
Abstract
Three new flavonoid glycosides—soyaflavonosides A (1), B (2), and C (3)—together with 23 known ones were obtained from the 70% EtOH extract of Flos Sophorae (Sophora japonica, Leguminosae). Their structures were elucidated by chemical and spectroscopic methods. Among the known isolates, 14, 18, 20, 22, and 26 were isolated from the Sophora genus for the first time; 12, 19, 24, and 25 were obtained from the species firstly. Moreover, NMR data for compounds 18 and 26 are reported for the first time here. Meanwhile, compounds 4, 8–13, 15, 16, 19, 21, and 22 presented obvious inhibitory effects on TG accumulation in HepG2 cells. Analysis of the structure–activity relationship indicated that all of the quercetin glycosides examined in this study possess significant activity that is not significantly influenced by the amount of glycosyl present, whereas increasing the amount of glycosyl reduced the activities of isorhamnetin glycosides and orobol. In addition, a high dose (30 μmol/l) of kaempferol was found to inhibit HepG2 cell growth, while a low dose (10 μmol/l) was observed to decrease TG accumulation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
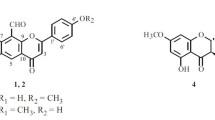
During the course of our continuing investigations of the bioactive compounds in the 70% ethanol–water extract of Flos Sophorae (Sophora japonica, Leguminosae) [1–4], three new flavonoid glycosides—termed soyaflavonosides A (1), B (2), and C (3)—were obtained, together with 23 known isolates: (±)-eriodictyol (4) [5], (2R,3R)-(+)-taxifolin (5) [6], kaempferol (6) [6], kaempferol 3-O-rutinoside (7) [7], quercetin 3-O-rutinoside (8) [8], quercetin-3-O-(2G-glucosyl)-rutinoside (9) [9], quercetin 3-O-(2″,6″-di-O-α-l-rhamnopyransoyl)-β-d-glucopyranoside (10) [10], quercetin 3-O-β-d-xylopyranosyl(1→3)-O-α-l-rhamnopyranosyl(1→6)-O-β-d-glucopyranoside (11) [11], quercetin 7-O-rhamnoside (12) [12], 3-O-α-l-rhamnopyranosyl(1→6)-β-d-glucopyranosyl quercetin 7-O-α-l-rhamnopyranoside (13) [13], quercetin 3-O-α-l-rhamnopyranosyl(1→6)-β-d-glucopyranoside-4′-O-β-d-glucopyranoside (14) [14], isorhamnetin 3-O-β-glucopyranoside (15) [15], isorhamnetin 3-O-rutinoside (16) [15], isorhamnetin 3-O-[2-O-β-glucopyranosyl-6-O-α-rhamnopyranosyl]-β-glucopyranoside (17) [16], quercetin 3-O-α-rhamnosyl(1′′′′→6″)-β-sophoroside-7-O-α-l-rhamnopyranoside (18), 8-methoxykaempferol 3-sophoroside (19) [17], 5-deoxyisorhamnetin 3-O-α-l-rhamnopyranosyl(1‴→6″)-β-d-glucopyranoside (20) [18], orobol (21) [19], orobol 4′-O-β-glucopyranoside (22) [20], genistein 4′-α-l-rhamnopyranosyl(1→2)-β-d-glucopyranoside (23) [21], genistin (24) [22], 3′-O-methylorobol-7-O-glycoside (25) [23], and cudracusisoflavone A (26). Among the known isolates, 14, 18, 20, 22, and 26 were isolated from Sophora genus for the first time; 12, 19, 24, and 25 were obtained from the species firstly. Moreover, the NMR data for compounds 18 and 26 are reported for the first time here. In addition, because many flavonoids show pharmacological activity (including antioxidant, antiinflammatory, anticancer, hypoglycemic, and hypolipidemic effects [24]), we evaluated the inhibitory effects of the above flavonoids on triglyceride (TG) accumulation using a model of sodium oleate (SO)-induced hepatic steatosis in HepG2 cells in order to evaluate Flos Sophorae as a potential lipid-lowering agent.
Results and discussion
From the 70% ethanol–water extract of Flos Sophorae, a series flavonoids, 1–26 were obtained (Figs. 1, 2).

