading to heart failure. It is a major cause of end-stage death among DM patients [4]. The exact cause of DCM is unclear because of its complexity and multifaceted pathogenesis. Efficacy of current clinical treatment, which focuses on glucose and blood pressure regulation, lipid management, and heart failure suppression, is low. Therefore, investigating the pathogenesis of DCM and actively seeking new drugs to effectively prevent and treat DCM is crucial.
It has been confirmed that DCM is closely associated with a chronic inflammatory response [5]. Monocytes-macrophages are the primary immune cells in diabetic cardiomyopathy and possess the ability to polarize phenotypically. Depending on their activation status and function, macrophages are classified classically (M1 type) and alternatively (M2 type) activated. M1 macrophages are responsible for producing multiple pro-inflammatory cytokines. Some of these cytokines and chemokines include tumor necrosis factor-alpha (TNF-α), interleukin 6 (IL-6), and inducible nitric oxide synthase (iNOS). These cytokines and chemokines act as a response to inflammation. In contrast, M2 macrophages suppress inflammation and repair tissues by secreting immunosuppressive cytokines, among them are the enzyme arginase 1 (Arg1), the cytokine interleukin 4 (IL-4), and the cytokine interleukin 10 (IL-10).
Glycosylation end products (AGEs), are polymers that are formed when proteins, lipids, nucleic acids, and glucose all react together in the absence of enzymes. Under physiological circumstances, the formation of AGEs is a slow process, and they tend to build up in the skin, cartilage, and pericardial fluid in only trace amounts. In contrast, the accumulation of AGEs is accelerated illnesses such as diabetes, obesity, and metabolic syndrome are examples of these [6]. The development and accumulation of AGEs are one of the characteristic pathological changes of DM. Moreover, it is also an essential inducer of the inflammatory response. AGEs accumulate in large quantities in the body as a result of accelerated glycosylation reactions or impaired clearance of glycation products in the intermediate and advanced stages of diabetes. This causes AGEs to bind to receptors on the surface of macrophages, which promotes polarization of macrophages towards the M1 and results in the release of pro-inflammatory mediators, both of which can eventually cause damage to myocardial tissue or fibrosis. In addition, recent research has shown that AGEs have the ability to bind to toll-like receptors 4 (TLR4) on myocardial cell surface in diabetic mice. This in turn recruits the downstream signaling molecule myeloid differentiation factor 88 (MyD88), which could activate nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways, mediate immune inflammation, and eventually lead to DCM [7]. AGEs-mediated immune inflammation is involved in DCM pathogenesis, which is difficult to be cleared once they have accumulated in cells and formed covalent bonds. Therefore, searching for a therapeutic strategy to antagonize AGEs-mediated immune inflammation could be a practical approach to preventing and treating DCM.
Rhein, a natural anthraquinone compound, is extracted from the herb Rheum officinale Baill. It is responsible for various bioactivities, such as anti-inflammatory, antineoplastic, improving insulin resistance, and promoting microcirculation [8,9,10,11]. Previous studies have described that rhein can improve insulin resistance by upregulating the production of insulin receptor substrate-1 (IRS-1), phosphatidylinositol 3-kinase (PI3K), and Akt Ser473 phosphorylation in DM mice model. Rhein also enhances glucose uptake and utilization by adipose tissue by upregulating peroxisome proliferator-activated receptor γ (PPARγ) and expression of glucose transporter type 4 (GluT-4) mRNA among diabetic models [12]. Additionally, rhein was able to inhibit the secretion of inflammatory factors such as IL-6, IL-1β, and TNF-α in RAW264.7 cells when those cells were stimulated with lipopolysaccharide (LPS) [13]. Thus, rhein has the potential efficacy in managing diabetic complications by improving insulin resistance and suppressing inflammation in diabetes. However, whether rhein can exert its protective effect against DCM through anti-inflammatory has not been reported.
Therefore, an in vitro (i.e., AGEs-stimulated macrophage conditioned medium induced myocardial H9C2 cell injury model) and an in vivo model of diabetic animals were established and administered using rhein to investigate whether rhein has a cardioprotective effect against DCM by alleviating the inflammatory immune response. This study could provide an experimental and theoretical basis for preventing and treating DCM.
Materials and methods
Materials
Rhein (C15H8O6) was obtained from Macklin Biochemical Company (Shanghai, China). AGEs were obtained from Biosynthesis Company (Beijing, China). Aminoguanidine (AG) was purchased from Sigma Company (USA). Dimethylsulfoxide (DMSO) was purchased from Beyotime (Shanghai, China). MTT and Cycloheximide (CHX) were purchased from Abcam. Hyclone was the source for both Dulbecco’s Modified Eagle medium (DMEM) and fetal bovine serum (FBS) (Waltham, MA, USA). Prostaglandin E2 (PGE2) ELISA kit was from FineTest (Wuhan, China). ExCell Bio Company (Shanghai, China) provided the TNF-α and IL-1β ELISA kit that was used in this study. An assay kit for nitrite oxide (NO) was provided by the Nanjing Jiancheng Institute (Jiangsu, China). The Beyotime Institute of Biotechnology (Shanghai, China) provided the staining kits for Hoechst 33258 as well as Rhodamine 123, the compounds BAY11-7082, SP 600125, and SB 202190 and NF-κB Activation and Nuclear Translocation Assay Kit. TUNEL staining kit was obtained from Nanjing Jiancheng (Nanjing, China). OxiSelect™ AGEs kit was from Cell Biolabs (San Diego, USA). The primary and secondary antibodies were from CST (Beverly, USA).
Cell culture
Both the RAW264.7 macrophage cell line and the H9C2 Rat myocardial cell line were obtained from Oricell® (Guangzhou, China). Cells were cultured in DMEM (4500 mg/l glucose) containing 10% FBS and 1% penicillin–streptomycin at 37 °C in the cell incubator.
Treatment of H9C2 cardiac cells with macrophage-conditioned medium
AGEs at a concentration of 300 μg/ml were used to stimulate RAW264.7 cells for a period of 24 h. The activated macrophage cells could release inflammatory factors into the medium. And the supernatant liquid from RAW264.7 cells was collected as AGEs/conditioned medium (CM). After that, H9C2 cells were stimulated with AGEs/CM with or without rhein (at concentrations of 5, 10, or 20 μM) for a period of 48 h (Fig. 2A).
Cell viability assay
RAW264. 7 cells were added into a 96-well plate at a density of 8 × 103 per well and left there for 24 h before being stimulated by AGEs at a concentration of 300 μg/ml with or without rhein at concentrations of 5, 10, or 20 μM. After 24 h of being seeded at a density of 5 × 103 cells per well in a 96-well plate, the H9C2 cells were grouped and stimulated by AGEs/CM with or without rhein (5, 10, 20 μM) for another 48 h. This process was repeated three times. The MTT assay was used to determine the cell survival rate. In a nutshell, the supernatants of the cells were discarded and replaced with new cell culture medium that contained 0.5 mg/ml of MTT and was incubated at 37 °C for 2–4 h. Then add DMSO into cells and formazen absorbance was measured at 570 nm.
NO assay
RAW264.7 cells were seeded in a 48-well plate (1.5 × 104 cells/well) for the first 24 h. After that, the cells were stimulated by AGEs at 300 μg/ml with or without rhein at 5, 10 or 20 μM for another 24 h. The amount of NO in the cells was measured using a commercial kit. To sum up, we took the medium from the cells and combined it with Griess reagent at a 1:3 ratio. After that, the cells were left to rest in an incubator at room temperature for 10 min. In addition, a measurement of 540 nm was obtained for the optical density of the solution.
