
Abstract
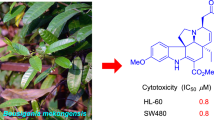
Two new bisindole alkaloids, leucophyllinines A (1) and B (2) consisting of eburnane and quebrachamine-type skeletons were isolated from the bark of Leuconotis eugeniifolia, and their structures were elucidated on the basis of spectroscopic data. Leucophyllinines A and B showed antiplasmodial activities against Plasmodium falciparum 3D7.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In our search for new bioactive principles from tropical plants in Malaysia and Indonesia [1,2,3,4], we have reported alkaloids from Hunteria zeylanica, Leuconotis griffithii, Cassia siamea, Dysoxylum densiflorum, and Tabernaemontana divaricata with potent biological activities [5,6,7,8,9,10,11,12,13,14,15,16]. Recently, an alkaloid isolated from Leuconotis griffithii, bisleuconothine A, was found to show cytostatic activity against A549 cells by autophagosome formation via inhibition of AKT–mTOR signaling [15, 17] and leucophyllidine from Leuconotis eugenifolius showed iNOS inhibitory activity and decreased iNOS protein expression dose-dependently [18]. Leuconotis eugeniifolia (Wall. ex G.Don) A.DC. is a member of the Apocynaceae family found in Sumatra, Borneo, and peninsular Malaysia. Medicinally, the latex of this plant was once used for the treatment of worm infections and yaws by applying it to infected skin [19]. The bark and leaves have been known to produce various skeletal alkaloids depending on the area where the plants were distributed. Our research on the bark of L. eugeniifolia resulted in the isolation of two new compounds, leucophyllinines A and B (1 and 2, Fig. 1) together with melaxillarinine [20]. In this paper, we describe the isolation and structure elucidation of 1 and 2, and their antiplasmodial activities against Plasmodium falciparum 3D7.

Structures of 1, 2 and melaxillarinine
Results and discussions
Structure elucidation of leucophyllinines A and B (1 and 2)
Leucophyllinine A (1), [α]16D-88 (c 1.0, MeOH), showed a molecular ion peak at m/z 593 (M)+ in the ESIMS, and the molecular formula C38H49N4O2 was established by HRESIMS [m/z 593.3846 (M)+, Δ − 1.0 mmu]. IR absorptions of 1 implied the presence of hydroxy and/or amine (3194 cm−1) functionality. Analysis of the 1H and 13C NMR data (Table 1) and the HSQC spectrum of 1 revealed the presence of four sp3 quaternary carbons, two sp3 methines, sixteen sp3 methylenes, two methyls, six sp2 methines, and eight sp2 quaternary carbons.
The planar structure of 1 was deduced from analyses of the 2D NMR data, including the 1H–1H COSY, HSQC, and HMBC spectra (Fig. 2). The 1H–1H COSY spectrum revealed the connectivity of eight partial structures a (C-9–C-12), b (C-5–C-6), c (C-3, C-14–C-15), d (C-16–C-17), e (C-18–C-19), f (C-5′–C-6′), g (C-3′, C-14′–C-15′), h (C-16′–C-17′), i (C-18′–C-19′) as shown in Fig. 2. These partial structures were classified into two units, A and B.

Selected 2D NMR correlations for leucophyllinine A (1)
In unit A, the connectivity of partial structure b and the indole ring (C-2, C-7–C-13, and N-1) was revealed by the HMBC correlations of H-5 and H-9 to C-7 (δC 104.5) and H-6a to C-2 (δC 134.0). HMBC correlations from H3-18 to C-20 (δC 35.5), H-19b to C-15 (δC 24.3), C-17 (δC 43.3) and C-21 (δC 60.2), and H-21 to C-2 and C-7, established the connections among C-15, C-17, C-19, and C-21 through C-20 and the connection between C-21 and C-2. HMBC cross-peaks of H-21 to C-3 (δC 44.6), H-5 to C-3 and C-21 suggested the linkage between C-3, C-5, and C-21 through a nitrogen atom. The 1H and 13C NMR chemical shifts of C-16 (δC 48.4; δH 5.60) were characteristic of an eburnane skeleton.