The new flavonoids (1–3) obtained from Flos Sophorae

The known flavonoids (4–26) obtained from Flos Sophorae
Soyaflavonoside A (1) was isolated as a yellow powder that exhibited negative optical rotation: \([\alpha ]_{\text{D}}^{25}\) −33.3° (MeOH). Its molecular formula was revealed to be C38H48O25 by negative HRESI-TOF–MS analysis (m/z 903.2358 [M–H]−, calcd for C38H47O25, 903.2429). The characteristic absorptions in its IR spectrum suggested the presence of hydroxyl (3383 cm−1) and α,β-unsaturated ketone (1651 cm−1) moieties, an aromatic ring (1605, 1512, 1446 cm−1), and an O-glycosidic linkage (1072 cm−1). The sugars in 1 were found to be d-glucose, d-xylose, and l-rhamnose by acid hydrolysis with 1 M HCl [1]. The 1H, 13C NMR spectra (Table 1) and various 2D NMR spectra, including 1H 1H COSY, HSQC, and HMBC spectra, indicated that the aglycon of 1 was quercetin [δ 6.14 (1H, d, J = 1.0 Hz, H-6), 6.33 (1H, d, J = 1.0 Hz, H-8), 6.85 (1H, d, J = 9.0 Hz, H-5′), 7.53 (1H, d, J = 2.0 Hz, H-2′), 7.54 (1H, dd, J = 2.0, 9.0 Hz, H-6′)]. Meanwhile, there were four glycoside groups in 1: two β-d-glucopyranosyl groups [δ 4.58 (1H, d, J = 7.5 Hz, H-1‴), 5.52 (1H, d, J = 7.5 Hz, H-1″)], one α-l-rhamnopyranosyl group [δ 4.34 (1H, br. s, H-1′′′′)], and one β-d-xylopyranosyl group [δ 4.23 (1H, d, J = 7.5 Hz, H-1′′′′′)]. Furthermore, in the HMBC experiment, long-range correlations were observed between δ H 5.52 (H-1″) and δ C 132.7 (C-3); δ H 4.58 (H-1‴) and δ C 82.3 (C-2″); δ H 4.34 (H-1′′′′) and δ C 66.7 (C-6″); and δ H 4.23 (H-1′′′′′) and δ C 81.0 (C-3′′′′). Consequently, the structure of soyaflavonoside A was determined as quercetin 3-O-β-d-xylopyranosyl(1′′′′′→3′′′′)-O-α-l-rhamnopyranosyl(1′′′′→6″)-β-sophoroside (1).
Soyaflavonoside B (2) was obtained as a pale yellow powder that exhibited negative optical rotation: \([\alpha ]_{\text{D}}^{25}\) −42.2° (MeOH). The molecular formula, C27H30O15, of 2 was determined from Q-TOF-ESI–MS analysis (m/z 593.1521 [M–H]−, calcd for C27H29O15, 593.1518). The 1H NMR (Table 2) spectrum indicated that the aglycon of 2 was 5,7,3′4′-tetrahydroxy isoflavone [δ 6.17 (1H, br. s, H-6), 6.32 (1H, br. s, H-8), 6.88 (1H, dd, J = 2.0, 9.0 Hz, H-6′), 7.07 (1H, d, J = 2.0 Hz, H-2′), 7.08 (1H, d, J = 9.0 Hz, H-5′), 8.32 (1H, s, H-2)]. There were two sugar moieties in 2 [δ 5.06 (1H, d, J = 7.5 Hz), 5.18 (1H, br. s)]. Furthermore, using acid hydrolysis and HPLC analysis, the presence of d-glucose and l-rhamnose in 2 was revealed [1]. 1H 1H COSY indicated the presence of the partial structure highlighted with bold lines in Fig. 3 in 2. Finally, in the HMBC experiment, long-range correlations were observed between following protons and carbons: δ H 5.06 (H-1″) and δ C 144.8 (C-4′); and δ H 5.18 (H-1‴) and δ C 76.9 (C-2″). Based on the above evidence, the structure of soyaflavonoside B was elucidated as 5,7,3′4′-tetrahydroxyisoflavone 4′-O-[α-l-rhamnopyranosyl(1→2)]-β-d-glucopyranoside (2).

The main 1H 1H COSY and HMBC correlations for 1–3
Soyaflavonoside C (3), \([\alpha ]_{\text{D}}^{25}\) −53.9° (MeOH), was isolated as a pale yellow powder. Its molecular formula, C28H32O15 (m/z 607.1671 [M–H]−, calcd for C28H31O15, 607.1668), was determined by Q-TOF-ESI–MS. While 3 had the same 5,7,4′-trihydroxy-3′-methoxy isoflavone-4′-O-β-d-glucopyranosyl [δ 3.88 (3H, s, 3′-OCH3), 5.11 (1H, d, J = 7.5 Hz, H-1″), 6.21 (1H, br. s, H-6), 6.34 (1H, br. s, H-8), 7.03 (1H, br. d, ca. J = 8 Hz, H-6′), 7.16 (1H, d, J = 8.0 Hz, H-5′), 7.20 (1H, br. s, H-2′), 8.11 (1H, s, H-2), 12.75 (1H, br. s, 5-OH)] moiety as 26, it also had an α-l-rhamnopyranosyl group [δ 1.24 (3H, d, J = 6.0 Hz, 6‴-CH3), 5.38 (1H, br. s, H-1‴)], based on its 1H and 13C NMR (Table 3) spectra. Acid hydrolysis of 3 with 1 M HCl led to the detection of d-glucose and l-rhamnose [1]. The 13C NMR signal for the 2-position of β-d-glucose was shifted downfield compared to the corresponding signal for 26 [δ 74.9 (C-2″) for 26; 78.4 (C-2″) for 3, both in CD3OD], indicating that α-l-rhamnopyranyl was connected at the 2-position of β-d-glucopyranosyl, as certified by the long-range correlation between δ H 5.38 (H-1‴) and δ C 78.4 (C-2″) observed in the HMBC experiment. Based on the above evidence, soyaflavonoside C was elucidated as 5,7,4′-trihydroxy-3′-methoxyisoflavone 4′-O-[α-l-rhamnopyranosyl(1→2)]-β-d-glucopyranoside (3).
