
Abstract
Four new xanthones, named schomburgones C-F (1‒4), along with six known xanthones (5‒10) were isolated from the stems of Garcinia schomburgkiana. Their structures were determined by spectroscopic analysis especially 1D and 2D NMR spectroscopies. The isolated compounds were evaluated for their cytotoxicity against five human cancer cell lines. Furanoxanthones 4‒6 showed potent cytotoxicity against four cell lines (KB, HeLa S3, MCF-7 and Hep G2) with IC50 values in the range of 0.18‒9.95 µM.
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Garcinia schomburgkiana (Clusiaceae) is an edible plant distributed in Thailand. It has been traditionally used for the treatment of diabetes, cough, laxative, menstrual disturbances and expectorant [1]. Previous chemical and biological studies on chemical constituents of Garcinia showed the presence of xanthones, biphenyls, flavonoids, triterpenoids, depsidones, and phloroglucinols, some of which exhibited significant cytotoxicity [2,3,4]. Herein, we reported four new xanthones, named schomburgones C-F (1‒4), along with six known xanthones (5‒10) which were isolated from the stems of this plant. The structures of all isolated compounds were elucidated using spectroscopic methods especially 1D and 2D NMR spectroscopies and compared with their 1H and 13C NMR spectroscopic data from the literature. Previous researches revealed that xanthones from Garcinia displayed potent cytotoxicity [5]. Therefore, all the isolated compounds (1–10) were evaluated against human cancer cell lines, including KB and HeLa S3 cells. The tested compounds with IC50 values lower than 10 µM against these two cells were further evaluated against MCF-7, Hep G2, and HT-29 cells.
Experimental
General experimental procedures
1D and 2D NMR spectra were recorded on Bruker 400 AVANCE spectrometer. HRESIMS spectra were obtained using a Bruker MICROTOF model mass spectrometer. IR data was obtained using Nicolet 6700 FT-IR spectrometer using KBr discs. UV–visible absorption spectra were taken on UV-2550 UV–Vis spectrometer (Shimadzu, Kyoto, Japan). The optical rotation was determined using Jasco P-1010 Polarimeter.
Plant material
The stems of G. schomburgkiana were collected from Bang Ramat Road, Khwaeng Bang Ramat, Khet Taling Chan, Bangkok Thailand, in October 2019. The plant material was identified by Dr. Suttira Sedlak, a botanist at the Walai Rukhavej Botanical Research Institute, Mahasarakham University, and a specimen retained as a reference (Khumkratok no. 92-08).
Extraction and isolation
The air-dried stems of G. schomburgkiana (5.0 kg) were macerated with CH2Cl2 over a period of 5 days at room temperature (2 × 10 L). Removal of the solvent under reduced pressure provided CH2Cl2 crude extract (85.0 g) which was further separated by column chromatography over silica gel and eluted with a gradient of hexane–EtOAc (100% hexane, 80%, 60%, 40% and 20% hexane–EtOAc, each 5 L) to give ten fractions (A‒J). Fraction D (3.5 g) was purified by a Sephadex LH-20 column (250 g) with 80% CH2Cl2–MeOH (2 L) and further applied to a radial chromatography (chromatotron) with 90% hexane–EtOAc (200 mL) to afford compounds 1 (8.5 mg) and 2 (3.0 mg). Fraction F (1.2 g) was purified by a chromatotron with 80% hexane–EtOAc (200 mL) to obtain compounds 5 (7.4 mg), 6 (5.4 mg), and 8 (6.2 mg). Compounds 3 (8.8 mg), 4 (6.7 mg), and 10 (6.2 mg) were obtained from fraction H (3.5 g) by a Sephadex LH-20 column (250 g) with 80% CH2Cl2–MeOH (2 L) followed by a chromatotron with 70% hexane–EtOAc (200 mL). Finally, Fraction J (2.5 g) was applied to a Sephadex LH-20 column (250 g) using 80% CH2Cl2–MeOH (2 L) to provide compounds 7 (5.7 mg) and 9 (7.2 mg).
Schomburgone C (1) Yellow amorphous powder; UV (CHCl3) λmax (log ε): 336 (0.2), 266 (0.7), and 211 (0.8) nm.; IR νmax (KBr): 3386, 2915, 1648, and 1428 cm−1; for 1H (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) spectroscopic data, see Table 1; HRESIMS m/z 499.2077 [M + Na]+ (calcd. for C26H32O6Na, 499.2097).
Schomburgone D (2) Yellow amorphous powder; \({[a]}_{D}^{20}\)+35.5 (c 1.0, MeOH); UV (CHCl3) λmax (log ε): 327 (0.1), 285 (0.2), and 217 (0.5) nm.; IR νmax (KBr): 3396, 2919, 1646, and 1467 cm−1; for 1H (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) spectroscopic data, see Table 1; HRESIMS m/z 483.1767 [M + Na]+ (calcd. for C28H28O6Na, 483.1784).