ELISA assay for TNF-α and PGE2
In a 48-well plate, 1.5 × 104/well RAW264.7 cells were cultured for 24 h before being stimulated by AGEs with or without rhein for 8 h (TNF-α assay) or 24 h (cell viability assay) (PGE2 assay). According to the experimental protocols of ELISA kits, the contents of TNF-α and PGE2 were measured and determined to be present in the supernatants.
Hoechst 33258 apoptosis assay
For the Hoechst 33258 apoptosis assay of rhein against AGEs/CM-induced cytotoxicity, H9C2 cells were co-treated with AGEs/CM and rhein as described previously for 48 h. Following a wash with PBS, the cells were fixed with cold paraformaldehyde at a concentration of 4% for 20 min before being incubated with Hoechst 33258 solution at 37 °C for 8 min. After performing three washes in PBS, fluorescent images were captured using a microscope equipped with a fluorescence mode.
Rhodamine 123 staining assay
H9C2 cells were stimulated by AGEs/CM with or without rhein for 48 h. After that, a Rhodamine 123 solution of 20 μM was added into the cells, and they were left to incubate at 37 °C and out of the light for 30 min. A fluorescence microscope was used to take the photographs of the fluorescent images.
Immunofluorescence assay
For intracellular staining of NF-κB p65, H9C2 cells were inoculated onto poly-l-lysine-coated coverslips in a 48-well plate (2 × 104/well) for 24 h. Cells were fixed with 4% paraformaldehyde as well as perforated with 0.2% Triton X-100 for 15–20 min at 37 °C following treatment with AGEs/CM with or without rhein for 4 h. Primary antibody against NF-κB p65 (1:250) was incubated with the cells for 12 h at 4 °C after they were blocked with PBST containing 5% BSA for 2 h. After being washed three times, the cells were left to incubate with a secondary antibody for 2 h at ordinary temperature in dark place. For nuclear staining, DAPI was used as a counterstain. A fluorescence microscope was used to examine the immunofluorescence images.
Western blot analysis
H9C2 cells or the hearts of rats were collected after treatment and lysed in RIPA buffer on ice for 30 min. To separate the components of the whole cell lysates, they were centrifuged at 12,000 rpm for 8 min. Proteins from the nucleus and the cytoplasm were isolated using a Nuclear Extraction kit. Bradford assay was used to quantify protein concentrations. The same amount of protein samples were added to the SDS-PAGE system for electrophoresis separation and then electrotransferred to the PVDF membranes. The protein bands were incubated with primary antibody at 4 °C for more than 12 h with minimal shaking after being blocked with non-fat milk at 5% (w/v) for 1 h. After being washed with PBST three times, the PVDF membranes were then treated with a secondary antibody at room temperature for a period of 2 h. After performing three separate washes, ECL was subsequently applied to the PVDF membranes to visualize the prorein bands.
Cellular thermal shift assay (CETSA)
For cell lysate CETSA experiment, H9C2 cells were collected and freeze-thawed five times using liquid nitrogen. The cell lysates were grouped into two aliquots, with one serving as the control and the other being treated with rhein (20 μM) for 4 h at 4 °C. Then, the lysates were heated at indicated temperatures (44–62 °C, respectively) for 5 min followed by cooling at room temperature. The protein bans were detected by immunoblot.
Quantification of mRNA expression by real time-PCR
AGEs/CM would be used to stimulate H9C2 cells for thirty minutes with or without the addition of rhein. Utilizing the Boxbio® RNA Extraction Kit, total RNA was successfully isolated. In order to conduct reverse transcription, we utilized the RevertAid First Strand cDNA Synthesis Kit (Fermentas). Moreover, for the purpose of amplification, The SeqPlex™ RNA Amplification Kit was utilized. RT-PCR was performed on the ABI 7500 system (MA, USA). The following is a list of the sequences of primers: TRAF6 forward primer, 5′-TTC CAG AAG TGCCA GGT TAA TAC-3′; reverse primer, 5′-CAA GTG TCGTG CCA AGT GAT-3′ and GAPDH forward primer, 5′-GGT GAA GGTCG GTG TGA ACG-3′; reverse primer, 5′-CTC GCT CCTGG AAG ATG GTG-3′. There was a triplicate analysis of each sample. The relative expression of genes was normalized to GAPDH, and the following formula was employed to calculate the results: level of relative mRNA expression of the TRAF6 gene (folds of control) = 2−ΔΔCT.
Animal treatment
The Experimental Animal Ethics Committee of the First Affiliated Hospital of Guangzhou University of Chinese Medicine approved all animal experiments.
The Experimental Animal Centre of Guangzhou University of Chinese Medicine supplied 55 male Sprague–Dawley (SD) rats aged 4 weeks and weighing 70 ± 10 g (License number: SCXK(Guangdong)2018-0020). As the control group, we randomly selected 10 rats and fed them normal chow. Except for that, the remaining 45 were fed a high-sugar/fat diet (high-AGEs diet, containing 60.4% basal diet, 20% sucrose, 10% lard, 1.5% cholesterol, 8% egg yolk powder, and 0.1% sodium cholate). All animals were housed within the Guangzhou University of Chinese Medicine’s Animal Experimentation Centre. Moreover, All the rats were allowed to drink freely and were kept in sanitary conditions in the specific pathogen-free (SPF) environment at 22 ± 1 °C and relative humidity of 40% ± 5% under the circumstance of a 12 h’ light cycle. Six weeks after feeding, a small dose of streptozotocin (STZ) 30 mg/kg was administered intraperitoneally for three consecutive days to induce a rat model of type 2 DM. Then, the fasting blood glucose was determined on days 3 and 7 post-STZ injections. In type 2 diabetic rat models, fasting glucose levels of ≥ 16.7 mmol/l were considered at both time points. In this experiment, 36 rats were successfully modeled.
After successful diabetic modeling, the subjects were randomly assigned to the DCM model group, low-dose rhein (100 mg/kg), high-dose rhein (200 mg/kg), and empagliflozin (30 mg/kg) groups. Rhein and EMPA were dissolved in 1% carboxymethylcellulose sodium, respectively, before administration. Sodium carboxymethylcellulose was administered in equal amounts to the control and DCM groups. All rat groups were gavaged daily for 12 weeks.
Echocardiography assessment of cardiac functions
After the gavage, the rats were anesthetized using 1% pentobarbital sodium. Then, they were placed in a supine position on a rat board, with the anterior thoracic region and anterior abdomen shaved. The left ventricular end-diastolic internal diameter (LVIDd), left ventricular end-systolic internal diameters (LVIDs), left ventricular posterior wall thickness during diastole (LVPWd) and left ventricular posterior wall thickness during systole (LVPWs) of each group were measured with small animal ultrasonography. Fractional shortening (FS) and ejection fraction (EF) were calculated to evaluate the impact of rhein on the cardiac function of rats. The mean values were obtained after 10 successive cardiac cycles of measurement.
Biochemical parameters determination
Following the gavage, the rats were anesthetized, positioned supine, and blood was drawn from the abdominal aorta. After collecting whole blood into tubes containing sodium heparin anticoagulation, we then waited 20–30 min at ordinary temperature for the blood to become coagulated. The next step is to obtain the supernatant by centrifuging the blood at a rate of 4000 revolutions per minute for 10 min while maintaining a temperature of 4 °C. After that, the supernatant was stored inside a refrigerator at − 80 °C. Then, the animals were immediately executed and the hearts were quickly removed. The myocardial tissues were frozen in liquid nitrogen and stored in a refrigerator with a temperature of − 80 °C.
Based on the manufacturer’s instructions, the inflammatory factors (TNF-α and IL-1β) in blood specimens were discovered employing ELISA. Simultaneously, the concentrations of AGEs in myocardial tissue were measured adopting the OxiSelect™ AGEs ELISA kit.