To check for inhibitory effects of the isolated flavonoids on SO-induced TG accumulation, HepG2 cells were incubated with either orlistat (5 μmol/l) or one of the isolates (30 μmol/l) in the presence of SO for 48 h. As shown in Fig. 4, the intracellular TG content was significantly increased by the presence of 200 μmol/l SO as compared to the intracellular TG content of the control group (from 16.70 ± 0.40 μg/g protein to 101.53 ± 1.35 μg/g protein). However, orlistat as well as compounds 4, 8, 9, 10, 11, 12, 13, 15, 16, 19, 21, and 22 significantly weakened this SO-mediated lipid accumulation; these numbered compounds reduced the TG content by between 6.41 ± 1.77% and 14.14 ± 1.50% compared to the model group (i.e., the group with 200 μmol/l SO but none of the isolated flavonoids or orlistat). In addition, a dose-dependency study was conducted for several of the inhibitory isolates with different structures (as almost every quercetin glycoside examined in this work had a significant inhibitory effect on SO-mediated lipid accumulation, further systematic studies of these compounds should be conducted). The results showed that compounds 15, 16, 19, 21, and 22 reduced SO-induced TG accumulation in a dose-dependent manner (as shown in Fig. 5).

Effects of compounds 1–26 on SO-induced intracellular TG accumulation in HepG2 cells. The TG content is represented as the mass of TG per gram of protein in a and b, with the corresponding protein concentrations plotted in c and d. Cells were cultured in normal medium without SO in the normal group (Nor.). Each value represents the mean ± S.E.M., n = 5, ***p < 0.001, **p < 0.01, *p < 0.05 vs. model group (Mod.)

Dose dependencies of the TG-lowering effects of compounds 15, 16, 19, 21, and 22 in HepG2 cells. Cells were cultured in normal medium without SO in the normal group (Nor.). Each value represents the mean ± S.E.M., n = 5, ***p < 0.001, **p < 0.01, *p < 0.05 vs. model group (Mod.)
In an analysis of the structure–activity relationship for these compounds, all of the quercetin glycosides (compounds 8, 9, 10, and 11) were observed to exert significant activity at 30 μmol/l (shown in Fig. 4a). Quercetin has been reported to possess hypolipidemic properties [25–27], and rutin is a prodrug, as it can be metabolized to quercetin by the intestinal microflora in the colon [28]. Therefore, we speculate that quercetin glycosides can be metabolized in cells to quercetin (the active form), but further studies should be conducted to test the validity of this hypothesis.
As shown in Fig. 4b, upon comparing the TG clearance of the isorhamnetin glycosides, we observed that \({\mathbf{15}}\;\left( { 1 1. 7 7 \pm 1. 2 5\% } \right) > {\mathbf{16}}\;\left( { 7. 1 2 \pm 1. 6 6\% } \right) > {\mathbf{17}}\;\left( { 1. 4 8 \pm 1. 6 7\% } \right)\), leading us to speculate that the monoglycoside is more active than the diglycoside, but the polyglycoside is inactive. Moreover, by comparing 16 with 20 (which showed no obvious activity), we inferred that the presence of the 5-hydroxyl moiety is crucial to TG-lowering activity. After comparing 16 and 19 with 7 (which also showed no obvious activity), we surmised that methoxylation may also play a role in the activities of the flavone diglycosides. When orobol (21) was compared with its glycoside (22), it became apparent that 4′-O-glycosyl substitution reduces the activity of orobol from 8.39 ± 1.63 to 6.41 ± 1.77%.
Interestingly, while kaempferol (6) and kaempferol 3-O-rutinoside (7) have been reported to show antiobesity or antidiabetic effects in vivo [29–31], these two isolates did not show any obvious TG-lowering activity in vitro in this study. To our knowledge, kaempferol at a concentration of 20 μmol/l can significantly inhibit the growth of HeLa, HepG2, and A549 cancer cells [32]. Thus, it is no wonder that the cellular protein concentrations were significantly decreased upon treatment with 30 μmol/l kaempferol or kaempferol 3-O-rutinoside compared to the model group (see Fig. 4c) in our study. However, at the relatively safe concentration of 10 μmol/l with no significant reduction in cellular protein concentration (see Fig. 6b), kaempferol exhibited a clear TG-lowering effect (shown in Fig. 6a). This indicates that kaempferol can inhibit HepG2 cell growth in high doses but also that it decreases TG accumulation in low doses. More detailed studies should be conducted to verify this conclusion. It is worth noting that, apart from these two isolates, none of the isolates examined in the current study produced any notable changes in cell growth at any of the concentrations tested (see Fig. 4c, d).

Dose dependency of the TG-lowering effects of compound 6 in HepG2 cells. The TG content is represented as the mass of TG per gram of protein in a, with the corresponding protein concentrations plotted in b. Cells were cultured in normal medium without SO in the normal group (Nor.). Each value represents the mean ± S.E.M., n = 5, ***p < 0.001, **p < 0.01, *p < 0.05 vs. model group (Mod.)
Experimental
General
UV and IR spectra were recorded on a Varian (Palo Alto, CA, USA) Cary 50 UV–Vis spectrophotometer and a Varian 640-IR FT-IR spectrophotometer, respectively. Optical rotations were determined on a Rudolph (Hackettstown, NJ, USA) Autopol® IV automatic polarimeter. NMR spectra were determined on a Bruker (Rheinstetten, Germany) 500 MHz NMR spectrometer at 500 MHz for 1H and 125 MHz for 13C NMR (internal standard: TMS). Column chromatography (CC) was performed on macroporous D101 resin (Haiguang Chemical Co., Ltd., Tianjin, China), silica gel (74–149 μm, Qingdao Haiyang Chemical Co., Ltd., Qingdao, China), ODS (50 μm, YMC Co., Ltd., Tokyo, Japan), and Sephadex LH-20 (GE Healthcare Bio-Sciences, Uppsala, Sweden). Preparative high-performance liquid chromatography (prep-HPLC) using a Cosmosil 5C18-MS-II column (20 mm i.d. × 250 mm, Nakalai Tesque, Inc., Tokyo, Japan) was employed to separate the constituents.
