
Abstract
Purpose
The main aim of this work was to determine the efficiency and mechanism of iron (Fe)-modified calcite (FMCA) capping for preventing the release of phosphorus (P) from sediments.
Methods
The performance of phosphate removal by FMCAs with different iron loading amounts was investigated using batch experiments, the mechanism for the phosphate removal by FMCA was explored by X-ray photoelectron spectroscopy, and the effect of FMCA capping on P mobilization in sediments at various depths was studied using high-resolution dialysis and diffusive gradient in thin films (DGT).
Results
FMCA possessed better phosphate adsorption ability for aqueous phosphate than raw calcite, and the phosphate removal efficiency of FMCA increased with its loading amount of Fe. The replacement of Fe-bound hydroxyl groups with phosphate anions to the inner-sphere surface complexes and the precipitation of calcium phosphate were vital to the sorption of phosphate onto FMCA. FMCA capping could effectively inhibit the release of soluble reactive P (SRP) and labile P monitored by DGT (DGT-P) from sediment to the overlying water. The decrease of DGT-P and pore water SRP in the top sediment layer is very important for the control of sediment-P release into the overlying water. FMCA capping caused reductions of the concentrations of Fe2+ and labile Fe monitored by DGT in the overlying water and the top sediment, suggesting a low risk of Fe releasing from the FMCA capping layer.
Conclusion
Iron-modified calcite has the potential to be an active capping material to suppress sedimentary P release.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Phosphorus (P) is identified to be a primary limiting nutrient driving the growth of harmful algae in freshwater bodies such as lakes, reservoirs, and ponds, and excessive P loading from the internal source (IPS, i.e., sediment) is recognized as an important cause of eutrophication in these freshwater bodies when P input from external P sources such as municipal wastewater and rain runoff is effectively controlled (Schindler et al. 2008; Némery and Garnier 2016; Tong et al. 2017; Chen et al. 2018; Tu et al. 2019). To reverse internal P loading from sediments, several remediation technologies have been proposed, including ex situ remediation (sediment dredging and treatment and disposal of dredged sediment) (Oldenborg and Steinman 2019; Chen et al. 2020), oxygenation (Prepas and Burke 1997), in situ chemical precipitation of sedimentary P with aluminum salt (Hansen et al. 2003), in situ inert capping using non-active materials (e.g., sand) (Kim and Jung 2010), and in situ active capping (Akhurst et al. 2004; Berg et al. 2004; Yin and Kong 2015; Zou et al. 2017; Xiong et al. 2018; Bonaglia et al. 2019; Gu et al. 2019; Li et al. 2019b; Zhan et al. 2019). Among these techniques, sediment remediation based on in situ active capping is considered to be a very promising method for intercepting P release from sediment and has attracted widespread attention recently (Akhurst et al. 2004; Berg et al. 2004; Gibbs and Özkundakci 2011; Yin and Kong 2015; Zou et al. 2017; Xiong et al. 2018; Yu et al. 2018; Bonaglia et al. 2019; Gu et al. 2019; Li et al. 2019b; Zhan et al. 2019). The advantages of this method included application convenience, simple operation, high efficiency, and low cost (Gu et al. 2019; Wang et al. 2019; Zhan et al. 2020).
The choice of a suitable P-inactivating solid-phase agent (PIA) as an active covering material is a key to the use of the in situ active capping technology for the control of sedimentary P release (Spears et al. 2013; Mucci et al. 2018). An ideal active capping material should have the following advantages: environmental friendliness, high P immobilization efficiency, and low cost (Wang et al. 2012; Spears et al. 2013; Mucci et al. 2018). Up to now, a lot of PIAs have been investigated as active capping materials for the suppression of P liberated from IPS, such as bentonite (Gu et al. 2019), bentonite modified with zirconium (Lin et al. 2019a), attapulgite modified with thermal treatment (Yin and Kong 2015), CaO2-based material (Zhou et al. 2019, 2020), zeolite (Xiong et al. 2018; Gu et al. 2019), iron-modified zeolite (Zhan et al. 2019), aluminum-modified zeolite (Gibbs and Özkundakci 2011), activated carbon (Bonaglia et al. 2019), lanthanum-modified bentonite (Spears et al. 2013; Wang et al. 2017; Li et al. 2019b), aluminum-modified attapulgite (Yin et al. 2018), lanthanum/aluminum co-modified attapulgite (Yin et al. 2020), illite (Gu et al. 2019), lanthanum hydroxide (Lin et al. 2019b), ferrihydrite (Zou et al. 2017; Yu et al. 2018), industrial by-products (Spears et al. 2013), and calcite (Berg et al. 2004; Zou et al. 2017). However, further development of new, safe, high-efficiency, and low-cost active covering materials to suppress sedimentary P liberation is still imperative.
Calcite is a common mineral in the hydrosphere and lithosphere and has a certain affinity towards aqueous phosphate (Karageorgiou et al. 2007; Sø et al. 2011; Li et al. 2017). The utilization of calcite as an active covering material to manage P release from IPS has a number of advantages. For example, calcite is a non-toxic material and represents a common constituent of sediments (Berg et al. 2004; Sø et al. 2011; Li et al. 2017). Furthermore, calcite has the absolute advantage of a low cost. In addition, calcite capping could decrease the releasing efflux of soluble reactive P (SRP) from sediments into overlying waters (OL-WTs) (Berg et al. 2004; Lin et al. 2011; Zou et al. 2017). However, the efficiency of the control of SRP release from sediment by calcite capping is not very high (Berg et al. 2004; Lin et al. 2011; Zou et al. 2017). Thus, modification of calcite to enhance its phosphate immobilization ability is very necessary for the utilization of the calcite-based capping technology to effectively suppress sedimentary P liberation. Iron is an eco-friendly element (Li et al. 2019a), and iron oxide possesses high affinity towards phosphate in water (Ajmal et al. 2018; Hilbrandt et al. 2019; Lyngsie et al. 2019). If iron oxide is loaded onto the surface of calcite, the resultant Fe-coated calcites (FMCAs) may have higher phosphate immobilization ability than the raw calcite (RCA). Consequently, FMCAs may be more promising active capping materials for the suppression of P released from IPS than RCA.