Staining of myocardial tissue sections
After the animals were put to death in the manner outlined earlier, the heart tissue was harvested and then stored in paraformaldehyde at a concentration of 4% for one night. Then, the fixed tissues were placed in an embedding box. Following a series of alcohol gradients for dehydration, the heart tissues were encapsulated in paraffin blocks. The embedded tissue blocks were cut into thin segments of 4 µm and subjected to hematoxylin–eosin (HE) and masson staining, correspondingly, for the purpose of determining histopathological hypertrophy and fibrosis of the myocardium. TUNEL staining was performed based on the manufacturer's protocol to visualize apoptotic cells within the myocardial tissue. Photographs were taken using an optical microscope (HE and Masson staining magnified at 40×, and TUNEL staining magnified at 10×).
Statistical analysis
For analysis, SPSS 16.0 was utilized, and experimental data were expressed as Mean ± SD. from minimum of three separate experiments. The means values were being made an comparison by One-way ANOVA with Bonferroni’s post hoc test. P < 0.01 indicated a statistically significant difference, whereas P < 0.05 indicated an observable distinction.
Results
Rhein downregulated inflammatory factors in AGEs-induced RAW264.7 cells
Rhein is a kind of emodin-type hydroxy-anthraquinone, which mainly exists in traditional Chinese medicines like Rheum officinale Baill. Rhein is well acknowledged for its pharmacological activities, such as anti-inflammatory. Its chemical structure is shown in Fig. 1A.

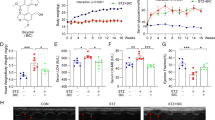
Rhein reduced the production of inflammatory factors in RAW264.7 that had been activated. RAW264.7 cells were treated with 300 μg/ml AGEs and 5, 10 or 20 μM concentrations of rhein. A The chemical structure and molecular weight of rhein are depicted. B The cell viability was measured using MTT. C Using a commercial kit, NO production was detected. D TNF-α levels were detected by ELISA. E PGE2 levels were measured by ELISA. F Western blotting was used to quantify the expression of iNOS and COX-2. N.S. no significance. ###P < 0.001 compared to the control group; *P < 0.05, **P < 0.01, ***P < 0.001 relative to AGEs group
We first tried to establish the AGEs-induced inflammatory macrophage model in a concentration- and time-dependent manner (Supporting Information Fig. S1A). With the increase of AGEs concentration, the production of NO in each group increased gradually. For 8 h-treated and 16 h-treated groups, there was no significance v.s. control. However, after the intervention of AGEs (300 μg/ml) for 24 h, the production of NO in RAW264.7 cells increased dramatically, which was about 4 times that of the control group. Supporting Information Fig. S1B showed that there was no significant decrease in the RAW264.7 cells viability treated with rhein below 20 μM for 24 h. With the increase of rhein concentration, the survival rate decreased significantly in a concentration-dependent manner.
Given the above results, we then evaluated the effect of rhein (5, 10, or 20 μM) 24 h of treatment with AGEs (300 μg/ml) reduced the viability of RAW264.7 cells. Consequently, it demonstrated a fact that the concentration range studied of rhein did not induce cell toxicity (Fig. 1B). We have proven that rhein reduced the expected increases in NO, TNF-α, and PGE2 in AGEs-induced RAW264.7 cells in a concentration-dependent sort of way (Fig. 1C–E). Similarly, protein bands analysis showed rhein treatment significantly reduced iNOS, and cyclooxygenase 2 (COX-2) interpretation in a concentration-dependent sort of way (P < 0.01 or P < 0.001) (Fig. 1F). In addition, our results suggested that the anti-inflammatory effect of rhein (20 μM) was comparable to that of the positive control drug aminoguanidine (AG), a selective AGEs inhibitor.
Rhein protected cardiac H9C2 cells against inflammation injury induced by AGEs/macrophage-conditioned medium
After validating that rhein had anti-inflammatory effect on AGEs-stimulated RAW264.7 cells, we further explored the effects of AGEs on cardiac H9C2 cells. Firstly, we need to establish a toxic cardiomyocytes model. Our supporting results had shown that 50–300 μg/mL AGEs had no cytotoxic effect on H9C2 cells (Supporting Information Fig. S2A). Moreover, 50–300 μg/mL AGEs had no effects on the production of NO and LDH in H9C2 cells (Supporting Information Fig. S2B, C), indicating that AGEs isn’t an ideal agent for inducing inflammatory injury in H9C2 cells. As inflammatory mediators secreted by activated RAW264.7 cells may promote cardiac fibrosis and apoptosis [14], and the dysfunction of resident macrophages or the inability of macrophages to communicate with myocardial cells, endothelial cells, and fibroblasts may result in abnormal repair, persistent injury, and heart failure, we tried to induce an inflammatory cell model that simulated the environment in vivo.
We successfully established a novel inflammatory injury H9C2 model by incubating cells with macrophage-conditioned media (CM). Supernatant collected from AGEs (300 μg/mL)-stimulated RAW264.7 cells was used as CM to incubate H9C2 cells for 48 h to induce injury (Fig. 2A). Initially, we examined the viability of H9C2 cells in the conditioned medium system using the MTT assay. The result showed that rhein treatment could prevent the AGEs/CM-induced decrease in cell viability in a concentration-dependent manner (P < 0.05) (Fig. 2B). The morphology of H9C2 cells was subsequently observed using an inverted microscope. The cells in the control group grew well, mainly in a shuttle shape. However, the AGEs/CM-induced H9C2 cells were irregularly polygonal or round, with elevated vacuolation and cellular debris. Rhein (5, 10, 20 μM) could gradually enhance the cell morphology (Fig. 2C). Additionally, Hoechst 33258 staining demonstrated that AGEs/CM significantly increased myocardial apoptosis compared to the control group (P < 0.01); nonetheless, the apoptosis rate was markedly decreased when H9C2 cells were treated with 5, 10 or 20 μM rhein (P < 0.05 or P < 0.01) (Fig. 2C, D). Rhodamine 123 staining also reflected the protective effect of rhein. Rhodamine 123 can enter the mitochondrial matrix selectively in normal cells based on the mitochondrial transmembrane potential (ΔΨm) and emit bright green fluorescence. However, in injury cells, Rhodamine 123 is published from mitochondria leading to a decline in mitochondrial membrane potential, resulting in a substantial decrease in the intensity of green fluorescence in mitochondria. In comparison to the control group, the model group's green fluorescence was considerably reduced (P < 0.001); whereas the injury H9C2 cells induced by AGEs/CM were blocked by rhein treatment (P < 0.05 or P < 0.01) (Fig. 2C, E). These results indicated that rhein had a protective effect against AGEs/CM-induced cardiomyocyte damage.

Rhein protected H9C2 cells from damage induced by AGEs/CM. A Diagram of the AGEs/conditioned macrophage medium experiment. B After 48 h of treatment with AGEs/CM with or without rhein, the viability of H9C2 cells was measured by MTT. C The morphology of H9C2 cells treated for 48 h with or without AGEs/CM was evaluated by microscopy. Hoechst 33258 staining and was used to assess cell apoptosis. Rhodamine 123 staining was used to assess the mitochondrial membrane potential (MMP) loss in injury cells. The arrows represent typical injury cells. The magnification was ×10; scale bar 100 μm. ###P < 0.001 compared to control group; *P < 0.05, **P < 0.01, ***P < 0.001 compared to AGEs/CM group
Rhein ameliorated AGEs/CM-induced inflammation response in H9C2 cells by inhibiting the IKKβ-IκB-NF-κB signaling pathway
NF-κB, as a primary transcription factor, plays a crucial role in the regulation of TNF-α, PGE2, and NO production the inflammatory reaction [15]. The phosphatation of NF-κB resulting inside its nuclear translocation but also activation of proinflammatory genes [16]. Immunofluorescence imaging revealed that AGEs/CM stimulated the translocation of NF-κB p65 from of the cytoplasm to the nucleus, and that this process was nearly blocked by concentration-dependent rhein treatment (Fig. 3A). As well as we noticed the rhein (5,10, 20 μM) could significantly inhibit the NF-κB p65 phosphorylation level induced by AGEs/CM in H9C2 cells (P < 0.001, Fig. 3B). In addition, the suppressive effect of rhein on NF-κB p65 nuclear translocation was verified by western blot analysis (Fig. 3C). We further investigated the impact of rhein on inducing IκB kinase α/β (IKKα/β) and inhibitor of NF-κB (IκB), two crucial upstream regulators of NF-κB activation. Our results indicated that rhein treatment (5, 10, 20 μM) could reverse the markedly elevated levels of phosphorylation IKKα/β and IκB ended up causing by AGEs/CM (P < 0.01 or P < 0.001) (Fig. 3D). Overall, these findings demonstrated that rhein might effectively impede the IKKβ/IκB/NF-κB inflammatory pathway activation induced by AGEs and CM.