In unit B, the connectivity of partial structure f and the indoline ring (C-2′, C-7′–C-13′, and N-1′) was revealed by the HMBC correlations of H-5′ to C-2′ (δC 102.4), H-6′ to C-7′ (δC 89.7), and C-8′ (δC 121.2). HMBC cross-peaks of H-5′ to C-2′, C-3′ (δC 57.1), and C-21′ (δC 63.0), and H-21′a to C-2′ and C-3′ established the connections among C-2′, C-3′, C-5′, and C-21′ through N-4′. The presence of an ethyl group at C-20′ was indicated by the HMBC correlations of H3-18′ to C-20′ (δC 32.4), and H-19′ to C-15′ (δC 31.1), C-17′ (δC 31.0), and C-21′. The connectivity of C-2′ and C-16′ was deduced by HMBC correlation of H-16′ to C-7′. The 13C chemical shifts of C-11′ and C-7′ (δC 158.7 and δC 89.7) indicated that they were attached to an oxygen atom. These data implied unit B possessed a quebrachamine skeleton.
Finally, unlike melaxillarinine [20] with a C-16 and C-12′ linkage, the linkage between units A and B of 1 at C-16 and C-10′ was suggested by the HMBC correlations of H-16 to C-9′ and C-11′, and H-17 to C-10′. Thus, the planar structure of leucophyllinine A (1) was assigned as a new eburnane–quebrachamine bisindole alkaloid with a bridge between C-16 of an eburnane skeleton and C-10′ of a quebrachamine skeleton as shown in Fig. 2.
The relative configuration of each monoterpene indole unit in 1 was assigned by NOESY correlations as shown in computer-generated 3D drawing (Fig. 3). In unit A, the NOESY correlations of H-21/H-17a and H-21/H-19a suggested that both H-21 and the ethyl group (C-18–C-19) are β-oriented, and the NOESY correlation of H-15a/H-16 and the 3JH-17a/H-16 (11.3 Hz) suggested that H-16 is α-oriented. In unit B, H-3′a/H-21′a, H-14′b/H-16′a, and H-21′a/H-15′a suggested that the 1-azabicyclo[3.3.1]nonane ring took a chair–chair conformation. If the orientation of the ethyl group is arbitrarily defined as \( \alpha \), the NOESY correlation of H-6′a/H-21′b suggested that the hydroxy group at C-7′ is β-oriented and the configuration of C-2′ is as shown in Fig. 3.

Selected NOESY correlations for units A and B in leucophyllinine A (1)
Finally, the conformation of 1 through the C-16–C-10′ bond was investigated by using molecular mechanics calculations. The global minimum obtained through Monte Carlo conformational search, as shown in Fig. 4, is consistent with the observed NOESY correlations of H-21/H-9′. In addition, theoretical calculation of the total molecular energy as a function of the dihedral angle of N-1–C-16–C-10′–C-11′ showed two energy minima (at 140°, corresponding to the global minimum shown in Fig. 4, and at 305°). However, the conformational change among these conformers could not be observed by molecular dynamics calculation.

Selected NOESY correlations for leucophyllinine A (1)
Leucophyllinine B (2), [α]26D-111 (c 1.0, MeOH), showed a molecular ion peak at m/z 607 (M)+ in the ESIMS, and the molecular formula C39H51N4O2 was established by HRESIMS [m/z 607.4011 (M)+, Δ − 0.1 mmu], indicating the presence of an additional methyl group relative to 1. Analysis of the 1H and 13C NMR data (Table 1) showed that the main differences between 1 and 2 were the presence of an additional signal at δH 2.69, δC 52.9 in 2, which was deduced to be the methoxy group at C-7′ by the HMBC correlations of the methoxy protons to C-7′. Analysis of the NOESY spectral data also suggested that 2 had the same configuration as 1 for both units A and B. The methoxy group (δH 2.69) was shifted to a higher magnetic field because of the anisotropic effect of the indole ring of unit A suggesting that 2 had 16S*, 20R*, 21R*, 2′S*, 7′R*, 20′R*.