Plant material
The Flos Sophorae was collected from Tangshan City, Hebei Province, China, and identified by Dr. Li Tianxiang. A voucher specimen has been deposited at the Academy of Traditional Chinese Medicine of Tianjin University (no. 20120909).
Extraction and isolation
The extraction and purification procedures of Flos Sophorae were described previously [1,2,3,4]. The EtOAc layer (SoE, 78.0 g), EtOAc precipitate (197.0 g), along with H2O (213.1 g), 95% EtOH (SoH, 122.1 g), acetone (8.2 g) eluents from D101 resin column chromatography were obtained.
SoE (50.0 g) was subjected to SiO2 gel CC [CHCl3 → CHCl3–MeOH (100:2 → 100:3 → 100:5, v/v) → CHCl3–MeOH–H2O (10:3:1, v/v/v, lower layer) → MeOH], and seven fractions (SoE 1–7) were obtained. SoE 4 (3.3 g) was isolated by SiO2 gel CC [PE-EtOAc(10:1 → 5:1 → 3:1 → 1:1 → 1:3 → 1:5 → 1:7 → 1:10, v/v) → MeOH] to afford 17 fractions (SoE 4-1–4-17). SoE 4-11 (223.2 mg) was purified by prep-HPLC [MeOH–H2O (50:50, v/v)] to yield (±)-eriodictyol (4, 10.7 mg), kaempferol (6, 40.0 mg), and orbol (21, 7.8 mg). SoE 5 (7.0 g) was separated by ODS CC [MeOH–H2O (30:70 → 40:60 → 50:50 → 60:40 → 70:30 → 80:20 → 100:0, v/v)], and twelve fractions (SoE 5-1–5-12) were obtained. SoE 5-4 (136.1 mg) was further isolated by prep-HPLC [CH3CN–(H2O + 1% HAc) (20:80, v/v)] to afford (2R,3R)-(+)-taxifolin (5, 8.0 mg). SoE 6 (7.5 g) was subjected to ODS CC [MeOH–H2O (10:90 → 45:55 → 50:50 → 60:40 → 70:30 → 80:20 → 100:0, v/v)], and twelve fractions (SoE 6-1–6-12) were obtained. SoE 6-3 (1462.0 mg) was isolated by prep-HPLC [MeOH–H2O (45:55, v/v)] to afford SoE 6-3-1–6-3-11. SoE 6-3-6 (37.0 mg) and 6-3-7 (54.8 mg) were further purified by prep-HPLC [CH3CN–H2O (21:79, v/v)] to give 3′-O-methylorobol-7-O-glycoside (25, 11.5 mg). SoE 6-3-10 (129.8 mg) was separated by Prep-HPLC [CH3CN–H2O (25:75, v/v)] to afford cudracusisoflavone A (26, 10.6 mg). Isorhamnetin 3-O-β-glucopyranoside (15, 11.7 mg) was obtained by prep-HPLC [CH3CN–H2O (23:77, v/v)] from SoE 6-3-11 (76.9 mg). SoE 7 (7.0 g) was subjected to ODS CC [MeOH–H2O (10:90 → 45:55 → 50:50 → 60:40 → 70:30 → 80:20 → 100:0, v/v)], and twelve fractions (SoE 7-1–7-12) were obtained. SoE 7-3 (1318.7 mg) was separated by prep-HPLC [MeOH–H2O (30:70 → 40:60 → 45:55 → 100:0, v/v)] to give 20 fractions (SoE 7-3-1–7-3-20). SoE 7-3-13 (35.5 mg) was further purified by prep-HPLC [CH3CN–H2O (23:77, v/v)] to afford genistin (24, 8.1 mg). Orobol 4′-O-β-glucopyranoside (22, 28.5 mg) was obtained from SoE 7-3-17 (100.0 mg) by prep-HPLC [CH3CN–H2O (23:77, v/v)] isolation.