Rhein’s impact on IKKβ-IκB-NF-κB signaling pathway in AGEs/CM-induced H9C2 cells. A NF-κB p65 nuclear translocation was observed by fluorescence microscopy. Red and blue fluorescence represent NF-κB p65 subunit and nuclear DAPI staining respectively (scale bar 100 μm). B NF-κB p65 phosphorylation was ascertained by Western blot analysis. C Western blot examination of NF-κB p65 nuclear translocation was performed. D Western blot evaluation was performed for IKKβ and IκB phosphorylation. ###P < 0.001 compared to the control group; **P < 0.01, ***P < 0.001 compared to the AGEs/CM group
Rhein inhibited NF-κB pathway activation by downregulating TRAF6 expression
The TLR4 pathway is central to the activation of reactions in response to AGEs or pro-inflammatory stimulators [17]. The tumor necrosis factor receptor-associated factor 6 (TRAF6) is a crucial protein mediator in TLR4 pathway and can transduce signals from upstream molecules such as the myeloid distinctiveness primary response gene 88 (MyD88) and interleukin-1 receptor-associated kinase 1 (IRAK1). Activated TRAF6 recruits other proteins, including transforming growth factor-β-activated kinase-1 (TAK1) and TAK1-binding protein 2 (TAB2), to form protein kinase complexes. The protein kinase complexes further stimulate phosphorylation of IKK and IκB degradation in order to activate NF-κB [18, 19]. Western blot analysis showed that rhein treatment inhibited the production of TRAF6 induced by AGEs/CM in a concentration-dependent manner (P < 0.001, Fig. 4D), without affecting consequently expression of MyD88, IRAK1and TAB2 (Fig. 4A). In addition, after H9C2 cells were treated with AGEs/CM with or without rhein for 30 min, RT-PCR was used to determine if the changes in TRAF6 protein expression were mirrored by a change in mRNA expression. We found mRNA expression of TRAF6 was not affected by rhein treatment (Fig. 4E) and suggested that rhein might regulate TRAF6 expression through post-translational modifications.

Rhein inhibited NF-κB pathway activation by downregulating TRAF6 expression. A Utilizing western blot, the expression levels of MyD88, IRAK1, and TAB2 were determined. B Rhein (20 μM) promoted TRAF6 protein degradation in a time-dependent manner. C The amount of TRAF6 mRNA was measured by RT-PCR. D H9C2 cells were treated with AGEs/CM with or without rhein for 1 h, and TRAF6 expression was measured by Western blot analysis. E The changes in TRAF6 mRNA were determined by RT-PCR. N.S. no significance. #P < 0.05 compared to the control group; ***P < 0.001 compared to the AGEs/CM group
In order to further verify that rhein does not inhibit the expression of TRAF6 by regulating the transcription level, we used 20 μM protein synthesis inhibitor cycloheximide (CHX) to suppress newly synthesized proteins, and found that rhein (20 μM) significantly accelerated TRAF6 degradation in a time-dependent fashion (Fig. 4B). And we found that rhein treatment alone without AGEs/CM did not affect the gene transcription level of TRAF6 in H9C2 cells (Fig. 4C). Thus, we speculated that rhein may down-regulate TRAF6 expression by promoting protein degradation rather than inhibiting its synthesis.
Rhein inhibited JNK MAPK activation and JNK MAPK could crosstalk with the NF-κB pathway by targeting IκB
MAPKs pathways are preserved signaling cascades in which a variety of protein kinases transmit signals from the cell membrane to a nucleus. For the purpose of examining the effect of rhein on the activation of the MAPKs pathway, we measured the levels of phosphorylation and total expression of the c-jun N-terminal kinase (JNK), p38, and extracellular regulated protein kinase (ERK) MAPKs in AGEs/CM-induced H9C2 cells with or without rhein treatment (5, 10, 20 μM). In a concentration-dependent manner, rhein significantly decreased the phosphorylation levels of JNK and p38 MAPKs (P < 0.01 or P < 0.001), and yet did not affect ERK MAPK phosphorylation expression (Fig. 5A).

Rhein inhibited the activation of JNK MAPK and JNK MAPK could crosstalk with the NF-κB pathway by targeting IκB in AGEs/CM-induced H9C2 cells. A On a Western blot, the phosphorylation and total expression of JNK, p38, and ERK were examined. B H9C2 cells were exposed to AGEs/CM with or without rhein (20 μM), 5 μM Bay 117082 (an NF-κB inhibitor), 20 μM SP 600125 (a JNK inhibitor) or 5 μM SB 202190 (a p38 inhibitor) for 48 h, and NO production was measured. C AGEs/CM have been used to induce H9C2 cells with or without 5 μM Bay 117082 and 20 μM SP 600125 for 4 h, phosphorylation and overall expression of NF-κB and JNK MAPK were performed by western blot. D H9C2 cells were incubated with AGEs/CM with or without 20 μM SP 600125 for for 1 h (p-IKKβ assay) or 4 h (p-IκB assay). E H9C2 cell lysates were incubated with rhein (20 μM) or vehicle followed by the cellular thermal shift assay (CETSA). Western Blot was used to detect the expression of associated proteins. N.S. no significance. ##P < 0.01, ###P < 0.001 compared to the control group; *P < 0.05, **P < 0.01, ***P < 0.001 compared to the AGEs/CM group; &&&P < 0.001 with respect to the SB 202190 treatment group
We utilized pathway inhibitors to even further investigate the impact of rhein on JNK/p38 MAPKs pathway. The results showed that when AGEs/CM-induced H9C2 cells were subjected to 5 μM Bay 117082 (a NF-κB pathway inhibitor) and 20 μM SP 600125 (a JNK MAPK pathway inhibitor), the increased NO induced by AGEs/CM was significantly inhibited (Fig. 5B). These findings suggested that both of the two pathways were involved in the inflammation response induced by AGEs/CM. However, we found that NO production induced by AGEs/CM was not significantly changed when exposed to rhein (20 μM) coexist Bay 117082 or SP 600125 (Fig. 5B). These results demonstrated that the downregulation of NO production by rhein was blocked when the NF-κB or JNK MAPK pathway was inhibited. In addition, on the condition that H9C2 cells were stimulated with 5 μM SB 202190 (a p38 MAPK pathway inhibitor), the NO production induced by AGEs/CM was not significantly reduced, and rhein (20 μM) still showed the obvious anti-inflammatory effect (P < 0.001), which demonstrated rhein's anti-inflammatory mechanism did not primarily involve regulation of p38 MAPK pathway (Fig. 5B). Therefore, we came to the conclusion that rhein's anti-inflammatory effects were realized via the NF-κB and JNK MAPK pathways.