Since iron oxide is very effective for phosphate removal from water (Ajmal et al. 2018; Hilbrandt et al. 2019; Lyngsie et al. 2019), the phosphate removal ability for FMCA may vary with its Fe loading amount. Understanding the difference of phosphate sorption performance among FMCAs with different Fe loading amounts is absolutely critical to determining the efficiency and mechanism of FMCA capping to inhibit the liberation of sedimentary P. Furthermore, knowing the impact of FMCA capping on the mobilization of P in sediments at various depths is also critical in determining the efficiency and mechanism of FMCA capping for the interception of sediment-P release. However, there are few studies on the efficiency evaluation of the control of P release from IPS by FMCA capping based on the effect of iron loading amount on the phosphate removal and the influence of FMCA capping on the mobilization of P in sediments at various depths. In addition, there is still a lack of enough understanding about the mechanism for the interception of P release from IPS by the FMCA capping.
This research aimed to evaluate the efficiency of FMCA capping for preventing sedimentary P liberation as well as to elucidate the controlling mechanism. For this purpose, the performances of phosphate elimination from water by FMCAs with different loading amounts of iron were comparatively investigated, the mechanism for the phosphate removal by FMCA was explored, and the impact of FMCA capping on the mobilization of P in sediments at various depths was studied. Results of this investigation will provide a solid basis for the use of FMCA as an active covering material to control the release of P from IPS in freshwater systems.
2 Materials and methods
2.1 Materials
The sediment samples utilized in this research were collected from a small-sized, closed, and shallow freshwater body, which is situated in Pudong New District, Shanghai City, China. Before its use, the collected sediment samples were air-dried, after which they were crushed, passed through a standard 100 mesh (0.15 mm) sieve, and completely mixed. The raw calcite with a particle size of less than 0.075 mm (through a standard 200 mesh sieve) was used in this research, and it was purchased from a factory situated in Changxing County, Zhejiang Province, China. The reagents utilized in this study were acquired from Sinopharm Chemical Reagent Co., Ltd., and all these chemicals were analytical pure. The devices of high-resolution dialysis (HR-Peeper) and ZrO-Chelex diffusive gradient in thin films (ZrO-Chelex DGT) adopted in the present research were acquired from Nanjing EasySensor Environmental Technology Co., Ltd.
2.2 Preparation and characterization of FMCAs
The preparation process of FMCAs was listed as follows. Firstly, 100 mL of deionized water (DW) was added into each of four 500-mL conical bottles, and 2, 4, 6, and 8 g of FeCl3·6H2O were also added into these conical bottles, respectively. Then, 20 g of calcite was introduced into each of these conical bottles. After that, all the conical bottles were placed on a constant temperature water bath oscillator and shaken at a shaking speed of 150 rpm and a fixed temperature of 298 K for 24 h. After the completion of the reaction, the solid materials were gathered by the centrifugal separation method, and then, they were cleaned with DW for three times. Afterwards, the solid materials were dried at 105 °C in an oven. After drying, the solid materials were ground and kept in self-sealing bags for the further studies. Hereafter, the resulting FMCAs under the preparation conditions of the FeCl3·6H2O/RCA mass ratios of 0.1, 0.2, 0.3, and 0.4 were named as FMCA1, FMCA2, FMCA3, and FMCA4, respectively.
The crystal phases of RCA and FMCAs were revealed by employing X-ray diffraction analysis (XRD) (Ultima IV, Rigaku Corporation, Japan). The chemical compositions of RCA and FMCAs were determined by employing X-ray fluorescence spectrometer (XRF) (XRF-1800, Shimadzu Corporation, Japan). An ASAP2460-specific surface area and aperture analyzer (Micromeritics Instrument Corporation, USA) was employed to acquire the nitrogen adsorption/desorption isotherms of RCA and FMCAs at 77 K. On the basis of the corresponding experimental isotherm data, the Brunauer-Emmett-Teller (BET) specific surface areas (SSAs), total pore volumes (TPVs), and pore size (PS) distributions of RCA and FMCAs were further computed. The chemical states of FMCA4 samples without and with the phosphate sorption at solution pH 7 were analyzed by employing X-ray photoelectron spectroscopy (XPS) (AXIS UltraDLD, Shimadu-Kratos Corporation). All the values of binding energy (BE) were calibrated by adopting the value of BE for C 1s at 284.8 eV, and all the XPS spectra were fitted by XPSPeak41 software.
2.3 Batch sorption experiments
Batch experiment tests were carried out to explore the influence of reaction time on the phosphate sorption onto RCA and FMCAs and to make an investigation on the influence of FMCA dosage and initial concentration of phosphate on the sorption of phosphate onto FMCAs. All the batch sorption experiments were implemented in duplicate in 50-mL conical flasks, which contained 25-mL of phosphate solution and a certain mass of RCA or FMCA. The conical flasks were placed on a stable temperature water bath oscillator and shaken at 150 rpm and 298 K for a defined contact time. After the sorption reaction was completed, the water samples were acquired by centrifugation, and the concentration level of phosphate in the as-obtained supernatant was determined by the spectrophotometry based on the molybdenum blue reaction. All the results were reported as the mean values and standard deviations. With regard to the study concerning the impact of reaction time, the initial pH value was 7, the initial concentration of phosphate was 10 mg L−1, the sorbent dosage was 25 mg, and the time of reaction was set to be 1, 2, 4, 6, 8, 16, 24, 48, and 76 h, respectively. In the sorbent dosage influence experiment, the dosages of FMCAs were set to be 10, 30, 50, 60, 80, and 100 mg, the time of reaction was 24 h, the initial pH of solution was 7, and the initial concentration of phosphate was 10 mg L−1. Regarding the research on the influence of initial phosphate concentration, the dosage of FMCA was set to be 25 mg, the initial pH of solution was 7, the initial concentrations of phosphate were 1, 2, 4, 6, 8, and 10 mg L−1, and the reaction time was 24 h. The phosphate sorption quantity (Q, mg P g−1) and removal efficiency (RE, %) were computed according to Eq. (1) and Eq. (2), respectively.
where ci and cf represent the aqueous phosphate concentrations prior to and after the sorption of phosphate, respectively (mg P L−1); V stands for the volume of phosphate solution (mL); M is the mass of the added sorbent (mg).