SoH (72 g) was isolated by SiO2 gel CC [CHCl3 → CHCl3–MeOH (100:2 → 100:5 → 100:7, v/v) → CHCl3–MeOH–H2O (10:3:1 → 7:3:1 → 6:4:1, v/v/v, lower layer) → MeOH], and 19 fractions (SoH 1–19) were obtained. SoH 11 (1.5 g) was subjected to Sephadex LH-20 CC [CHCl3–MeOH (1:1, v/v)] to give eleven fractions (SoH 11-1–11-11). SoH 12 (3.2 g) was separated by prep-HPLC [MeOH–H2O (20:80 → 40:60 → 50:50 → 60:40 → 100:0, v/v)], affording 21 fractions (SoH 12-1–12-21). Isorhamnetin 3-O-rutinoside (16, 20.3 mg) was obtained from SoH 12-15 (238.7 mg) by prep-HPLC [CH3CN–(H2O + 1% HAc) (23:77, v/v)] purification. SoH 12-17 (143.1 mg) was separated by prep-HPLC [CH3CN–(H2O + 1% HAc) (23:77, v/v)] to yield quercetin 7-O-rhamnoside (12, 72.4 mg), respectively. SoH 13 (5.0 g) was subjected to ODS [MeOH–H2O (10:90 → 20:80 → 30:70 → 40:60 → 50:50 → 60:40 → 70:30 → 100:0, v/v)], and fifteen fractions (SoH 13-1–13-15) were obtained. SoH 13-12-6 (8.4 mg) and 13-12-7 (32.0 mg) were separated by prep-HPLC [MeOH–(H2O + 1% HAc) (45:55, v/v)], yielding soyaflavonoside C (3, 4.3 mg) and genistein 4′-α-l-rhamnopyranosyl(1→2)-β-d-glucopyranoside (23, 10.3 mg), respectively. SoH 14 (6.0 g) was separated by ODS CC [MeOH–H2O (30:70 → 40:60 → 50:50 → 60:40 → 70:30 → 80:20 → 100:0, v/v)] to give ten fractions (SoH 14-1–14-10). SoH 14-4 (147.7 mg) was further purified by prep-HPLC [CH3CN–(H2O + 1% HAc) (15:85, v/v)]; as a result, quercetin 3-O-rutinoside (8, 16.6 mg) and 8-methoxykaempferol 3-sophoroside (19, 12.0 mg) were obtained. Soyaflavonoside B (2, 54.2 mg) together with kaempferol 3-O-rutinoside (7, 43.1 mg) and 5-deoxyisorhamnetin 3-O-α-l-rhamnopyranosyl(1‴→6″)-β-d-glucopyranoside (20, 11.2 mg) were obtained from SoH 14-5 (900.0 mg) by prep-HPLC [CH3CN–(H2O + 1% HAc) (15:85, v/v)] separation. SoH 17 (300 mg) was isolated by prep-HPLC [MeOH–(H2O + 1% HAc) (15:85, v/v)] to give SoH 17-1–17-4. SoH 17-2 (28.7 mg) was further purified by prep-HPLC [CH3CN–(H2O + 1% HAc) (15:85, v/v)] to give 3-O-α-l-rhamnopyranosyl(1→6)-β-d-glucopyranosyl quercetin 7-O-α-l-rhamnopyranoside (13, 6.5 mg) and isorhamnetin 3-O-[2-O-β-glucopyranosyl-6-O-α-rhamnopyranosyl]-β-glucopyranoside (17, 6.0 mg). SoH 17-3 (27.5 mg) was purified by prep-HPLC [CH3CN–(H2O + 1% HAc) (17:83, v/v)] to yield quercetin 3-O-β-d-xylopyranosyl(1→3)-O-α-l-rhamnopyranosyl(1→6)-O-β-d-glucopyranoside (11, 23.5 mg). SoH 18 (5.5 g) was subjected to ODS CC [MeOH–H2O (30:70 → 40:60 → 60:40 → 80:20 → 100:0, v/v)] to yield thirteen fractions (SoH 18-1–18-13). SoH 18-3 (1.1 g) was separated by prep-HPLC [CH3CN–(H2O + 1% HAc) (13:87, v/v)], and nine fractions (SoH 18-3-1–18-3-9) were obtained. SoH 18-3-3 (21.3 mg) was identified as quercetin 3-O-α-rhamnosyl(1′′′′ → 6″)-β-sophoroside-7-O-α-rhamnopyranoside (18). SoH 18-3-4 (20.0 mg) was purified by prep-HPLC [MeOH–(H2O + 1% HAc) (35:65, v/v)] to give soyaflavonoside A (1, 14.1 mg). Quercetin-3-O-(2G-glucosyl)-rutinoside (9, 302.0 mg) was yielded from SoH 18-3-5 (348.5 mg) by prep-HPLC [MeOH–(H2O + 1% HAc) (35:65, v/v)] preparation. SoH 18-3-6 (140.0 mg) was purified using the same prep-HPLC conditions as used for SoH 18-3-5 to give quercetin 3-O-α-l-rhamnopyranosyl(1→6)-β-d-glucopyranoside-4′-O-β-d-glucopyranoside (14, 6.5 mg). SoH 18-3-7 (32.0 mg) was further isolated by prep-HPLC [CH3CN–(H2O + 1% HAc) (15:85, v/v)] to yield quercetin 3-O-(2″,6″-di-O-α-l-rhamnopyranosyl)-β-d-glucopyranoside (10, 8.5 mg).
Soyaflavonoside A (1): yellow powder; \([\alpha ]_{\text{D}}^{25}\) –33.3° (c 0.63, MeOH); UV λ max (MeOH) nm (log ε): 254 (4.22), 351 (4.10); IR ν max (KBr) cm−1: 3383, 2922, 2696, 1651, 1605, 1512, 1446, 1360, 1300, 1200, 1169, 1072, 988, 812; 1H and 13C NMR data, see Table 1; HR-ESI-TOF–MS: negative-ion mode m/z 903.2358 [M-H]− (calcd for C38H47O25, 903.2429).
Soyaflavonoside B (2): pale yellow powder; \([\alpha ]_{\text{D}}^{25}\) −42.2° (c 0.72, MeOH); UV λ max (MeOH) nm (log ε): 258 (4.33); IR ν max (KBr) cm−1: 3483, 2933, 2711, 1653, 1621, 1576, 1509, 1446, 1367, 1275, 1194, 1133, 1060, 926, 818; 1H and 13C NMR data, see Table 2; HR-ESI-TOF–MS: negative-ion mode m/z 593.1521 [M-H]− (calcd for C27H29O15, 593.1518).