We then sought a link between the NF-κB pathway and the JNK MAPK pathway. Moreover, we have observed that the expression of p-NF-κB was significantly inhibited (P < 0.001) when AGEs/CM-induced H9C2 cells were then treated with an inhibitor of JNK MAPK, while p-JNK expression was not affected by NF-κB inhibitor treatment (Fig. 5C). These results showed that decreased activity of JNK MAPK could inhibit NF-κB pathway activation in AGEs/CM-induced H9C2 cells. Therefore, we concluded that JNK MAPK might be an upstream signal for the NF-κB pathway. To prove this hypothesis, we investigated the influence of JNK MAPK on the activation of IKKβ and IκB and found JNK MAPK inhibitor treatment could markedly reduce p-IκB expression (P < 0.01) without altering the expression of p-IKKβ (Fig. 5D). The results indicated JNK/MAPK might exhibit crosstalk with the NF-κB pathway on the level of IκB. What’s more, CETSA showed that rhein (20 μM) could significantly protect IκB from temperature-dependent denaturation, suggesting that rhein had a direct interaction with IκB (Fig. 5E).
Effect of rhein on cardiac function in rats having DCM
Echocardiography is an essential tool for evaluating structural and functional alterations in the heart and has definite advantages in the early detection and diagnosis of diabetic cardiomyopathy. As illustrated in Fig. 6 and Table 1, the LVIDd and LVIDs were significantly enlarged in the model group compared to the control group (P < 0.001). Moreover, the ejection fraction (EF) and fractional shortening (FS) were also considerably reduced in the model rats compared to the control rats (P < 0.001), along with the enlargement of the left ventricular internal diameter. Theses indicated impaired left ventricular systolic function and severe left ventricular dilatation among the model rats. The above parameters (LVIDd, LVIDs, EF, FS) were differentially enhanced after 12 weeks of rhein or engramine gavage. What’s more, compared with the model group, rhein (200 mg/kg) group and EMPA (30 mg/kg) group showed significant differences (P < 0.05 or P < 0.01). Additionally, no apparent differences were found between the LVPWd and LVPWs of rats among the groups.

Representative M-mode echocardiography in each group after continuous gavage for 12 weeks. A The representative echocardiography in control group; B The representative echocardiography in model group; C The representative echocardiography in low-dose rhein (100 mg/kg) group; D The representative echocardiography in high-dose rhein (200 mg/kg) group; E The representative echocardiography in EMPA (30 mg/kg) group
Effects of rhein on biochemical indicators and myocardial histopathology among rats with DCM
After 12 weeks of being gavaged, the plasma of the rats was analyzed to determine the production of inflammatory factors (TNF-α and IL-1β). The results further indicated that the production of TNF-α and IL-1β in the representative sample was higher than in the group that served as the control, while rhein could dose-dependently decrease the inflammatory factor levels. In particular, there was a substantial disparity between the model group and the high-dose rhein group (200 mg/kg) (P < 0.001). The positive drug EMPA (30 mg/kg) failed to significantly reduce IL-1β levels compared to the model group (Fig. 7A, B). The AGEs content was further examined in the myocardium of rats in each group, which illustrated that the model group had content that was over three times higher than that of the control group (P < 0.001). In contrast, the amount of AGEs that were found in rats that were given a high dose of rhein (200 mg/kg) was 1.66 ± 0.75 µg/mg protein, comparable to that of the control group, but with a notable difference (P < 0.05). Moreover, the impact of high-dose rhein (200 mg/kg) in reducing AGEs was similar to that of EMPA (Fig. 7C). In addition, we detected the content of iNOS and COX-2 protein in rat myocardial samples. The western blot results (Fig. 7D) showed that compared with control group, iNOS and COX-2 in the myocardium of model group increased by 1.35 times and 1.62 times respectively, and the differences were significant (P < 0.05). After high-dose rhein (200 mg/kg) treatment, the expression of these two proteins was significantly reduced (P < 0.05 or P < 0.01). Compared with positive drugs EMPA, the treatment of high-dose rhein (200 mg/kg) was equivalent.

Effects of rhein on biochemical indexes and myocardial histopathology in DCM model rats. All the groups were gavaged for 12 weeks. Rhein groups were gavaged using 100 mg/kg and 200 mg/kg rhein daily. The EMPA group was set of related with 30 mg/kg EMPA per day, while the control and model groups were gavaged with 1% sodium carboxymethylcellulose per day. A Effect of rhein on plasma TNF-α levels in model rats; B Effect of rhein on plasma IL-1β levels in model rats; C Effect of rhein on AGEs content in the myocardium of the model rats; D Effect of rhein on iNOS and COX-2 expression in the myocardium of the model rats; E Effect of rhein on the histopathology and apoptosis of the myocardium in model rats. HE and Masson staining magnified at ×40, scale bar 20 μm; and TUNEL staining magnified at ×10, scale bar 100 μm. n = 5. ###P < 0.001 compared to the control group; *P < 0.05 compared to the model group
HE staining (Fig. 7E) showed that the myocardial tissues of the control rats were uniformly stained. In addition, there was no significant inflammatory cell infiltration between the myocardium and the myocardial fibers. The rats in the DCM model had discombobulated and loose myocardium, enlarged cardiomyocytes, and elevated fibroblasts with infiltrating inflammatory cells. After 12 weeks of rhein (100–200 mg/kg) intervention, myocardial cell swelling was significantly reduced. Additionally, myocardial fiber breakage has been improved and aligned in the same direction, as well as inflammatory cell infiltration was lowered, particularly in the 200 mg/kg rhein group. The EMPA group also significantly enhanced the myocardial histopathological changes in the model rats. Staining with Masson (Fig. 7E) demonstrated that the myocardial fibers in the control group were properly arranged. Only a small number of evenly distributed blue collagen fibers were visible between the myocardium. In way of comparison, in the myocardial tissue of the model rats, a significant number of blue collagen fibers were placed between the myocardium, where they were unevenly distributed, disorganized, and wrapped around the myocardial cells. This indicated that myocardial fibers were proliferating in the model rats. After 12 weeks of rhein administration, blue collagen fibril deposits inside the myocardial tissue were markedly reduced, and the effect of rhein treatment at 200 mg/kg was approximately similar to that of 30 mg/kg of EMPA. These findings suggested that rhein could successfully inhibit myocardial tissue pathology in DCM-model rats, enhance the inflammatory environment of myocardial tissue, keep improving myocardial extracellular matrix remodeling, and reduce myocardial tissue fibrosis. Additionally, TUNEL staining was used to observe the apoptosis of cardiomyocytes. According to the consequence, it revealed that (Fig. 7E) in the myocardial tissues of the control group, there were no obvious TUNEL-positive cells, and only a small number of green fluorescent spots. In contrast, many TUNEL-positive cells (i.e., apoptotic cells) showing green fluorescence were observed in the myocardium of rats belonging to the model group. In comparison with the model group, the green fluorescence in rhein-treated myocardial tissues of rats progressively decreased. This indicated that rhein could suppress cardiomyocyte apoptosis.
Rhein had similar effect on TRAF6-NF/κB and JNK pathway in vivo
In vitro experiments had suggested that rhein could inhibit the TRAF6-NF/κB and JNK signaling pathway, we also detected this pathway in animal. Figure 8 showed that in rats treated with rhein for 12 weeks, the level of TRAF6, p-NF-κB and p-JNK isolated from of hearts were down-regulated, and there was a statistically signifcant diference between the group that received the high dose of rhein and the model (P < 0.001). This indicated that rhein had similar effect on TRAF6-NF/κB and JNK pathway in vivo.