2.4 Sediment remediation experiments
We conducted sediment incubation experiments to explore the effect of RCA and FMCA capping on the mobilization of phosphorus in sediment using a series of glass bottles with an internal diameter of about 10 cm and a volume of about 1800 mL. In order to build sediment cores, the completely mixed sediment was introduced into the glass bottle, and the height of the sediment core was 60 mm. Then, three types of sediment treatments were designed (two glass bottles for each treatment) as follows. The sediment core in column I was subjected to an identical condition without adding capping materials, and this column was named as control column. The sediment core in column II was evenly capped with 20 g calcite (2.55 kg m−2), and this column was named as RCA capping column. The sediment core in column III was evenly capped with 20 g FMCA1 (2.55 kg m−2), and this column was named as FMCA capping column. After that, a mixed solution of NaCl/NaHCO3/CaCl2 was prepared, and the concentration levels of NaCl, NaHCO3, and CaCl2 in the as-prepared solution were 10, 1, and 1 mmol L−1, respectively. Then, the pH level of the as-prepared solution was adjusted to 7.5, and the concentration level of DO in the as-prepared solution was reduced to less than 0.5 mg L−1 by employing the sodium sulfite oxidation method (Kim et al. 2003). After that, the uncapped and capped sediment cores were covered with the as-prepared solution, and then, the bottle mouth was sealed by the rubber plug and white vaseline in order to make all the sediment cores in an anoxic environment. Then, all the sediment cores were away from light and were incubated under indoor temperature. On day 73, the DO concentrations and pH levels of OL-WT in all the columns were monitored by employing a portable DO monitoring device and pH meter, respectively. In the meanwhile, water samples were taken out from all the columns, and the concentration levels of SRP in the water samples were monitored by employing the spectrophotometry based on the molybdenum blue reaction. On day 86, the devices of HR-Peeper and ZrO-Chelex DGT were deployed into the sediment cores of different columns. After 24 h of balanced reaction, the probes of ZrO-Chelex DGT in all the columns were collected from the sediment cores and the sediments clung to the DGT surfaces were cleaned by DW. Then, the probes of ZrO-Chelex DGT were used to analyze the concentrations of DGT labile P and Fe (hereafter abbreviated as DGT-P and DGT-Fe, respectively) in the profiles of OL-WT and sediment. The procedure for measuring DGT-P and DGT-Fe in the sediment/OL-WT profile was introduced in detail in a previous literature (Liu et al. 2018). After 48 h of balanced reaction, the probes of HR-Peeper were collected from the sediment cores and the sediments adhered to the HR-Peeper surfaces were eliminated with tissue. Afterwards, the aqueous solution was taken out from each cell of the HR-Peeper device collected, and these water samples were kept in a series of 4-mL centrifuge tubes, respectively. To avoid being oxidized, 20 μL of HCl solution with a concentration of 100 mmol L−1 was then introduced into each of the collected centrifuge tubes. The molybdenum-blue spectrophotometry was adopted to measure the concentration levels of SRP in the aqueous solutions of the centrifuge tubes on a microplate spectrophotometer at a detecting wavenumber of 630 nm. The o-phenanthroline spectrophotometry was employed to determine the concentration levels of Fe2+ in the aqueous solutions of the centrifuge tubes on a microplate spectrophotometer at a detecting wavenumber of 470 nm.
3 Results and discussion
3.1 Characterization of FMCAs
The XRD patterns of RCA and FMCAs are shown in Fig. 1. From Fig. 1, it was observed that the main X-ray diffraction peaks of RCA were in accordance to those of pure calcite (JCPDS 01-086-2334). This suggests that the RCA used in this study was chiefly composed of calcite. The XRD patterns of FMCAs showed that the main X-ray diffraction peaks of FMCA1, FMCA2, FMCA3, and FMCA4 were also consistent with those of pure calcite (JCPDS 01-086-2334). This reveals that the modification of RCA with Fe salts did not lead to an obvious change of the crystal phase of RCA. That is to say, the as-prepared FMCAs contained calcite. Table 1 shows the chemical compositions of RCA and FMCAs. The XRF data indicate that the major elementary component of RCA is Ca with small amounts of Mg, Al, Si, and Fe, while the main elementary components of FMCA1, FMCA2, FMCA3, and FMCA4 are Ca and Fe with small amounts of Mg, Al, and Si. This further confirms that the RCA utilized in this work is mainly composed of calcite. This also suggests that the as-prepared FMCAs are mainly composed of calcite and iron oxide. Furthermore, it is noted that the content of Fe in FMCAs increases in the order of FMCA1 < FMCA2 < FMCA3 < FMCA4, which suggests that the more the quantity of iron salt added during the preparation process, the more the amount of Fe loaded on the surface of calcite. The nitrogen adsorption/desorption curves of RCA and FMCAs are given in Fig. 2. The calculated BET surface areas and total volumes are also listed in Fig. 2. It was shown that the nitrogen adsorption isotherms of RCA and FMCAs all belonged to type IV isotherm with a hysteresis loop at a high value of relative pressure (P/P0), which suggests the presence of mesopores (2–50 nm) (Liu et al. 2019; Xu et al. 2020). Moreover, the content of nitrogen adsorbed increased sharply with the increase of P/P0 at a low P/P0 value, demonstrating the presence of micropores (< 2 nm) (Lu et al. 2020). In addition, the isotherms of RCA and FMCAs raised quickly near P/P0 = 1, suggesting the presence of macropores (> 50 nm) (Qiu and Duan 2019). Thus, the RCA and FMCAs employed in this research have a wide pore size distribution and possess micropores, mesopores, and macropores. The average PSs of RCA, FMCA1, FMCA2, FMCA3, and FMCA4 were calculated to be 16.0, 7.75, 4.77, 3.88, and 3.85 nm, respectively. Obviously, the modification of RCA with Fe reduced the mean PS of the solid material. Furthermore, the TPVs of RCA, FMCA1, FMCA2, FMCA3, and FMCA4 were found to be 0.00305, 0.0171, 0.0215, 0.0310, and 0.0365 cm3 g−1, respectively, and the BET SSAs of RCA, FMCA1, FMCA2, FMCA3, and FMCA4 were determined to be 1.11, 8.64, 18.9, 33.0, and 38.7 m2 g−1, respectively. These results indicate that the modification of RCA with Fe tremendously increases the TPV and SSA of the solid material, and the more the amount of iron salt added during the preparation process, the more the TPV and SSA of the as-synthesized FMCA. It should be noted that a high specific surface area of an adsorbent may be favorable for pollutant adsorption from aqueous solution. The volumes of micropores and mesopores in FMCAs were much larger than that in RCA, and that in FMCAs increased in the order of FMCA1 < FMCA2 < FMCA3 < FMCA4. This demonstrates that the generated iron oxide on the surface of RCA has plenty of micropores and mesopores.