Schomburgone E (3) Yellow amorphous powder; UV (MeOH) λmax (log ε): 359 (0.2), 274 (0.5), and 232 (0.5) nm.; IR νmax (KBr): 3442, 2965, 1648, and 1494 cm−1; for 1H (400 MHz, Acetone d6) and 13C NMR (100 MHz, Acetone-d6) spectroscopic data, see Table 1; HRESIMS m/z 369.0955 [M + Na]+ (calcd. for C18H18O7Na, 369.0950).
Schomburgone F (4) Yellow amorphous powder; \({[a]}_{D}^{20}\)‒65.5 (c 1.0, MeOH); UV (MeOH) λmax (log ε): 336 (0.2) and 258 (0.5) nm.; IR νmax (KBr): 3423, 2923, 1652, and 1579 cm−1; for 1H (400 MHz, Methanol-d4) and 13C NMR (100 MHz, Methanol-d4) spectroscopic data, see Table 1; HRESIMS m/z 437.1582 [M + Na]+ (calcd. for C23H26O7Na, 437.1576).
Cytotoxicity assay
All isolated compounds (1–10) were subjected to cytotoxic evaluation against KB (human epidermoid carcinoma), HeLa S3 (human cervical carcinoma), HT-29 (human colon adenocarcinoma), MCF-7 (human breast adenocarcinoma) and Hep G2 (human liver carcinoma) cell lines employing the MTT colorimetric method by incubating cells for 72 h, as described previously [6]. The results are expressed as the mean values of three independent experiments. Doxorubicin was used as the positive control.
Results and discussion
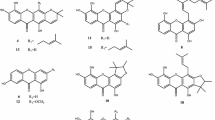
Phytochemical investigation of CH2Cl2 crude extract from the stems of G. schomburgkiana led to the isolation of four new xanthones, schomburgone C-F (1‒4) together with six known xanthones (5‒10) (Fig. 1), formoxanthone C (5) [7], 2-deprenylrheediaxanthone B (6) [8], cycloderivativexanthone (7) [9], toxyloxanthone B (8) [10], 1,3,5,6-tetrahydroxyxanthone (9) [11], 1,5,6-trihydroxy-3-methoxyxanthone (10) [12]. The structures of the known compounds were determined and confirmed by comparison of their 1H and 13C NMR spectroscopic data with those previously published data.
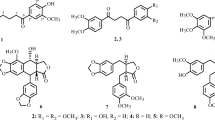
Schomburgone C (1) was obtained as a yellow amorphous powder. Its molecular formula was determined as C29H32O6 by the positive HRESIMS measurement through the ion peak at m/z 499.2077 [M + Na]+ (calcd. for C29H32O6Na, 499.2097). The UV spectrum displayed absorption bands at λmax 336, 266 and 211 nm. The IR spectrum showed absorption bands for a phenolic hydroxyl and a hydrogen-bonded carbonyl groups at 3386 and 1648 cm−1. The 1H NMR spectrum showed the signals of a hydrogen-bonded hydroxyl proton, two aromatic protons, a hydroxyl proton, and a methoxy proton which appeared as five singlets at δH 13.39 (1H, s, OH-1), 6.86 (1H, s, H-5), 6.34 (1H, s, H-2), 6.25 (1H, s, OH-6), and 3.90 (3H, s, OCH3-3), respectively. The signals at δH 8.03 (1H, d, J = 10.2 Hz, H-1″), 5.83 (1H, d, J = 10.2 Hz, H-2″), and 1.50 (6H, s, H-4″ and H-5″) in the spectrum were indicated of a pyran ring. In addition, a geranyl group showed signals at δH 5.19 (1H, t, J = 6.8 Hz, H-2′), 5.04 (1H, t, J = 6.8 Hz, H-6'), 3.45 (2H, d, J = 7.1 Hz, H-1′), 2.04 (2H, m, H-5′), 1.97 (2H, m, H-4′), 1.85 (3H, s, H-10′), 1.60 (3H, s, H-9′), and 1.54 (3H, s, H-8′). The 1H and 13C NMR spectroscopic data of 1 (Table 1) were shown to be similar to those of the known xanthone, virgataxanthone B [12], except for the hydroxyl group at C-3 was replaced by a methoxy group in 1. In the HMBC correlations (Fig. 2), the methoxy proton at δH 3.90 showed a cross-peak with δC 163.8 (C-3), an aromatic proton at δH 6.34 showed cross-peak with δC 107.6 (C-4) and 104.2 (C-9a), and a hydrogen-bonded hydroxyl proton at δH 13.39 showed cross-peaks with δC 162.3 (C-1), 104.2 (C-9a), and 94.4 (C-2). In addition, a methylene proton at δH 3.45 showed cross-peaks with δC 163.8 (C-3), 154.1 (C-4a), and 135.5 (C-3'), indicated that a geranyl group was attached to C-4. Thus, the complete assignment of schomburgone C was determined as 1.