Rhein had similar effect on TRAF6-NF/κB and JNK pathway in vivo. All the groups were gavaged for 12 weeks. Rhein groups were gavaged using 100 mg/kg and 200 mg/kg rhein daily. The EMPA group was set of related with 30 mg/kg EMPA per day, while the control and model groups were gavaged with 1% sodium carboxymethylcellulose per day. A Representative western blots of phosphorylated NF-κB (p65), TRAF6 isolated from the rats hearts of different groups. B Representative western blots of phosphorylated JNK isolated from the rats hearts of different groups. C Rhein down-regulated the protein expression of TRAF6, p-NF-κB and p-JNK in the heart. Data were expressed as the mean ± S.D.; n = 5; ###P < 0.001 compared to the control group; ***P < 0.001 compared to the model group
Discussion
DCM is a heart-specific disease associated with primary structural and functional damage to the heart due to disturbances in glucose metabolism. DCM is a common diabetes complication that leads to heart failure and death. Multiple factors complicate the pathogenesis of DCM. Numerous studies have shown that the inflammatory immune response is intimately associated with the pathological process of DCM. Autoantibodies, macrophages, and inflammatory factors are involved in the myocardial injury process in patients with T2DM. Moreover, chronic hyperglycemia and hyperlipidemia in T2DM patients could affect the aldose reduction pathway and the accumulation of AGEs. This triggers the immune system and produces an inflammatory response [20]. DCM patients suffer from a prolonged and persistent state of chronic inflammation, polarizing the monocytes in the cardiovascular periphery toward classically activated macrophages (e.g., M1-type macrophages) to engulf necrotic cardiomyocytes. M1-type macrophages could secrete large amounts of inflammatory factors around the myocardium, including TNF-α and IL-1β [21]. In turn, inflammatory factors secreted by macrophages exacerbate cardiomyocyte injury, leading to cardiomyocyte hypertrophy and apoptosis while boosting the development of myocardial interstitial fibrosis and inflammation progression. Recent studies have revealed that AGEs can mediate chronic inflammation through the TLR4-MD2-MYD88 pathway [7, 22, 23]. In our study, we explored whether AGEs combined with inflammatory factors secreted by macrophages (macrophage conditioned medium, CM) at the cellular level could cause cardiomyocyte injury through chronic inflammation regulated by NF-κB and MAPK pathways. Additionally, we studied the protective effect of rhein on cardiomyocytes.
In our research, we discovered that rhein could inhibit macrophage cell line (RAW264.7 cells) activation by concentration-dependently downregulating TNF-α, PGE2, and NO. We concluded that further study of rhein as an anti-inflammation drug candidate was supported. To characterize the possible mechanisms of rhein’s anti-inflammatory effect, we examined the well-known inflammatory pathway TLR4-TRAF6-NF-κB [24,25,26]. TRAF6 has a vital role in signal transduction not only for TNF receptor superfamily, but rather the IL-1 receptor/TLR superfamily owing to its special receptor-binding specificity [27]. Under conditions of AGEs/CM stimulation, TRAF6 is activated by signals from upstream and recruited protein mediators to form kinase complexes. These kinase complexes activate IKKβ and facilitate the phosphorylation and ensuing degradation of IκB to free NF-κB [28,29,30]. Subsequently, dissociated NF-κB leaves the cytoplasm and goes into the nucleus and binds promoter sites to activate gene expressions. In our study, we found rhein significantly downregulated TRAF6 expression to suppress the phosphorylation of IKKβ, followed by inhibiting p-IκB and p-NF-κB activation. In addition, the results showed that rhein decreased TRAF6 protein expression in a concentration-dependent manner, but had no effect on TRAF6 mRNA expression. So we hypothesized that rhein might regulate TRAF6 expression through post-translational modifications. Since the ubiquitin-proteasomal pathway and autophagy-lysosomal pathway are the two highly effective protein degradation pathways, and central to the regulation of signaling proteins in many cellular processes [31], we hypothesized that rhein may regulate TRAF6 degradation through the ubiquitin–proteasome or autophagy-lysosomal pathways. Which pathway is involved requires further study. According to previous reports, TRAF6 could also activate MAP kinase kinase 6 (MKK6) through phosphorylation. Phosphorylated MKK6 could activate the MAPKs family (JNK, p38, and ERK1/2), stimulate activation of activating transcription factor 1 (ATF-1), and regulate cell proliferation, differentiation, transformation, and death [32,33,34]. In the latest examination, we discovered that rhein might also inhibit the phosphorylation of JNK/p38 MAPKs in H9C2 cells induced by AGEs/CM, while not affecting ERK MAPK, which was responsive to growth factor stimulation. JNK/p38 MAPK plays crucial roles in the regulation of specific cell types' survival, proliferation, differentiation, and migration. Such processes are essential for tissue homeostasis and inflammatory response [35,36,37,38,39]. According to our research, the anti-inflammatory effect of rhein is mediated by JNK MAPK pathways. Recent research demonstrates that the JNK pathway regulates cytochrome C release as part of the mitochondrial death signaling pathway [40]. Biochemical characterization and genetic studies revealed that imbalances of JNK MAPK activity were implicated in immune disorders and cardiovascular diseases [41]. Therefore, JNK MAPK may be the critical target of rhein in inflammatory-mediated diabetic cardiomyopathy disease.
Meanwhile, we found NF-κB and JNK MAPK pathways were both affected by rhein in AGEs/CM-induced H9C2 cells, which suggested a connection between the two pathways. Recent research suggests that inhibiting JNK activation reduces the degradation of IκB-α, and yet had no influence on IKK phosphorylation in TNF-α induced HepG2 cells [42]. In our study, we found downregulation of JNK MAPK activity could inhibit phosphorylation levels of NF-κB and IκB without affecting IKKβ changes. We verified this phenomenon using pathway inhibitors, investigating phosphorylated protein levels. Our results supported the conclusion of the previous study. In a word, we found rhein could attenuate AGEs/CM-induced myocardial inflammation through inhibition of the JNK MAPK as wellas NF-κB pathways, and their crosstalk.
The combination of a high sugar/fat diet and low-dose STZ intraperitoneal injection is ideal for constructing a T2DM rat model. The previous study reported that STZ-induced changes in cardiomyocyte ultrastructure among diabetic rats at 8–12 weeks after the first STZ injection, and changes in cardiac function at 6–14 weeks [43]. In this research, rats were fed a high-sugar and high-fat diet for six weeks, followed by an intraperitoneal injection of 30 mg/kg STZ at a low dosage. Twelve weeks of a high-sugar and fat diet were administered to the T2DM rats. After 12 weeks of treatment, the results showed that the LVIDd and LVIDs were significantly enlarged in the DCM model group, the EF and FS were dramatically reduced, and myocardial tissue was more fibrotic and infiltrated with inflammatory cells.The reproducibility of this method in preparing a rat model of diabetic cardiomyopathy was confirmed by our results, consistent with previous study. However, the LVPWd and LVPWs in the DCM model group had no significant differences compared to the control group. It could be associated with the short modeling time of our experiment and the small number of rats counted within each group. What’s more, both 200 mg/kg rhein and 30 mg/kg EMPA could significantly ameliorate the EF and pathological ventricular dilatation, improve pathological changes in myocardial tissue, depresse inflammatory infiltration, inhibit myocardial cell apoptosis, and reduce the content of AGEs within myocardial tissues of DCM model rats. And we found that rhein had similar effect on TRAF6-NF/κB and JNK pathway in vivo. This suggested that rhein could exert cardioprotective effects through anti-inflammation, which is consistent with our results in vitro described above. In the next step, we will further conduct in-depth studies in vivo to decipher the relationship between TLR4-TRAF6-NF-κB/MAPKs pathway and the multiple pathological alterations in DCM and the interventional role of rhein.