Soyaflavonoside C (3): pale yellow powder; \([\alpha ]_{\text{D}}^{25}\) −53.9° (conc. 0.25, MeOH); UV λ max (MeOH) nm (log ε): 260 (4.37); IR ν max (KBr) cm−1: 3383, 2932, 1651, 1610, 1579, 1514, 1455, 1364, 1310, 1266, 1206, 1177, 1134, 1049, 913, 817; 1H and 13C NMR data, see Table 3; HR-ESI-TOF–MS: negative-ion mode m/z 607.1671 [M-H]− (calcd for C28H31O15, 607.1668).
Quercetin 3-O-α-rhamnopyranosyl(1′′′′→6″)-β-sophoroside-7-O-α-l-rhamnopyranoside (18): yellow powder; 1H NMR (DMSO-d 6, 500 MHz): δ 6.44 (1H, br. s, H-6), 6.80 (1H, br. s, H-8), 7.73 (1H, br. s, H-2′), 6.92 (1H, d, J = 8.0 Hz, H-5′), 7.67 (1H, br. d, ca. J = 8 Hz, H-6′), 12.61 (1H, br. s, 5-OH), 5.62 (1H, d, J = 7.5 Hz, H-1″), 3.53 (1H, dd, J = 7.5, 9.0 Hz, H-2″), 3.51 (1H, t, J = 9.0 Hz, H-3″), 3.14 (1H, m, overlapped, H-4’’), 3.28 (1H, m, overlapped, H-5″), [3.28 (1H, m, overlapped), 3.64 (1H, br. d, ca. J = 12 Hz), H2-6″], 4.61 (1H, d, J = 7.5 Hz, H-1‴), 3.06 (1H, dd, J = 7.5, 9.0 Hz, H-2‴), 3.16 (1H, t, J = 9.0 Hz, H-3‴), 3.14 (1H, m, overlapped, H-4‴), 3.08 (1H, m, H-5‴), [3.46 (1H, m, overlapped), 3.56 (1H, br. d, ca. J = 12 Hz), H2-6‴], 4.36 (1H, br. s, H-1′′′′), 3.36 (1H, br. d, ca. J = 3 Hz, H-2′′′′), 3.23 (1H, dd, J = 3.0, 9.0 Hz, H-3′′′′), 3.07 (1H, t, J = 9.0 Hz,, H-4′′′′), 3.21 (1H, m, H-5′′′′), 0.94 (3H, d, J = 6.0 Hz, 6′′′′-CH3), 5.56 (1H, br. s, H-1′′′′′), 3.85 (1H, br. d, ca. J = 3 Hz, H-2′′′′′), 3.65 (1H, dd, J = 3.0, 9.0, H-3′′′′′), 3.31 (1H, t, J = 9.0 Hz, H-4′′′′′), 3.46 (1H, m, H-5′′′′′), 1.14 (3H, d, J = 6.0 Hz, 6′′′′′–CH3); 13C NMR (DMSO-d 6, 125 MHz): δ 155.9 (C-2), 133.0 (C-3), 177.4 (C-4), 160.8 (C-5), 99.2 (C-6), 161.5 (C-7), 94.6 (C-8), 156.7 (C-9), 105.5 (C-10), 120.6 (C-1′), 112.8 (C-2′), 147.1 (C-3′), 150.0 (C-4′), 115.3 (C-5′), 123.2 (C-6′), 98.3 (C-1″), 81.9 (C-2″), 76.4 (C-3″), 69.5 (C-4″), 75.7 (C-5″), 66.1 (C-6″), 103.5 (C-1‴), 74.2 (C-2‴), 76.4 (C-3‴), 69.6 (C-4‴), 76.8 (C-5‴), 60.7 (C-6‴), 100.4 (C-1′′′′), 70.2 (C-2′′′′), 70.4 (C-3′′′′), 71.7 (C-4′′′′), 68.1 (C-5′′′′), 17.5 (6′′′′-CH3), 98.3 (C-1′′′′′), 69.8 (C-2′′′′′), 70.2 (C-3′′′′), 71.6 (C-4′′′′′), 70.0 (C-5′′′′′), 17.8 (6′′′′′-CH3). HRESI-TOF–MS: negative-ion mode m/z 931.2726 [M–H]− (calcd for C40H31O25, 931.2726).