XRD patterns of a RCA, b FMCA1, c FMCA2, d FMCA3, and e FMCA4

N2 adsorption/desorption isotherms of a RCA, b FMCA1, c FMCA2, d FMCA3, and e FMCA4; f pore size distributions of RCA, FMCA1, FMCA2, FMCA3, and FMCA4
3.2 Comparison of phosphate removal by FMCAs with different Fe loading amounts and removal mechanism of phosphate by FMCA
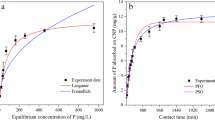
The influence of contact time on the removal of phosphate by RCA, FMCA1, FMCA2, FMCA3, and FMCA4 is given in Fig. 3a. From Fig. 3a, it was observed that the phosphate removal efficiency of RCA was very low (0.50–1.50%) at the reaction time of ≤ 8 h, but it slightly rose to 5.36–7.33% as the reaction time increased from 8 to 16–24 h, and greatly increased to 80.2% with an increase of reaction time from 24 to 76 h. This indicates that the RCA employed in this study has low phosphate removal ability at a relatively high concentration of phosphate during an early reaction stage, but it has high removal ability for phosphate in the later reaction stage. Previous studies have shown that the phosphate removal mechanism of calcite depended upon the initial concentration of phosphate in aqueous solution and reaction time, and the elimination of phosphate by calcite was primarily controlled by adsorption at a relatively low concentration of phosphate and a reactively short reaction time (Sø et al. 2011; Li et al. 2017). The maximum phosphate monolayer adsorption capacity for calcite shown in a previous literature (Sø et al. 2011) was found to be 1.4 μmol m-2. Based on this data, the theoretical maximum phosphate monolayer adsorption capacity for RCA can be determined to be about 0.05 mg g−1 because the surface area of RCA was 1.11 m2 g−1. In addition, the desorption of phosphate from the phosphate-adsorbed calcite can be complete in less than 0.5 h (Sø et al. 2011). Thus, RCA is expected to have a low adsorption capacity for aqueous phosphate, and the interaction between the adsorbed phosphate and the RCA surface is expected to be weak. Previous studies also have shown that the elimination of phosphate by calcite at a high concentration of phosphate mainly involves two mechanisms: adsorption and precipitation (Sø et al. 2011; Liu et al. 2012; Li et al. 2017). In other words, the removal process of phosphate at a high concentration from water by calcite has the following features: an initial rapid adsorption of phosphate on calcite, followed by the precipitation of calcium phosphate on calcite (Sø et al. 2011; Liu et al. 2012; Li et al. 2017). The amounts of phosphate sorbed onto RCA were calculated to be 0.05–0.15 mg g−1 at the reaction time of ≤ 8 h according to the phosphate removal efficiency, and these values were very close to the predicted maximum phosphate adsorption capacity for RCA (0.05 mg g−1). Thus, we deduced that the removal of phosphate by the RCA utilized in this research at a relatively high concentration of phosphate (e.g., 10 mg L−1) in the early reaction stage was primarily dominated by the surface adsorption mechanism, while the uptake of phosphate by the RCA employed in this work during the later reaction was primarily dominated by the calcium phosphate precipitation mechanism. From Fig. 3a, it also can be observed that the phosphate removal efficiencies of FMCA1, FMCA2, FMCA3, and FMCA4 tended to increase on the whole with the increase of reaction time. Furthermore, the removal efficiencies of phosphate by FMCA1, FMCA2, FMCA3, and FMCA4 were higher than that of RCA at reaction time of ≤ 24 h, and the difference in the efficiency of phosphate removal between FMCA and RCA increased with the increase of the Fe loading amount on the surface of calcite. This means that the modification of RCA with Fe enhances its phosphate removal ability during the early reaction stage, and this enhancement will increase as the loading amount of Fe on RCA increases. Previous literatures have observed that iron oxide possesses strong affinity towards aqueous phosphate, and the ligand-exchange reaction between phosphate and surface hydroxyl group to form the inner-sphere Fe-O-P bonding mainly controls the uptake of phosphate by iron oxide (Cao et al. 2016; Lin et al. 2020; Qiu et al. 2020). Thus, we deduced that the improvement of phosphate sorption by the modification of RCA with Fe could mainly thank to the generation of inner-sphere Fe-phosphate complexes through the ligand-exchange reaction of Fe-bound hydroxyl groups with phosphate. At a relatively short reaction time (≤ 8 h), the major mechanisms for the elimination of phosphate by FMCAs are expected to be the surface adsorption of phosphate onto calcite and Fe species. At a relatively long reaction time (≥ 8 h), phosphate could be removed by FMCAs mainly through the surface adsorption of phosphate onto calcite, the surface adsorption of phosphate onto iron species, and the precipitation of calcium phosphate.

a Effect of contact time on phosphate removal by RCA and FMCAs; impact of FMCA dosage on b phosphate removal efficiency and c amount of phosphate removed by FMCA; d influence of initial phosphate concentration on the removal of phosphate by FMCAs; e sorption isotherms of phosphate on FMCAs
The effects of FMCA dosage on the efficiencies of phosphate removal by FMCAs and the amounts of phosphate sorbed onto FMCAs are presented in Fig. 3b and c, respectively. As shown in Fig. 3b, when the FMCA dosage rose from 0.4 to 4.0 g L−1, the efficiencies of phosphate removal by FMCA1, FMCA2, FMCA3, and FMCA4 at reaction time of 24 h rose from 3.31 to 42.7%, 6.44 to 62.5%, 8.45 to 88.5%, and 10.5 to 94.2%, respectively. This means that a high FMCA dosage will be conductive to the elimination of phosphate from water, and the addition of sufficient dose of FMCA into water could reduce the concentration of aqueous phosphate to a low level. From Fig. 3b, the phosphate removal efficiency of FMCAs at sorbent dosages of 1.2–4.0 g L−1 increased in the order of FMCA1 < FMCA2 < FMCA3 < FMCA4, which further suggests that a higher loading amount of Fe on RCA will be beneficial to the elimination of phosphate from water. As depicted in Fig. 3c, as the sorbent dosage was in the range of 0.4 and 4.0 g L−1, the amounts of phosphate sorbed onto FMCA1, FMCA2, FMCA3, and FMCA4 at reaction time of 24 h fluctuated between 0.827 and 1.24, 1.56 and 1.69, 2.11 and 2.68, and 2.36 and 3.11 mg P g−1, respectively. This demonstrates that phosphate can be effectively accumulated on the surface of FMCA, and the maximum quantity of phosphate sorbed onto FMCA could come up to 3.11 mg P g−1.