Conclusion
In summary, the results of our study provided evidence that rhein exhibited excellent anti-inflammatory effects in AGEs/CM-induced H9C2 cells and marked cardioprotective effects in DCM rats, including improving the pathological changes of cardiac structure and function, and inhibiting inflammatory cell infiltration. In addition, we provided evidence that JNK MAPK exhibited crosstalk with the NF-κB pathway in AGEs/CM-induced H9C2 cells by targeting IκB. The mechanism of rhein anti-inflammation effect might involve the negative regulation of the TRAF6/NF-κB and JNK MAPK pathways, and crosstalk between the two pathways (Fig. 9).

Mechanism Diagram. Rhein alleviated H9C2 cells inflammation injury stimulated by AGEs/macrophage conditioned medium. The protective mechanism of rhein may involve the negative regulation of TRAF6/NF-κB, JNK MAPK pathways and the crosstalk between the two pathways
Data availability statement
All data are available from the corresponding authors upon request.
Abbreviations
- AG:
-
Aminoguanidine
- AGEs:
-
Advanced glycation end products
- CM:
-
Macrophage conditioned medium
- COX-2:
-
Cyclooxygenase 2
- DCM:
-
Diabetic cardiomyopathy
- EF:
-
Ejection fraction
- EMPA:
-
Empagliflozin
- ERK:
-
Extra cellular regulated protein kinase
- FS:
-
Fractional shortening
- IKK:
-
IκB kinase
- iNOS:
-
Inducible nitric oxide synthase
- IκB:
-
Inhibitor of NF-κB
- LVIDd:
-
Left ventricular end-diastolic internal diameter
- LVIDs:
-
Left ventricular end-systolic internal diameters
- LVPWd:
-
Left ventricular posterior wall thickness during diastole
- LVPWs:
-
Left ventricular posterior wall thickness during systole
- JNK:
-
C-jun n-terminal kinase
- MAPK:
-
Mitogen-activated protein kinase
- MyD88:
-
Myeloid differentiation primary response gene 88
- MMP:
-
Mitochondrial membrane potential
- NF-κB:
-
Nuclear factor-κB
- PGE2 :
-
Prostaglandin E2
- STZ:
-
Streptozotocin
- T2DM:
-
Type 2 diabetes mellitus
- TLR4:
-
Toll-like receptors 4
- TNF-ɑ:
-
Tumor necrosis factor ɑ
- TRAF6:
-
The tumor necrosis factor receptor-associated factor 6
References
Kumar S, Mittal A, Babu D, Mittal A (2021) Herbal medicines for diabetes management and its secondary complications. Curr Diabetes Rev 17(4):437–456. https://doi.org/10.2174/1573399816666201103143225
Sarraju A, Spencer-Bonilla G, Rodriguez F, Mahaffey KW (2021) Canagliflozin and cardiovascular outcomes in Type 2 diabetes. Future Cardiol 17(1):39–48. https://doi.org/10.2217/fca-2020-0029
Shah A, Isath A, Aronow WS (2022) Cardiovascular complications of diabetes. Expert Rev Endocrinol Metab 17(5):383–388. https://doi.org/10.1080/17446651.2022.2099838
Murtaza G, Virk H, Khalid M et al (2019) Diabetic cardiomyopathy—a comprehensive updated review. Prog Cardiovasc Dis 62(4):315–326. https://doi.org/10.1016/j.pcad.2019.03.003
Sun Y, Ding SZ (2021) NLRP3 inflammasome in diabetic cardiomyopathy and exercise intervention. Int J Mol Sci 22(24):13228. https://doi.org/10.3390/ijms222413228
Bodiga VL, Eda SR, Bodiga S (2014) Advanced glycation end products: role in pathology of diabetic cardiomyopathy. Heart Fail Rev 19(1):49–63. https://doi.org/10.1007/s10741-013-9374-y
Wang Y, Luo W, Han J et al (2020) MD2 activation by direct AGE interaction drives inflammatory diabetic cardiomyopathy. Nat Commun 11(1):2148. https://doi.org/10.1038/s41467-020-15978-3
Sun H, Luo GW, Chen DH, Xiang Z (2016) A comprehensive and system review for the pharmacological mechanism of action of rhein, an active anthraquinone ingredient. Front Pharmacol 7:247. https://doi.org/10.3389/fphar.2016.00247
Lai WW, Yang JS, Lai KC et al (2009) Rhein induced apoptosis through the endoplasmic reticulum stress, caspase- and mitochondria-dependent pathways in SCC-4 human tongue squamous cancer cells. In Vivo 23(2):309–316. https://doi.org/10.1089/hum.2008.138
Malaguti C, Vilella CA, Vieira KP et al (2008) Diacerhein downregulate proinflammatory cytokines expression and decrease the autoimmune diabetes frequency in nonobese diabetic (NOD) mice. Int Immunopharmacol 8(6):782–791. https://doi.org/10.1016/j.intimp.2008.01.020
Pei R, Jiang Y, Lei G et al (2021) Rhein derivatives, a promising pivot? Mini Rev Med Chem 21(5):554–575. https://doi.org/10.2174/1389557520666201109120855
Li Q, Su J, Jin SJ et al (2018) Argirein alleviates vascular endothelial insulin resistance through suppressing the activation of Nox4-dependent O2- production in diabetic rats. Free Radic Biol Med 121:169–179. https://doi.org/10.1016/j.freeradbiomed.2018.04.573
Wen Q, Miao J, Lau N et al (2020) Rhein attenuates lipopolysaccharide-primed inflammation through NF-κB inhibition in RAW264.7 cells: targeting the PPAR-γ signal pathway. Can J Physiol Pharmacol 98(6):357–365. https://doi.org/10.1139/cjpp-2019-0389
Lafuse WP, Wozniak DJ, Rajaram M (2020) Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair. Cells 10(1):51. https://doi.org/10.3390/cells10010051
Lentsch AB, Ward PA (1999) Activation and regulation of NFkappaB during acute inflammation. Clin Chem Lab Med 37(3):205–208. https://doi.org/10.1515/CCLM.1999.038
Singh S, Singh TG (2020) Role of nuclear factor kappa B (NF-κB) signalling in neurodegenerative diseases: an mechanistic approach. Curr Neuropharmacol 18(10):918–935. https://doi.org/10.2174/1570159X18666200207120949
Prantner D, Nallar S, Vogel SN (2020) The role of RAGE in host pathology and crosstalk between RAGE and TLR4 in innate immune signal transduction pathways. FASEB J 34(12):15659–15674. https://doi.org/10.1096/fj.202002136R
Brown J, Wang H, Hajishengallis GN, Martin M (2011) TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res 90(4):417–427. https://doi.org/10.1177/0022034510381264
Nelson PT, Soma LA, Lavi E (2002) Microglia in diseases of the central nervous system. Ann Med 34(7–8):491–500. https://doi.org/10.1080/078538902321117698
Wilson AJ, Gill EK, Abudalo RA et al (2018) Reactive oxygen species signalling in the diabetic heart: emerging prospect for therapeutic targeting. Heart 104(4):293–299. https://doi.org/10.1136/heartjnl-2017-311448
Varga ZV, Giricz Z, Liaudet L et al (2015) Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy. Biochim Biophys Acta 1852 2:232–242. https://doi.org/10.1016/j.bbadis.2014.06.030
Sumneang N, Apaijai N, Chattipakorn SC, Chattipakorn N (2021) Myeloid differentiation factor 2 in the heart: bench to bedside evidence for potential clinical benefits? Pharmacol Res 163:105239. https://doi.org/10.1016/j.phrs.2020.105239
Liu ZW, Wang JK, Qiu C et al (2015) Matrine pretreatment improves cardiac function in rats with diabetic cardiomyopathy via suppressing ROS/TLR-4 signaling pathway. Acta Pharmacol Sin 36(3):323–333. https://doi.org/10.1038/aps.2014.127