Cudracusisoflavone A (26): pale yellow powder; yellow powder; 1H NMR (CD3OD, 500 MHz): δ 8.11 (1H, s, H-2), 6.22 (1H, br. s, H-6), 6.35 (1H, br. s, H-8), 7.22 (1H, br. s, H-2′), 7.21 (1H, d, J = 8.0 Hz, H-5′), 7.06 (1H, br. d, ca. J = 8 Hz, H-6′), 4.94 (1H, d, J = 7.5 Hz, H-1″), 3.53 (1H, dd, J = 7.5, 9.0 Hz, H-2″), 3.49 (1H, t, J = 9.0 Hz, H-3″), 3.42 (1H, t, J = 9.0 Hz, H-4″), 3.43 (1H, m, H-5″), [3.72 (1H, dd, J = 4.5, 12.0 Hz), 3.88 (1H, br. d, ca. J = 12 Hz), H2-6″], 3.90 (3H, s, 3′-OCH3); 13C NMR (CD3OD, 125 MHz): δ 155.4 (C-2), 124.2 (C-3), 181.9 (C-4), 163.8 (C-5), 100.2 (C-6), 166.2 (C-7), 95.0 (C-8), 159.6 (C-9), 106.2 (C-10), 127.2 (C-1′), 114.9 (C-2′), 150.6 (C-3′), 148.0 (C-4′), 117.9 (C-5′), 122.9 (C-6′), 102.7 (C-12″), 74.9 (C-2″), 77.9 (C-3″), 71.4 (C-4″), 78.2 (C-5″), 62.5 (C-6″), 56.8 (3′-OCH3). HR-ESI-TOF–MS: negative-ion mode m/z 461.1102 [M–H]− (calcd for C22H21O11, 461.1089).
Acid hydrolysis for 1–3
A solution of the new compounds 1–3 (2.0 mg of each) in 1 M HCl was heated under reflux for 3 h. The reaction mixture was pretreated and analyzed by HPLC method as our previous report [1]. This led to the identification of d-glucose and l-rhamnose from 1–3 and d-xylose from 1 by comparing their retention times and optical rotations with those of authentic samples, (I) t R 13.5 min (d-glucose, positive optical rotation); (II) t R 8.9 min (d-xylose, positive optical rotation); (III) t R 7.2 min (l-rhamnose, negative optical rotation).
Evaluating inhibitory effects on TG accumulation in SO-induced HepG2 cells
The isolated flavonoids were screened for inhibitory effects on TG accumulation in SO-induced HepG2 cells using the classical model of SO-induced hepatic steatosis, as described previously [33]. Briefly, stock cultures of HepG2 cells were grown in 10% (v/v) fetal bovine serum (FBS, Mediatech, Manassas, VA, USA)/Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 1% antibiotics (penicillin and streptomycin, Thermo Fisher Scientific) at 37 °C in an atmosphere of 95% air and 5% CO2. The TG accumulation model was induced by 200 µmol/l SO for 48 h at a cell sowing density of 70,000 cells/ml on 48-multiwell plates. During this period, 30 µmol/l of the isolate of interest or 5 µmol/l orlistat (positive control, Sigma–Aldrich, St. Louis, MO, USA) were added to the culture medium in the presence of SO, respectively. In the dose-dependency study, cells were treated with particular concentrations (1, 10, and 30 µmol/l) of several isolates using the same method. At the end of the treatment, the cells were rinsed with phosphate-buffered saline and the intracellular TG contents were determined using a commercial assay kit (BioSino Bio-technology and Science Inc., Beijing, China) at 492 nm. Protein concentrations were examined using a BCA Protein Assay Kit (Thermo Fisher Scientific) at 562 nm according to the manufacturer’s protocols. Each TG value was normalized to protein content.
Statistical analysis
Values are expressed as the mean ± S.E.M. All statistical analysis of data for different groups was performed with SPSS 11.0 (SPSS Inc., Chicago, IL, USA). Significant differences between groups were tested for with one-way analysis of variance (ANOVA), and Tukey’s studentized range test was used for post hoc evaluations. p < 0.05 was considered to indicate statistical significance.
References
Zhang Y, Qu L, Liu L, Li X, Liu E, Han L, Fang S, Gao X, Wang T (2015) New maltol glycosides from Flos Sophorae. J Nat Med 69:249–254
Liu L, Li X, Chen Y, Sun J, Liu E, Zhang Y (2014) Study on phenolic acids from Flos Sophorae. Tianjin Zhongyiyao Daxue Xuebao 33:39–41
Liu L, Wang T, Li X, Shi P, Liu E, Zhang Y (2014) Isolation and identification of chemical constituents from Flos Sophorae II. Liaoning Zhongyiyao Daxue Xuebao 16:51–53
Liu L, Li X, Chen Y, Shi P, Qu L, Zhang Y (2014) Isolation and identification of chemical constituents from Flos Sophorae I. Tianjin Zhongyiyao Daxue Xuebao 33:230–233
Pan J, Zhang S, Yan L, Tai J, Xiao Q, Zou K, Zhou Y, Wu J (2008) Separation of flavanone enantiomers and flavanone glucoside diastereomers from Balanophora involucrata Hook. f. by capillary electrophoresis and reversed-phase high-performance liquid chromatography on a C18 column. J Chromatogr A 1185:117–129
Abdullah NH, Salim F, Ahmad R (2016) Chemical constituents of Malaysian U. cordata var. ferruginea and their in vitro α-glucosidase inhibitory activities. Molecules 21:525/1–525/11
Sawabe A, Nesumi C, Morita M, Matsumoto S, Matsubara Y, Komemushi S (2005) Glycosides in African dietary leaves, Hibiscus sabdariffa. J Oleo Sci 54:185–191