The influence of initial phosphate concentration on the sorption of phosphate on FMCAs is given in Fig. 3d. It was shown that the phosphate sorption capacity for FMCA tended to increase with the increase of initial phosphate concentration, and it also tended to increase with the augment of the loading amount of Fe on RCA. From Fig. 3d, it also can be seen that the maximum amount of phosphate sorbed onto FMCA could reach 4.99 mg P g−1, which was relatively high. This is beneficial to the practical application of FMCA as an active capping material for the management of P released from sediment. The mechanism of phosphate sorption can be revealed by sorption isotherm models. The experimental data in Fig. 3d were further analyzed by three common isotherm models including the Langmuir (Eq. (3)), Freundlich (Eq. (4)), and Dubinin-Radushkevich (D-R) (Eq. (5)) isotherm models (Langmuir 1916; Freundlich 1926; Mahmoud et al. 2020), and the results are given Fig. 3e and Table 2.
where qf is the amount of phosphate sorbed on a sorbent after sorption (mg g−1); cf is the concentration level of aqueous phosphate after sorption (the unit is mg L−1 for the Langmuir and Freundlich models, and it is mol L−1 for the D-R model); Qm is the maximum quantity of phosphate sorbed onto a sorbent predicted from the Langmuir isotherm model (mg g−1); kL (L mg−1) and kF (mg(1-1/n) L1/n g−1) are the constants of Langmuir and Freundlich models, respectively; 1/n is also the Freundlich constant; QDR is the maximum content of phosphate sorbed on a sorbent predicted from the D-R isotherm equation (mg g−1); kDR is the constant of D-R (mol2 kJ−2); E is the average free energy of sorption (kJ mol−1).
The coefficients of correlation (R2) of the Freundlich and D-R isotherm models were higher than that of the Langmuir isotherm model as shown in Table 2. Thus, the Freundlich and D-R isotherm models are the better-fitted models for the sorption of phosphate on FMCAs than the Langmuir isotherm model. The calculated 1/n values between 0 and 1 suggested favorable sorption of phosphate on FMCAs (Lin and Zhan 2012). The calculated kF value for the sorption of phosphate on FMCAs increased in the order of FMCA1 < FMCA2 < FMCA3 < FMCA4, further confirming the increase in the phosphate sorption ability with the increase of the Fe loading amount on calcite. The value of E can be used to distinguish the mechanisms of physisorption and chemisorption, and the E values in the range of 1–8 kJ mol−1 and > 8 kJ mol−1 refer to the physisorption and chemisorption types, respectively (Chabani et al. 2006; Meghdadi 2018; Abo Markeb et al. 2019; Mahmoud et al. 2020). The calculated E values for phosphate sorption onto FMCA1, FMCA2, FMCA3, and FMCA4 were 18.0, 13.5, 14.3, and 15.5 kJ mol−1, respectively, which were higher than 8 kJ mol−1. This reveals that chemisorption mechanism plays an important role in the uptake of phosphate by FMCAs. That is to say, the forming of inner-sphere Fe-O-P bonding by means of the replacement of Fe-bound hydroxyl groups (Fe-OH) with phosphate and the precipitation of calcium phosphate are expected to play a significant role in the sorption of phosphate on FMCAs.
XPS is a valuable method to explore the interaction between sorbents and sorbates during the sorption process (Jiang et al. 2018). In order to clarify the mechanism for the sorption of phosphate onto FMCA, the FMCA4 samples without and with the sorption of phosphate were comparatively characterized by the XPS analysis, and the results are given in Fig. 4. As shown in Fig. 4a, FMCA4 had the XPS peaks of Ca 2p, O 1s, C 1s, and Fe 2p on the survey spectrum. This could be attributed to the fact that FMCA4 is mainly composed of calcite and iron oxide. After the sorption of phosphate, the XPS peaks of Ca 2p, O 1s, C 1s, and Fe 2p were also present on the survey spectrum of the P-loaded FMCA4. This indicates that the P-loaded FMCA4 still contained calcite and iron oxide. Based on the experimental data listed in Fig. 4a, the atomic concentrations on the surfaces of raw and P-loaded FMCA4 were computed, and the results are listed in Table 3. It was shown that the atomic concentration of P increased from 0 to 0.59% after the sorption of phosphate on FMCA4, which indicates that phosphate has been loaded onto the surface of FMCA4 after the phosphate sorption. As depicted in Fig. 4b, the XPS peaks of Fe 2p1/2 and Fe 2p3/2 were situated at 724.58 and 710.98 eV, respectively, which were very close to those of hydroxide ferric oxide (Zhang et al. 2018; Wei et al. 2019; Lin et al. 2020). This further indicates the formation of Fe-OH on the surface of calcite after the modification of RCA with Fe. After the sorption of phosphate on FMCA4, the XPS peaks of Fe 2p1/2 and Fe 2p3/2 shifted to lower binding energy values, which confirms the occurrence of the exchange of Fe-bound hydroxyl groups (Fe-OH) with phosphate anions to generate the Fe-O-P inner-sphere complexation (Zhang et al. 2018; Lin et al. 2020). As described in Fig. 4c, the XPS peaks of Ca 2p1/2 and Ca 2p3/2 for the fresh FMCA4 were situated at 350.68 and 347.08 eV, respectively, which were very close to those of calcite. This further confirms that the Ca species in FMCAs is in form of calcite (Wei et al. 2019; Dong et al. 2020). After the sorption of phosphate on FMCA4, the XPS peaks of Ca 2p1/2 and Ca 2p3/2 slightly shifted to the lower values of binding energy. This confirms the forming of strong chemical bonds (Ca-O-P) between calcium and phosphate after the uptake of phosphate by FMCAs (Jiang et al. 2018; Dong et al. 2020). From Fig. 4d, it can be seen that the XPS peak of P 2p in FMCA4 was situated at 133.15 eV, which was higher than the BE value of P 2p in phosphate-adsorbed magnetic cationic hydrogel though cationic attraction mechanism (131.9 eV)(Dong et al. 2017). However, the XPS peak of P 2p in FMCA4 (133.15 eV) was close to that in phosphate-bound by iron oxide through an inner-sphere Fe-O-P complexation mechanism (133.08–133.60 eV) (Mallet et al. 2013; Lin et al. 2020). Thus, the interaction between phosphate and Fe/Ca is expected to be stronger than the electrostatic attraction, and the formation of chemical bonds between Fe/Ca and phosphate plays a significant role in the uptake of phosphate by FMCAs. Therefore, the XPS analysis results confirm that the formation of Fe-O-P inner-sphere complexes by way of a ligand exchange reaction and the precipitation of calcium phosphate could play an important role in the sorption of phosphate onto FMCAs.