The configuration of melaxillarinine has been incompletely elucidated. Compared with the 1H and 13C NMR data of 1, the signals of H2-3′, H2-5′ and H2-14′ in melaxillarinine were shifted to a higher magnetic field because of the anisotropic effect of the indole ring, suggesting the relative configuration of melaxillarinine to be 16S*, 20R*, 21R*, 2′S*, 7′R*, 20′R*. The absolute configuration of melaxillarinine was then assigned by comparing the experimental CD spectrum with the calculated CD spectra [21]. As shown in Fig. 5, the calculated CD spectra of the isomer with 16S, 20R, 21R, 2′S, 7′R, 20′R configurations shows a similar CD pattern to the experimental melaxillarinine. Thus the absolute configuration of melaxillarinine was proposed as 16S, 20R, 21R, 2′S, 7′R, 20′R.

Experimental and calculated CD (BP86 and M06-L level of theory) spectra, and a stable conformer for melaxillarinine
Considering their biogenesis relationships, the absolute configuration of 1 and 2 should be the same as melaxillarinine, i.e. 16S, 20R, 21R, 2′S, 7′R, 20′R. Nevertheless, the possibility of 1 having 16S, 20R, 21R, 2′R, 7′S, 20′S could not be completely excluded. However, the calculated CD spectra of the isomer with 16S, 20R, 21R, 2′S, 7′R, 20′R (Fig. 6, isomer 2) showed a closer similarity to the experimental CD of 1, relative to the 16S, 20R, 21R, 2′R, 7′S, 20′S (Fig. 6, isomer 1). Therefore, their absolute configuration of 1 was proposed as 16S, 20R, 21R, 2′S, 7′R, 20′R (isomer 2). The absolute configuration of compound 2, which showed a similar CD spectrum to 1, was determined to be the same as 1.

Experimental and calculated CD (BP86 and M06-L level of theory) spectra for leucophyllinine A (1). Isomer 1: 16S, 20R, 21R, 2′R, 7′S, 20′S and isomer 2: 16S, 20R, 21R, 2′S, 7′R, 20′R
A plausible biogenetic path for leucophyllinines A (1) and B (2) is proposed as shown in Scheme 1. A series of dimers might be derived by intermolecular coupling of imine intermediate a generated from the corresponding alkaloid such as eburnamine, with the corresponding alkaloid such as 11-hydroxyquebrachamine, followed by an intramolecular cyclization reaction of imine intermediate b generated from the hydroxylated alkaloid, as shown in Scheme 1.

A plausible biogenetic path of leucophyllinine A (1) and B (2)
Biological activity of leucophyllinines A and B (1 and 2)
Against P. falciparum 3D7 strain, 1 and 2 with a C-16–C-10′ bridge showed potent in vitro antimalarial activity [Table 2: half-maximal (50%) inhibitory concentration (IC50) = 32.7 and 2.6 μM, respectively], whereas melaxillarinine with a C-16–C-12′ bridge showed low activity (IC50 > 50 μM; growth inhibition rate = 31.0% at 50 μM). Furthermore, the cytotoxic activity was evaluated using two human cell lines, HEK293T cells (relatively sensitive to cytotoxicity) and HepG2 cells (relatively resistant to cytotoxicity). The cytotoxic activity of 1 and 2 was low [Table 2: half-maximal (50%) cytotoxic concentration (CC50) \(\geqq\,50\) and 26.9 µM for HEK293T cells and each > 50 µM for HepG2 cells, respectively]. In addition, the most active compound, compound 2, also had the highest selectivity [Table 2: selective index (SI) = 10.3 and > 19.2 for HEK293T and HepG2 cells, respectively]. These results suggested that the biological activity of the C-16–C-10′ bridge is stronger than that of the C-16–C-12′ bridge.