Youssef ME, Abdelrazek HM, Moustafa YM (2021) Cardioprotective role of GTS-21 by attenuating the TLR4/NF-κB pathway in streptozotocin-induced diabetic cardiomyopathy in rats. Naunyn Schmiedebergs Arch Pharmacol 394(1):11–31. https://doi.org/10.1007/s00210-020-01957-4
Feng B, Chen S, Gordon AD, Chakrabarti S (2017) miR-146a mediates inflammatory changes and fibrosis in the heart in diabetes. J Mol Cell Cardiol 105:70–76. https://doi.org/10.1016/j.yjmcc.2017.03.002
Shi H, Zhou P, Ni YQ, Wang SS et al (2021) In vivo and in vitro studies of Danzhi Jiangtang capsules against diabetic cardiomyopathy via TLR4/MyD88/NF-κB signaling pathway. Saudi Pharm J 29(12):1432–1440. https://doi.org/10.1016/j.jsps.2021.11.004
Kobayashi T, Walsh MC, Choi Y (2004) The role of TRAF6 in signal transduction and the immune response. Microbes Infect 6(14):1333–1338. https://doi.org/10.1016/j.micinf.2004.09.001
Chen X, Yu M, Xu W et al (2021) Rutin inhibited the advanced glycation end products-stimulated inflammatory response and extra-cellular matrix degeneration via targeting TRAF-6 and BCL-2 proteins in mouse model of osteoarthritis. Aging 13(18):22134–22147. https://doi.org/10.18632/aging.203470
Bonizzi G, Karin M (2004) The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 25(6):280–288. https://doi.org/10.1016/j.it.2004.03.008
Hayden MS, Ghosh S (2008) Shared principles in NF-kappaB signaling. Cell 132(3):344–362. https://doi.org/10.1016/j.cell.2008.01.020
Heintz L, Meyer-Schwesinger C (2021) The intertwining of autophagy and the ubiquitin proteasome system in podocyte (patho)physiology. Cell Physiol Biochem 55(S4):68–95. https://doi.org/10.33594/000000432
Wu Q, Feng Y, Ouyang Y et al (2021) Inhibition of advanced glycation endproducts formation by lotus seedpod oligomeric procyanidins through RAGE-MAPK signaling and NF-κB activation in high-AGEs-diet mice. Food Chem Toxicol 156:112481. https://doi.org/10.1016/j.fct.2021.112481
Zhu P, Ren M, Yang C et al (2012) Involvement of RAGE, MAPK and NF-κB pathways in AGEs-induced MMP-9 activation in HaCaT keratinocytes. Exp Dermatol 21(2):123–129. https://doi.org/10.1111/j.1600-0625.2011.01408.x
Nonaka K, Kajiura Y, Bando M et al (2018) Advanced glycation end-products increase IL-6 and ICAM-1 expression via RAGE, MAPK and NF-κB pathways in human gingival fibroblasts. J Periodontal Res 53(3):334–344. https://doi.org/10.1111/jre.12518
Jeong YH, Kim Y, Song H et al (2014) Anti-inflammatory effects of α-galactosylceramide analogs in activated microglia: involvement of the p38 MAPK signaling pathway. PLoS ONE 9(2):e87030. https://doi.org/10.1371/journal.pone.0087030
Lee KM, Bang JH, Han JS et al (2013) Cardiotonic pill attenuates white matter and hippocampal damage via inhibiting microglial activation and downregulating ERK and p38 MAPK signaling in chronic cerebral hypoperfused rat. BMC Complement Altern Med 13:334. https://doi.org/10.1186/1472-6882-13-334
Rincón M, Davis RJ (2009) Regulation of the immune response by stress-activated protein kinases. Immunol Rev 228(1):212–224. https://doi.org/10.1111/j.1600-065X.2008.00744.x
Weston CR, Davis RJ (2002) The JNK signal transduction pathway. Curr Opin Genet Dev 12(1):14–21. https://doi.org/10.1016/s0959-437x(01)00258-1
Zhao H, Cheng L, Liu Y et al (2014) Mechanisms of anti-inflammatory property of conserved dopamine neurotrophic factor: inhibition of JNK signaling in lipopolysaccharide-induced microglia. J Mol Neurosci 52(2):186–192. https://doi.org/10.1007/s12031-013-0120-7
Yang Z, Lv J, Lu X et al (2018) Emulsified isoflurane induces release of cytochrome C in human neuroblastoma SHSY-5Y cells via JNK (c-Jun N-terminal kinases) signaling pathway. Neurotoxicol Teratol 65:19–25. https://doi.org/10.1016/j.ntt.2017.12.001
Zuo G, Ren X, Qian X et al (2019) Inhibition of JNK and p38 MAPK-mediated inflammation and apoptosis by ivabradine improves cardiac function in streptozotocin-induced diabetic cardiomyopathy. J Cell Physiol 234(2):1925–1936. https://doi.org/10.1002/jcp.27070
Ruan J, Qi Z, Shen L et al (2015) Crosstalk between JNK and NF-κB signaling pathways via HSP27 phosphorylation in HepG2 cells. Biochem Biophys Res Commun 456(1):122–128. https://doi.org/10.1016/j.bbrc.2014.11.045
Thompson EW (1988) Structural manifestations of diabetic cardiomyopathy in the rat and its reversal by insulin treatment. Am J Anat 182(3):270–282. https://doi.org/10.1002/aja.1001820308
Acknowledgements
The support of the experimental platform was generously provided by the Lingnan Medical Research Center of Guangzhou University of Chinese Medicine, and we are extremely thankful for this.
Funding
This study was funded by the National Natural Science Foundation of China for Young People (Nos. 82104621, 82204809) and Guangdong Basic and Applied Basic Research Foundation (No. 2021A1515220066).
Author information
Authors and Affiliations
Contributions
S-ML and S-YZ conceived and designed the experiments. S-YZ carried out the experiments and wrote the manuscript. H-HZ participated in animal experiments. B-H provided the regents. CS and W-JP helped revise the article. All authors have read and agreed to the published version of the manuscript. S-YZ is the first author of this article. Correspondence should be addressed to S-YZ and S-ML.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethics statement
The First Affiliated Hospital of Guangzhou University of Chinese Medicine's Experimental Animal Ethics Committee approved all of the experiments performed on animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
11418_2023_1741_MOESM1_ESM.tif
Supplementary file1 Fig. S1 RAW264.7 cells were treated with AGEs or rhein alone in a concentration or time-dependent manner. (A) RAW264.7 cells were treated with AGEs (from 0 to 350 μg/ml) for 8, 16, and 24 h respectively. NO production was measured. (B) RAW264.7 cells were treated with rhein (from 0 to 100 μM) for 24 h. cell viability was measured. N.S.: no significance. #P < 0.05, ###P < 0.001 compared to the control group (TIF 2847 KB)
11418_2023_1741_MOESM2_ESM.tif
Supplementary file2 Fig. S2 AGEs isn’t an ideal agent for inducing inflammatory injury in H9C2 cells. H9C2 cells were treated with AGEs (50–500 μg/ml) for 48 h, and the cell viability was then detected by MTT. Cell viabilities were similar between AGEs-treated H9C2 cells and DMEM-treated cells. (B–C) The releases of NO and LDH were detected by Griess and LDH Cytotoxicity Assay. ##P < 0.01 compared to the control group (TIF 4777 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhao, SY., Zhao, HH., Wang, BH. et al. Rhein alleviates advanced glycation end products (AGEs)-induced inflammatory injury of diabetic cardiomyopathy in vitro and in vivo models. J Nat Med 77, 898–915 (2023). https://doi.org/10.1007/s11418-023-01741-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-023-01741-7