a Full XPS spectra, b Fe 2p spectra, and c Ca 2p spectra of FMCA4 samples before and after phosphate loading; d P 2p spectrum of phosphate-loaded FMCA4
3.3 Impact of FMCA capping on concentration of SRP in OL-WT
The effect of FMCA capping on the release of sedimentary P into OL-WT was investigated by adopting the traditional active sampling technique. The results found that the concentrations of OL-WT SRP in the control, RCA capping and FMCA capping columns on day 73 were 0.283 ± 0.028, 0.219 ± 0.013, and 0.013 ± 0.009 mg L−1, respectively, and the DO concentrations of OL-WT in the control, RCA capping, and FMCA capping columns on day 73 were 0.49 ± 0.04, 0.34 ± 0.05, and 0.37 ± 0.06 mg L−1, respectively. This demonstrates that, under anoxic condition, P could be released from sediment into OL-WT. This also indicates that RCA capping could only slightly inhibit the release of SRP from sediment into OL-WT under anoxic condition, with the SRP reduction efficiency of 22.6%. However, FMCA capping can significantly inhibit the release of SRP from sediment into OL-WT under anoxic condition, with the SRP reduction efficiency of 95.4%. Thus, from a viewpoint of the efficiency of the suppression of sedimentary P liberation into OL-WT, the use of FMCA as a capping material is more promising for the management of SRP released from IPS into OL-WT than that of RCA.
3.4 Impact of FMCA capping on concentrations of SRP and Fe2+ in profile of OL-WT and sediment
In order to further understand the impact of FMCA capping on the release of sedimentary P into OL-WT, the vertical distributions of SRP and Fe2+ in the sediment/OL-WT profiles for the control, RCA capping, and FMCA capping columns on day 86 were comparatively studied by employing an in situ passive sampling techniques, i.e., HR-Peeper, and the results are given in Fig. 5. For the control column, the concentration level of SRP in the interstitial water was higher than that in the overlying water. Furthermore, it increased with increasing depth in the sediment profile within a certain depth. This indicates that, under no capping condition, P could be released from the sediment solid into the overlying water, and the risk of SRP liberated from the sediment solid into the interstitial water tends to increase with increasing depth in the sediment profile within a certain depth. Moreover, the concentration level of interstitial water Fe2+ was larger than that of OL-WT Fe2+, and it rose with increasing depth in the sediment profile within a certain depth (Fig. 5b). This demonstrates that, under no capping condition, Fe could be liberated from the sediment solid into the overlying water, and an increase in the sediment depth increases the risk of Fe2+ liberated from the sediment solid to the interstitial water within a certain depth. Furthermore, from Fig. 5d, it was found that the concentration level of SRP in the profile of sediment and OL-WT had a fairly good linear relation with the Fe2+ concentration (R2 = 0.977). This confirms that the reductive dissolution of Fe(III) oxyhydroxides (FeOOH) and then the synchronous release of Fe2+ and SRP are critical to the mobilization of P from the sediment solid to the interstitial and overlying waters under no capping condition. The release of P from sediments due to the reduction and dissolution of Fe-P compounds has also been observed in previous studies (Gao et al. 2016; Yang et al. 2019; Norgbey et al. 2020; Wang et al. 2020; Yuan et al. 2020). From Fig. 5a, it also can be seen that RCA capping did not prevent the release of SRP from the sediment solid into the interstitial and overlying waters on day 86. It should be noted that the controlling efficiency of sediment-P liberation to OL-WT by RCA capping on day 73 was 22.6%, which was different to that on day 86. This may be attributed to the fact that although RCA capping could decrease the risk of sediment-P release during the early stage of RCA application, it could not control the release of P from sediment during the later stage of RCA application. In contrast to RCA capping, FMCA capping can effectively inhibit the liberation of SRP from sediment to interstitial and overlying waters (Fig. 5a). The reduction rates of OL-WT SRP by the FMCA capping layer were computed to be 16.7–60.0%, and the efficiencies of the reduction of interstitial water SRP in the top 20 mm sediments by the FMCA capping layer were found to be in the range of 48.2% and 93.9% (Fig. 5c). Under FMCA capping condition, an obvious stratification of interstitial water SRP appeared in the profile of sediment. This phenomenon was highlighted by the forming of a phosphorus static layer in the top sediment (average concentration: 0.0169 mg L−1; depth: 10 mm), followed by an active sediment layer (average concentration: 0.135 mg L−1; depth: 10 mm). Similar results have also been observed for sediment capping with lanthanum-modified bentonite (Wang et al. 2017) and zirconium-modified bentonite (Lin et al. 2019a). From Fig. 5b, it was observed that RCA capping had no negative impact on the concentration levels of Fe2+ in the overlying water and the interstitial water of the top 5 mm sediment but had a certain effect of interstitial water in the lower sediment, while FMCA capping greatly decreased the concentration level of Fe2+ in the overlying water and the top 20 mm sediment, with the reduction rates of 27.8–95.0% (Fig. 5c). This suggests that the Fe2+ species in the overlying and interstitial waters could be effectively removed by the FMCA capping layer, resulting in an accumulation of Fe on the surface of FMCA in the capping layer. This also indicates that sediment capping using FMCA does not give rise to an increase in the risk of Fe released from sediment to OL-WT. In the RCA capping column, the SRP concentration of overlying and interstitial waters had a fairly good linear relation with the concentration of Fe2+ (Fig. 5e), which indicates that the cycle of Fe-coupled P is an important mechanism governing the release of SRP from the sediment solid into the interstitial water under RCA capping condition. In the FMCA capping column, the concentration of interstitial water SRP in the active layer of sediment had a relatively good linear relation with the concentration of interstitial water Fe2+ (Fig. 5f), which means that the iron-coupled P cycle plays a significant role in the release of SRP from the active sediment layer to the interstitial water under FMCA capping condition.