Acknowledgement
We thank Prof. Masatsugu Kimura (Osaka City University, Osaka, Japan) for the kind gift of the 3D7 strain and Dr. Toru Okamoto and Prof. Yoshiharu Matsuura (Osaka University, Osaka, Japan) for kindly providing HEK293T cells. We also thank Mr. Yuji Toya and Dr. Kinya Uchihashi (Sysmex, Kobe, Japan) for the setting of the XN-30 analyzer and Ms. Toshie Ishisaka and Ms. Sawako Itagaki for their technical assistance.
This research was partially supported by the Ministry of Education, Culture, Sports, Science and Technology, Grant-in-Aid for Young Scientists (B) to AEN and YH [grant numbers 17K15472 and 15K18890, respectively], and Grants-in-Aid for Scientific Research (C) [grant number 16K08759] to TT, and by Sysmex Corporation to TH.
Experimental section
General experimental procedures
Optical rotations were measured on a JASCO DIP-1000 polarimeter. UV spectra were recorded on a Shimadzu UVmini-1240 spectrophotometer and IR spectra on a JASCO FT/IR-4100 spectrophotometer. High-resolution ESI MS were obtained on a LTQ Orbitrap XL (Thermo Scientific). 1H and 2D NMR spectra were measured on a 600 MHz spectrometer at 300 K, while 13C NMR spectra were on a 150 MHz spectrometer. The residual solvent peaks were used as internal standards (δH 3.31 and δC 49.0 for CD3OD). Standard pulse sequences were used for the 2D NMR experiments. Merck silica gel 60 (40–63 μm) was used for the column chromatography, and the separations were monitored by Merck silica gel 60 F254, or Merck silica gel RP C-18 F254 TLC plates.
Material
The barks of L. eugeniifolia were collected in Johor, Malaysia in August 2007. The botanical identification was made by Prof. A. Hamid A. Hadi, University of Malaya. Voucher specimens (Herbarium No. KL 5476) was deposited at the Herbarium of the Department of Chemistry, University of Malaya, Kuala Lumpur, Malaysia.
Extraction and isolation
The bark of L. eugeniifolia (920 g) was extracted with MeOH (3 × 25 L, each 24 h), then AcOH (3 × 25 L, each 24 h), and the extract (20.4 g) was treated with 3% tartaric acid (pH 2) and then partitioned with EtOAc. The aqueous layer was treated with saturated Na2CO3 (aq) to pH 10 and extracted with CHCl3 to give an alkaloidal fraction (1.88 g). The alkaloidal fraction was subjected to an amino silica gel column (CHCl3/MeOH, 49:1 → 0:1) to give leucophyllinine A (1, 155.2 mg, 0.0169%), and the fractions were further separated using a silica gel column (CHCl3/MeOH, 9:1 → 1:1, CHCl3/MeOH/H2O, 7:3:0.5) to give leucophyllinine B (2, 179.6 mg, 0.0195%) and using ODS HPLC (CH3CN/H2O, 21:79; formic acid 0.1%) to give melaxillarinine (6.8 mg, 0.0007%).
Leucophyllinine A
[α]16D-88 (c 1.0, MeOH); IR (Zn–Se) νmax 3194, 2935, 1624 cm−1; UV (MeOH) λmax (ε) 294 (8300), 224 (32,200), 207 (40,800) nm; CD (MeOH) λmax (Δε) 300 (− 2.99), 295 (− 2.04), 234 (− 17.40), 203 (46.30) nm; 1H and 13C NMR data, see Table 1; ESIMS m/z 593 (M)+; HRESIMS m/z 593.3846 (M)+ (calcd for C38H49N4O2, 593.3856).
Leucophyllinine B
[α]26D-111 (c 1.0, MeOH); IR (Zn–Se) νmax 3387, 1676, 1200, 1139 cm−1; UV (MeOH) λmax (ε) 293 (4000), 2265 (20,200), 210 (25,700) nm; CD (MeOH) λmax (Δε) 299 (− 3.16), 296 (− 2.72), 230 (− 24.77), 203 (33.26) nm; 1H and 13C NMR data, see Table 1; ESIMS m/z 607 (M)+; HRESIMS m/z 607.4011 (M)+ (calcd for C39H51N4O2, 607.4012).