Chemical structures of 1‒10
Schomburgone D (2) was obtained as a yellow amorphous powder and optically active \({[a]}_{D}^{20}\)+35.5 (c 1.0, MeOH). Its molecular formula was determined as C28H28O6 by the positive HRESIMS measurement through the ion peak at m/z 483.1767 [M + Na]+ (calcd. for C28H28O6Na, 483.1784). The UV, IR, 1H and 13C NMR data of 2 (Table 1) were closely related to those of 1. The major difference was the cyclization of geranyl group at C-4 to C-3 by ether linkage to form pyran ring in 2. the 1H NMR signals showed three methine protons at δH 6.81 (1H, d, J = 10.2 Hz, H-1′), 5.52 (1H, d, J = 10.2 Hz, H-2′), and 5.09 (1H, t, J = 6.3 Hz, H-6′), two methylene protons at δH 2.10 (2H, m, H-5′) and 1.80 (2H, m, H-4′), and three methyl protons at δH 1.66 (3H, s, H-9′), 1.57 (3H, s, H-8′), and 1.44 (3H, s, H-10′). The location of a pyran ring was deduced by HMBC correlations (Fig. 2) of methine protons at H-1′ to C-3 (δC 160.3), C-4a (δC 151.1), and C-3′ (δC 80.7), and H-2′ to C-4 (δC 100.4), C-3′ (δC 80.7), C-4′ (δC 41.8), and C-10′ (δC 27.2). From these data, the structure of schomburgone D was assigned as 2.
Schomburgone E (3) was obtained as a yellow amorphous powder. Its molecular formula was determined as C18H18O7 by the positive HRESIMS measurement through the ion peak at m/z 369.0955 [M + Na]+ (calcd. for C18H18O7Na, 369.0950). The UV spectrum displayed absorption bands at λmax 359, 274 and 232 nm. The IR spectrum exhibited the signals of phenolic hydroxyl groups and a hydrogen-bonded carbonyl group at 3442 and 1648 cm−1. The 1H NMR spectrum showed the presence of three aromatic proton signals at δH 7.60 (1H, d, J = 8.7 Hz, H-8), 6.96 (1H, d, J = 8.7 Hz, H-7), and 6.30 (1H, s, H-2), two methylene proton signals at δH 2.99 (2H, m, H-1′) and 1.80 (2H, m, H-2′), and two methyl proton signals at δH 1.33 (6H, s, H-4′ and H-5'). The 1D NMR data (Table 1) were closely to those of the known xanthone, 1,5,6-trihydroxy-3-methoxy-4-(3-hydroxyl-3-methylbutyl)xanthone [14], except for the methoxy group at C-3 was replaced by a hydroxyl group. In the HMBC correlations of 3 (Fig. 2), the aromatic proton at δH 6.30 showed cross-peaks with δC 163.4 (C-1) and 162.6 (C-3), the methylene proton at δH 2.99 showed cross-peaks with δC 162.6 (C-3), 156.2 (C-4a), and 72.2 (C-3'), indicated that a 3-hydroxyl-3-methylbutyl group was located at C-4. Accordingly, the structure of schomburgone E was determined as 3.
Schomburgone F (4) was obtained as yellow amorphous powder and optically active \({[a]}_{D}^{20}\)‒65.5 (c 1.0, MeOH). Its molecular formula was deduced as C23H26O7 by the positive HRESIMS measurement through the ion peak at m/z 437.1582 [M + Na]+ (calcd. for C23H26O7Na, 437.1576). The UV spectrum displayed absorption bands at λmax 336 and 258 nm. The IR spectrum showed O–H and C–O stretching bands at 3423 and 1652 cm−1, respectively. The 1H NMR spectrum showed the presence of two aromatic proton signals at δH 7.56 (1H, d, J = 8.4 Hz, H-8) and 6.86 (1H, d, J = 8.4 Hz, H-7), two methylene proton signals at δH 2.63 (2H, dd, J = 5.1 and 9.9 Hz, H-1′) and 1.68 (2H, t, J = 8.6 Hz, H-2′), a vinylic proton signal at δH 4.51 (1H, q, J = 6.5 Hz, H-2″), and five methyl proton signals at δH 1.61 (3H, s, H-5″), 1.40 (3H, d, J = 6.5 Hz, H-3″), 1.32 (3H, s, H-4″), and 1.25 (6H, s, H-4′ and H-5′). The 1H and 13C NMR data (Table 1) were nearly identical to those of the known xanthone, formoxanthone C (5) [7] except for the prenyl group at C-3 was hydrated to be a 3-hydroxyl-3-methylbutyl group in 4. In the HMBC correlations of 4 (Fig. 2), the methylene proton at δH 2.63 showed cross-peaks with δC 165.5 (C-1), 162.0 (C-3), and 71.6 (C-3') indicated that the 3-hydroxyl-3-methylbutyl unit was located at C-2. The absolute configuration at C-2″ was assigned as S based on the negative value of optical rotation and comparison with reference [7]. Thus, the structure of schomburgone F was defined as 4.