Vertical distributions of a SRP and b Fe2+ in different columns; c reduction rates of SRP and Fe2+ by FMCA capping; relationships between SRP and Fe2+ for d control, e RCA capping, and f FMCA capping columns
It is widely accepted that there are two basic steps for the upward migration of P from sediment into OL-WT (Boström et al. 1988; Lin et al. 2019c). The first step is the transport of P from sediment to interstitial water, and the second step is the diffusion of SRP from interstitial water to OL-WT (Boström et al. 1988; Lin et al. 2019c). Without capping, P could be liberated from sediment into the interstitial water via the reductive dissolution of iron(III) oxides/hydroxides, bringing about a high concentration level of interstitial water SRP in the sediment layer. The SRP in the interstitial water could be further transported into the overlying water through a molecular diffusion mechanism, thus bringing about the release of P from sediment to the overlying water and the high concentration level of SRP in the overlying water under anoxic condition. With RCA capping, on one hand, P could be liberated from sediment to the interstitial water through the reductive dissolution of iron(III) oxides and hydroxides, thereby resulting in a high concentration of SRP in the interstitial water; but on the other hand, the RCA capping layer may have a low adsorption ability for the interstitial water SRP at a low concentration, possibly leading to a slight influence on the concentration of interstitial water SRP. This may give a rise to a high concentration level of SRP in the interstitial water under RCA capping condition. The high concentration level of SRP in the interstitial water and the low phosphate adsorption capacity for the RCA capping layer may cause a high releasing flux of SRP from the RCA-capped sediment to the overlying water. This may eventually lead to the fact that the RCA capping layer had a low efficiency of the suppression of SRP liberation from sediment to the overlying water or cannot prevent the release of SRP from sediment into the overlying water compared with no capping. With FMCA capping, P still could be released from sediment to the interstitial water via the Fe-P coupling mechanism, thereby resulting in the increase of the interstitial water SRP concentration. However, the FMCA capping layer could effectively adsorb the SRP in the interstitial water due to the strong affinity of FMCA towards aqueous phosphate, bringing about the decrease of the interstitial water SRP concentration level. This will result in a low concentration of SRP in the interstitial water under FMCA capping condition, especially a very low concentration of SRP in the top sediment (i.e., the formation of a static layer in the top sediment). The very low concentration of interstitial water SRP in the top sediment and the good phosphate adsorption performance of the FMCA capping layer will generate the low releasing flux of SRP from the FMCA-capped sediment to the overlying water and the high controlling efficiency of sediment-P release to the overlying water by the FMCA capping layer. Thus, the forming of a static layer in the top sediment caused by the sorption of SRP on the FMCA capping layer should be of vital importance to the suppression of SRP released from IPS into OL-WT by the use of FMCA as an active capping material.
It should be noted that FMCA capping also could reduce the concentration level of interstitial water Fe2+ in the upper sediment, especially could significantly reduce the concentrations of interstitial water Fe2+ in the top 20 mm sediment layer, thereby giving rise to the accumulation of Fe on the FMCA capping layer. Ding et al. (2018) conducted an experimental research concerning the effect of lanthanum-modified bentonite clay (LMB) on the transport and transformation of Fe in sediment and found that the application of LMB could bring about the decrease of the concentration of Fe2+ in the interstitial water, thereby causing the accumulation of Fe in the LMB capping layer (Ding et al. 2018). This study also found that the adsorption of Fe on LMB is very important to the stabilization of LMB-bound P (Ding et al. 2018). Thus, the sorption of Fe on the FMCA covering layer may be important to the stabilization of FMCA-bound P. However, this inference still needed to be further confirmed.
3.5 Impact of FMCA capping on concentrations of DGT-P and DGT-Fe in sediment/OL-WT profile
In order to further illuminate the effect of FMCA capping on P release from sediment into OL-WT, the vertical distributions of DGT-P and DGT-Fe in the sediment/OL-WT profiles in the control, RCA capping, and FMCA capping columns on day 86 were comparatively studied by employing an in situ passive sampling techniques, i.e., ZrO-Chelex DGT, and the results are given in Fig. 6. A DGT-labile element in sediment not only comes from the dissolved fraction of the element in the interstitial water, but also comes from the labile fraction of the element in the sediment solid that can be dynamically released to resupply the element concentration in the interstitial water (Lin et al. 2017). The concentration of DGT-P in sediment could be used to reflect the risk of P release from sediment, and the combined use of DGT-P and DGT-Fe could be used to reflect the coupled cycle of P and Fe (Gao et al. 2016; Chen et al. 2019; Yuan et al. 2020). As shown in Fig. 6a and b, on the whole, the concentration levels of DGT-P and DGT-Fe in the sediment layers were greater than those in the overlying water, and they tended to rise with increasing depth in the sediment profile within a certain depth range. These results mean that P and Fe could be released from sediment into the interstitial water, and then, they could be transported into the overlying water. Furthermore, the concentration level of DGT-P in the profile of sediment and OL-WT in the control column had a fairly good linear relation with the concentration of DGT-Fe (Fig. 6d), which further confirms the simultaneous liberation of phosphorus and iron into the overlying and interstitial waters via the mechanism of Fe-coupling P cycle. Furthermore, the vertical distributions of DGT-P and DGT-Fe in the profiles of sediment and OL-WT in the RCA and FMCA capping columns were similar to those in the control column (Fig. 6a and b), and the concentrations of DGT-P in the RCA and FMCA capping columns had a fairly good linear relation with those of DGT-Fe (Fig. 6e, f). This demonstrates that P also could be released from the RCA-capped and FMCA-capped sediments under our experimental condition through the Fe-coupled P cycling mechanism. Compared with the control column, the concentrations of DGT-P in the overlying water and in the top 18 mm sediment for the RCA capping column were larger (Fig. 6a). This result suggests that the RCA capping cannot control the release of sediment-P into the interstitial water of the top sediment layer and to the overlying water during the later stage of RCA application. However, the concentration levels of DGT-P in the overlying water and the top 22 mm sediment under the condition of FMCA capping were lower than those under the condition of no capping, with the reduction efficiencies of 13.2–93.2% (Fig. 6a and c). This demonstrates that the FMCA capping could effectively inhibit the release of sediment-P into the interstitial water of the top sediment layer and to the overlying water. Similar to the effect of FMCA capping on the concentration of interstitial water SRP in the sediment layer, FMCA capping also led to the stratification of DGT-P in the sediment layer, i.e., the generation of a phosphorus static layer in the top sediment as well as a phosphorus active layer in the lower sediment. In addition to the reduction of DGT-P, the FMCA capping also could reduce the concentrations of DGT-Fe in the overlying water and the top 22 mm sediment layer, with the reduction rates of 12.6–95.9% (Fig. 6b, c). This further confirms that the FMCA capping cannot lead to the increase in the releasing risk of sedimentary Fe to the overlying water. This also further confirms the accumulation of Fe on the FMCA capping layer after its application.