CD calculations
The conformations were obtained using Monte Carlo analysis with MMFF94 force field and charges on Macromodel 9.1. CD calculations were performed in Turbomole 7.1 using RI-TD-DFT-M06-L/SVPD level of theory on RI-DFT-M06-L/TZVP optimized geometries and RI-TD-DFT-BP-86/SVPD level of theory on RI-DFT-D3-BP-86/TZVP optimized geometries.
Parasite strain and culture
Plasmodium falciparum laboratory strain 3D7 was obtained from Prof. Masatsugu Kimura (Osaka City University, Osaka, Japan). For the assessment of antimalarial activity of the compounds in vitro, the parasites were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 0.5 g/L l-glutamine, 5.96 g/L HEPES, 2 g/L sodium bicarbonate (NaHCO3), 50 mg/L hypoxanthine, 10 mg/L gentamicin, 10% heat-inactivated human serum, and red blood cells (RBCs) at a 3% hematocrit in an atmosphere of 5% CO2, 5% O2, and 90% N2 at 37 °C as previously described [22]. Ring-form infected RBCs were collected using the sorbitol synchronization technique [23]. Briefly, the cultured cells were collected by centrifugation at 840g for 5 min at room temperature, suspended in a fivefold volume of 5% d-sorbitol (Nacalai Tesque) for 10 min at room temperature, and then they were washed twice with RPMI 1640 medium to remove the d-sorbitol. The utilization of blood samples of healthy Japanese volunteers for the parasite culture was approved by the institutional review committee of the Research Institute for Microbial Diseases (RIMD), Osaka University (approval number: 22-3).
Antimalarial activity
Ring-form-synchronized parasites were cultured with 1, 2, and melaxillarinine at sequentially decreasing concentrations (50, 15, 5, 1.5, 0.5, 0.15, 0.05, and 0.015 µM) for 48 h for the flow cytometric analysis using an automated hematology analyzer, XN-30. The XN-30 analyzer was equipped with a prototype algorithm for cultured falciparum parasites (prototype; software version: 01-03, (build 16)) and used specific reagents (CELLPACK DCL, SULFOLYSER, Lysercell M, and Fluorocell M) (Sysmex, Kobe, Japan) [24]. Approximately 100 µL of the culture suspension diluted with 100 µL phosphate-buffered saline was added to a BD Microtainer MAP Microtube for Automated Process K2 EDTA 1.0 mg tube (Becton–Dickinson and Co., Franklin Lakes, NJ, USA) and loaded onto the XN-30 analyzer with an auto-sampler as described in the instrument manual (Sysmex). The parasitemia (MI-RBC%) was automatically reported [24]. Then 0.5% dimethyl sulfoxide alone or containing 5 µM artemisinin was used as the negative and positive control, respectively. The growth inhibition (GI) rate was calculated from the MI-RBC% according to the following equation:
The IC50 was calculated from GI (%) using GraphPad Prism version 5.0 (GraphPad Prism Software, San Diego, CA, USA) [25].