Nawwar M, Ayoub N, Hussein S, Hashim A, El-Sharawy R, Wende K, Harms M, Lindequist U (2012) Flavonol triglycoside and investigation of the antioxidant and cell stimulating activities of Annona muricata Linn. Arch Pharm Res 35:761–767
Aquino R, Behar I, D’Agostino M, De Simone F, Schettino O, Pizza C (1987) Phytochemical investigation on Mercurialis annua. Biochem Syst Ecol 15:667–669
Qu G, Ouyang J, Kong D, Du S (2008) Studies on chemical constituents of Primula maximowiczii Regel II. Zhongguo Yaoxue Zazhi 43:1300–1304
Onodera K, Hanashiro K, Yasumoto T (2006) Camellianoside, a novel antioxident glycoside from the leaves of Camellia japonica. Biosci Biotechnol Biochem 70:1995–1998
Awaad AS, Maitland DJ, Soliman GA (2006) Hepatoprotective activity of Schouwia thebica webb. Bioorg Med Chem Lett 16:4624–4628
Yoshimitsu H, Nishida M, Hashimoto F, Tanaka M, Sakata Y, Okawa M, Nohara Y (2007) Chromone and flavonol glycosides from Delphinium hybridum cv. “Belladonna Casablanca”. J Nat Med 61:334–338
Jaramillo K, Dawid C, Hofmann T, Fujimoto Y, Osorio C (2011) Identification of antioxidative flavonols and anthocyanins in Sicana odorifera fruit peel. J Agric Food Chem 59:975–983
Atay I, Kirmizibekmez H, Goren AC, Yesilada E (2015) Secondary metabolites from Sambucus ebulus. Turk J Chem 39:34–41
Lou H, Yuan H, Yamazaki Y, Sasaki T, Oka S (2001) Alkaloids and flavonoids from peanut skins. Planta Med 67:345–349
Ferreres F, Tomas-Barberan FA, Tomas-Lorente F, Nieto JL, Rumbero A, Olias JM (1989) 8-Methoxykaempferol 3-sophoroside, a yellow pigment from almond pollen. Phytochemistry 28:1901–1903
Veitch NC, Sutton PS, Kite GC, Ireland HE (2003) Six new isoflavones and a 5-deoxyflavonol glycoside from the leaves of Ateleiaherbert-smithii. J Nat Prod 66:210–216
Lin YL, Tsai WJ, Chen IS, Kuo YH (1998) Chemical constituents from Mucuna membranacea. J Chin Chem Soc 45:213–217
Kang YM, Lee NH (2011) A new isoflavone glycoside from the stems of Talia taquetii Schneider. Bull Korean Chem Soc 32:1048–1050
Sun A, Sun Q, Liu R (2007) Preparative isolation and purification of flavone compounds from Sophora japonica L. by high-speed counter-current chromatography combined with macroporous resin column separation. J Sep Sci 30:1013–1018
Zhang L, Zhang Y, Chen Y (2011) Isoflavones in leaves of Belamcanda chinensis. Tianran Chanwu Yanjiuyukaifa 23:69–71
Li H, Zhai F, Yang M, Li X, Wang P, Ma X (2012) A new benzofuran derivative from Flemingia philippinensis Merr. et Rolfe. Molecules 17:7637–7644
Formica JV, Regelson W (1995) Review of the biology of quercetin and related bioflavonoids. Food Chem Toxicol 33:1061–1080
Boots AW, Haenen GR, Bast A (2008) Health effects of quercetin: from antioxidant to nutraceutical. Eur J Pharmacol 585:325–337
Duchnowicz P, Broncel M, Podsędek A, Koter-Michalak M (2012) Hypolipidemic and antioxidant effects of hydroxycinnamic acids, quercetin, and cyanidin 3-glucoside in hypercholesterolemic erythrocytes (in vitro study). Eur J Nutr 51:435–443
Tang Y, Gao C, Xing M, Li Y, Zhu L, Wang D, Yang X, Liu L, Yao P (2012) Quercetin prevents ethanol-induced dyslipidemia and mitochondrial oxidative damage. Food Chem Toxicol 50:1194–1200
Lipkin M, Reddy B, Newmark H, Lamprecht SA (1999) Dietary factors in human colorectal cancer. Annu Rev Nutr 19:545–586
Chang CJ, Tzeng TF, Liou SS, Chang YS, Liu IM (2011) Kaempferol regulates the lipid-profile in high-fat diet-fed rats through an increase in hepatic PPARα levels. Planta Med 77:1876–1882
Habtemariam S (2011) A-glucosidase inhibitory activity of kaempferol-3-O-rutinoside. Nat Prod Commun 6:201–203
Muhammad A, Guerrero-Analco JA, Martineau LC, Musallam L, Madiraju P, Nachar A, Saleem A, Haddad PS, Arnason JT (2012) Antidiabetic compounds from Sarracenia purpurea used traditionally by the Eeyou Istchee Cree First Nation. J Nat Prod 75:1284–1288
Wang H, Gao M, Wang J (2013) Kaempferol inhibits cancer cell growth by antagonizing estrogen-related receptor α and γ activities. Cell Biol Int 37:1190–1196
Li J, Adelakun TA, Wang S, Ruan J, Yang S, Li X, Zhang Y, Wang T (2017) Inhibitory effects of constituents from the aerial parts of Rosmarinus officinalis L. on triglyceride accumulation. Molecules 22:E110. doi:10.3390/molecules22010110
Acknowledgements
This work was financially supported by grants from the National Natural Science Foundation of China (NSFC 81673703) and the program for Changjiang Scholars and Innovative Research Team in University (PCSIRT IRT_14R41).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Shi, W., Liu, L., Li, J. et al. Bioactive flavonoids from Flos Sophorae. J Nat Med 71, 513–522 (2017). https://doi.org/10.1007/s11418-017-1084-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-017-1084-7