The cytotoxicity of all isolated compounds against five human cancer cell lines (KB, HeLa S3, HT-29, MCF-7 and Hep G2) were shown in Table 2. Furanoxanthones 4‒6 showed potent cytotoxicity against four cell lines including KB, HeLa S3, MCF-7, and Hep G2 with IC50 values in the range of 0.18‒9.95 µM. The SAR study (Fig. 1 and Table 2) suggest that the presence of the ortho hydroxy groups at C-5 and C-6, and the trimethylfuran ring at C-3 and C-4 might improve the cytotoxicity as inferred from the comparison with their cytotoxicity of compounds 1‒10.

Key HMBC (arrow curves) and COSY (bold lines) correlations of 1–4
References
Mungmee C, Sitthigool S, Suttisri R, Buakeaw A (2012) Xanthones and biphenyls from Garcinia schomburgkiana wood and their cytotoxicity. Thai J Pharm Sci 36:6–9
Kaennakam S, Mudsing K, Rassamee K, Siripong P, Tip-Pyang S (2019) Two new xanthones and cytotoxicity from the bark of Garcinia schomburgkiana. J Nat Med 73:257–261
Sukandar ER, Siripong P, Khumkratok S, Tip-pyang S (2016) New depsidones and xanthone from the roots of Garcinia schomburgkiana. Fitoterapia 111:73–77
Sukandar ER, Kaennakam S, Aree T, Nöst X, Rassamee K, Bauer R (2020) Picrorhizones A-H, polyprenylated benzoylphloroglucinols from the stem bark of Garcinia picrorhiza. J Nat Prod 83:2102–2111
Vo HT, Nguyen NTT, Nguyen HT, Do KQ, Connolly JD, Maas G (2012) Cytotoxic tetraoxygenated xanthones from the bark of Garcinia schomburgkiana. Phytochemistry Lett 5:553–557
Kongkathip N, Kongkathip B, Siripong P, Sangma C, Luangkamin S, Niyomdecha M (2003) Potent antitumor activity of synthetic 1,2-naphthoquinones and 1,4-naphthoquinones. Bioorg Med Chem 11:3179–3191
Boonsri S, Karalai C, Ponglimanont C, Kanjana-opas A, Chantrapromma K (2006) Antibacterial and cytotoxic xanthones from the roots of Cratoxylum formosum. Phytochemistry 67:723–727
Rath G, Potterat O, Mavi S, Hostettmann K (1996) Xanthones from Hypericum roeperanum. Phytochemistry 43:513–520
Monache FD, Botta B, Nicoletti M, de Barros Coêlho JS, de Andrade Lyra FD (1981) Three new xanthones and macluraxanthone from Rheedia benthamiana Pl. Triana (guttiferae). J Chem Soc Perkin 1:484–488
Sukandar ER, Kaennakam S, Rassamee K, Ersam T, Siripong P, Tip-pyang S (2019) Tetrandraxanthones A-I, prenylated and geranylated xanthones from the stem bark of Garcinia tetrandra. J Nat Prod 82:1312–1318
Sia GL, Bennett GJ, Harrison LJ, Sim KY (1995) Minor xanthones from the bark of Cratoxylum cochinchinense. Phytochemistry 38:1521–1528
Nielsen H, Arends P (1979) Xanthone constituents of Hypericum androsaemum. J Nat Prod 42:301–304
Merza J, Aumond MC, Rondeau D, Dumontet V, Le Ray AM, Séraphin D (2004) Prenylated xanthones and tocotrienols from Garcinia virgata. Phytochemistry 65:2915–2920
Shen J, Yang JS (2006) Two new xanthones from the stems of Garcinia cowa. Chem Pharm Bull 54:126–128
Acknowledgements
This research was funded by the Faculty of Applied Science, King Mongkut’s University of Technology North Bangkok (KMUTNB), Contract no. 641069. We also thank Ms. Suttira Khumkratok, Walai Rukhavej Botanical Research Institute, Mahasarakham University, Mahasarakham 44000, Thailand for the identification and deposition of the plant material.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kaennakam, S., Sukandar, E.R., Juntagoot, T. et al. Four new xanthones and their cytotoxicity from the stems of Garcinia schomburgkiana. J Nat Med 75, 871–876 (2021). https://doi.org/10.1007/s11418-021-01527-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-021-01527-9