Cytotoxic activity
HEK293T and HepG2 (JCRB1054) cell lines were obtained from Prof. Yoshiharu Matsuura (Osaka University, Osaka, Japan) [26] and the Japanese Collection of Research Bioresources (JCRB, Osaka, Japan), respectively. The cells were cultured in Dulbecco’s modified Eagle’s medium [DMEM (1.0 g/L glucose) with l-glutamine and sodium pyruvate; Nacalai Tesque] supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco-BRL, Grand Island, NY, USA) in a humidified incubator with 5% CO2 at 37 °C. For the cytotoxic assay, the cells (5 × 103/well) were seeded in a 96-well plate. 1, 2, and melaxillarinine at gradually decreasing concentrations (50, 15, 5, 1.5, 0.5, 0.15, 0.05, and 0.015 µM) were added to the cell culture after 24 h and the cells were subsequently cultured for 48 h. Cell viability was measured using a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions. Briefly, 10 µL CCK-8 reagent was added to each well containing culture medium and incubated for 2 h under standard culture conditions. The absorbance of the sample was measured at 450 nm using a PowerWave HT microplate spectrophotometer (BioTek Instruments, Winooski, VT, USA). The cell viability was expressed as a percentage of the absorbance of the untreated control cells after subtracting the appropriate background intensity. The CC50 was calculated from the cell viability using GraphPad Prism version 5.0. The SI, which is the ratio between the antimalarial activity and cytotoxic activity, was calculated according to the flowing equation.
References
Nugroho AE, Hashimoto A, Wong CP, Yokoe H, Tsubuki M, Kaneda T, Hadi AHA, Morita H (2018) Ceramicines M-P from Chisocheton ceramicus: isolation and structure–activity relationship study. J Nat Med 72:64–72
Nugroho AE, Inoue D, Wong CP, Hirasawa Y, Kaneda T, Shirota O, Hadi AHA, Morita H (2018) Reinereins A and B, new onocerane triterpenoids from Reinwardtiodendron cinereum. J Nat Med 72:588–592
Nugroho AE, Matsumoto M, Sotozono Y, Kaneda T, Hadi AHA, Morita H (2018) Cycloartane triterpenoids with anti-melanin deposition activity. Nat Prod Commun 13:809–812
Kaneda T, Matsumoto M, Sotozono Y, Fukami S, Nugroho AE, Hirasawa Y, Hamid AHA, Morita H (2019) Cycloartane triterpenoid (23R, 24E)-23-acetoxymangiferonic acid inhibited proliferation and migration in B16-F10 melanoma via MITF downregulation caused by inhibition of both β-catenin and c-Raf–MEK1–ERK signaling axis. J Nat Med 73:47–58
Nugroho AE, Sugai M, Hirasawa Y, Hosoya T, Awang K, Hadi AHA, Ekasari W, Widyawaruyanti A, Morita H (2011) New antiplasmodial indole alkaloids from Hunteria zeylanica. Bioorg Med Chem Lett 21:3417–3419
Hirasawa Y, Hara M, Nugroho AE, Sugai M, Zaima K, Kawahara N, Goda Y, Awang K, Hadi AHA, Litaudon M, Morita H (2010) Bisnicalaterines B and C, atropisomeric bisindole alkaloids from Hunteria zeylanica, showing vasorelaxant activity. J Org Chem 75:4218–4223
Nugroho AE, Hirasawa Y, Kawahara N, Goda Y, Awang K, Hadi AHA, Morita H (2009) Bisnicalaterine A, a vobasine–vobasine bisindole alkaloid from Hunteria zeylanica. J Nat Prod 72:1502–1506
Oshimi S, Deguchi J, Hirasawa Y, Ekasari W, Widyawaruyanti A, Wahyuni TS, Zaini NC, Shirota O, Morita H (2009) Cassiarins C–E, antiplasmodial alkaloids from the flowers of Cassia siamea. J Nat Prod 72:1899–1901
Morita H, Oshimi S, Hirasawa Y, Koyama K, Honda T, Ekasari W, Indrayanto G, Zaini NC (2007) Cassiarins A and B, novel antiplasmodial alkaloids from Cassia siamea. Org Lett 9:3691–3693
Nugroho AE, Zhang W, Hirasawa Y, Tang Y, Wong CP, Kaneda T, Hadi AHA, Morita H (2018) Bisleuconothines B-D, modified eburnane–aspidosperma bisindole alkaloids from Leuconotis griffithii. J Nat Prod 81:2600–2604
Morita H, Nugroho AE, Nagakura Y, Hirasawa Y, Yoshida H, Kaneda T, Shirota O, Ismail IS (2014) Chrotacumines G-J, chromone alkaloids from Dysoxylum acutangulum with osteoclast differentiation inhibitory activity. Bioorg Med Chem Lett 24:2437–2439
Nugroho AE, Hirasawa Y, Wong CP, Kaneda T, Hadi AHA, Shirota O, Ekasari W, Widyawaruyanti A, Morita H (2012) Antiplasmodial indole alkaloids from Leuconotis griffithii. J Nat Med 66:350–353
Motegi M, Nugroho AE, Hirasawa Y, Arai T, Hadi AHA, Morita H (2012) Leucomidines A–C, novel alkaloids from Leuconotis griffithii. Tetrahedron Lett 53:1227–1230
Nugroho AE, Hirasawa Y, Hosoya T, Awang K, Hadi AHA, Morita H (2010) Bisleucocurine A, a novel bisindole alkaloid from Leuconotis griffithii. Tetrahedron Lett 51:2589–2592
Hirasawa Y, Shoji T, Arai T, Nugroho AE, Deguchi J, Hosoya T, Uchiyama N, Goda Y, Awang K, Hadi AHA, Shiro M, Morita H (2010) Bisleuconothine A, an eburnane-aspidosperma bisindole alkaloid from Leuconotis griffithii. Bioorg Med Chem Lett 20:2021–2024
Hirasawa Y, Dai X, Deguchi J, Hatano S, Sasaki T, Ohtsuka R, Nugroho AE, Kaneda T, Morita H (2019) New vasorelaxant indole alkaloids, taberniacins A and B, from Tabernaemontana divaricata. J Nat Med. https://doi.org/10.1007/s11418-019-01293-9
Wong CP, Seki A, Horiguchi K, Shoji T, Arai T, Nugroho AE, Hirasawa Y, Sato F, Kaneda T, Morita H (2015) Bisleuconothine A induces autophagosome formation by interfering with AKT-mTOR signaling pathway. J Nat Prod 78:1656–1662
Deguchi J, Shoji T, Nugroho AE, Hirasawa Y, Hosoya T, Shirota O, Awang K, Hadi AHA, Morita H (2010) Eucophylline, a tetracyclic vinylquinoline alkaloid from Leuconotis eugenifolius. J Nat Prod 73:1727–1729
Goh SH, Mohd Ali AR, Wong WH (1989) Alkaloids of Leuconotis griffithii and L. eugenifolia (Apocynaceae). Tetrahedron 45:7899–7920
Yan KX, Feng XZ (1997) Melaxillarinine, a new bisindole alkaloid from Melodinus axillaris. Chin Chem Lett 8:313–314
Nugroho AE, Morita H (2014) Circular dichroism calculation for natural products. J Nat Med 68:1–10
Trager W, Jensen J (1976) Human malaria parasites in continuous culture. Science 193:673–675
Lambros C, Vanderberg JP (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65:418–420
Tougan T, Suzuki Y, Itagaki S, Izuka M, Toya Y, Uchihashi K, Horii T (2018) An automated haematology analyzer XN-30 distinguishes developmental stages of falciparum malaria parasite cultured in vitro. Malar J 17:59
Tougan T, Toya Y, Uchihashi K, Horii T (2019) Application of the automated haematology analyzer XN-30 for discovery and development of anti-malarial drugs. Malar J 18:8
Aizawa S, Okamoto T, Sugiyama Y, Kouwaki T, Ito A, Suzuki T, Ono C, Fukuhara T, Yamamoto M, Okochi M, Hiraga N, Imamura M, Chayama K, Suzuki R, Shoji I, Moriishi K, Moriya K, Koike K, Matsuura Y (2016) TRC8-dependent degradation of hepatitis C virus immature core protein regulates viral propagation and pathogenesis. Nat Commun 7:11379–11390
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tang, Y., Nugroho, A.E., Hirasawa, Y. et al. Leucophyllinines A and B, bisindole alkaloids from Leuconotis eugeniifolia. J Nat Med 73, 533–540 (2019). https://doi.org/10.1007/s11418-019-01297-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-019-01297-5