Vertical distributions of a DGT-P and b DGT-Fe in different columns; c reduction rates of DGT-P and DGT-Fe by FMCA capping; relationships between DGT-P and DGT-Fe in sediments for d control, e RCA capping, and f FMCA capping columns
3.6 Implication for application
The present study observed that RCA capping could only slightly decrease the concentration of SRP in OL-WT during the early stage of RCA application or cannot control the release of SRP from sediment to OL-WT during the later stage of RCA application, while sediment capping using FMCA can suppress the release of SRP from sediment to OL-WT effectively. In addition, FMCA exhibited stronger phosphate adsorption ability for aqueous phosphate than RCA. Thus, FMCA is a more promising capping material for the control of P release from sediment than RCA from the point of view of the interception efficiency. For the application of FMCA as an active capping material to intercept sedimentary P liberation, the optimization of the mass ratio of Fe salt to RCA during the preparation process of FMCA is of the utmost importance. This is because the increase in the mass ratio of Fe salt to RCA during the FMCA preparation process could increase its phosphate adsorption capacity, but the price of the as-synthesized FMCA also increases. Our study found that the phosphate removal performance of FMCAs increased in the order of FMCA1 < FMCA2 < FMCA3 < FMCA4. However, the price of FMCAs also increases in the order of FMCA1 < FMCA2 < FMCA3 < FMCA4. Therefore, FMCA1 could be considered to be a good FMCA choice for the control of P release from IPS in freshwater bodies from the point of view of the remediation cost, while FMCA4 could be considered to be a good FMCA choice for the suppression of sedimentary P release from the standpoint of the controlling efficiency.
4 Conclusions
FMCA exhibited better phosphate adsorption ability for phosphate in water than RCA, and the phosphate removal efficiency of FMCA increased with its loading amount of Fe. High FMCA dosage is favorable for the elimination of phosphate from water, and the quantity of phosphate removed by FMCA increased with the increase of initial phosphate concentration. The generation of Fe-O-P inner-sphere complexes by way of a ligand exchange reaction and the precipitation of calcium phosphate could be of vital importance to the sorption of phosphate onto FMCA. FMCA capping not only led to the reductions in the concentrations of SRP and DGT-P in the overlying water, but also brought about the reductions in the concentrations of interstitial water SRP and DGT-P in the top sediment. The reduction of interstitial water SRP and DGT-P in the top sediment by the FMCA capping layer played a huge role in the suppression of the release of SRP from sediment to OL-WT. In addition, FMCA capping brought about the reductions of the concentrations of Fe2+ and DGT-Fe in the overlying water and the upper sediment, suggesting a low risk of Fe releasing from the FMCA capping layer. All results indicate that FMCA has potential for use in the remediation of P-contaminated sediments in freshwater bodies. However, full-scale testing is still needed before the use of FMCA as an active capping material to inhibit the release of P from IPS in freshwater bodies. In addition, it is also very necessary to evaluate the stabilization of P bound by FMCA after its application.
References
Abo Markeb A, Llimós-Turet J, Ferrer I, Blánquez P, Alonso A, Sánchez A, Moral-Vico J, Font X (2019) The use of magnetic iron oxide based nanoparticles to improve microalgae harvesting in real wastewater. Water Res 159:490–500
Ajmal Z, Muhmood A, Usman M, Kizito S, Lu J, Dong R, Wu S (2018) Phosphate removal from aqueous solution using iron oxides: adsorption, desorption and regeneration characteristics. J Colloid Interf Sci 528:145–155
Akhurst D, Jones GB, McConchie DM (2004) The application of sediment capping agents on phosphorus speciation and mobility in a sub-tropical dunal lake. Mar Freshwater Res 55:715–725
Berg U, Neumann T, Donnert D, Nüesch R, Stüben D (2004) Sediment capping in eutrophic lakes-efficiency of undisturbed calcite barriers to immobilize phosphorus. Appl Geochem 19:1759–1771
Bonaglia S, Rämö R, Marzocchi U, Le Bouille L, Leermakers M, Nascimento FJA, Gunnarsson JS (2019) Capping with activated carbon reduces nutrient fluxes, denitrification and meiofauna in contaminated sediments. Water Res 148:515–525
Boström B, Andersen JM, Fleischer S, Jansson M (1988) Exchange of phosphorus across the sediment-water interface. Hydrobiologia 170:229–244
Cao D, Jin XY, Gan L, Wang T, Chen ZL (2016) Removal of phosphate using iron oxide nanoparticles synthesized by eucalyptus leaf extract in the presence of CTAB surfactant. Chemosphere 159:23–31
Chabani M, Amrane A, Bensmaili A (2006) Kinetic modelling of the adsorption of nitrates by ion exchange resin. Chem Eng J 125:111–117
Chen M, Ding S, Chen X, Sun Q, Fan X, Lin J, Ren M, Yang L, Zhang C (2018) Mechanisms driving phosphorus release during algal blooms based on hourly changes in iron and phosphorus concentrations in sediments. Water Res 133:153–164
Chen Q, Chen J, Wang J, Guo J, Jin Z, Yu P, Ma Z (2019) In situ, high-resolution evidence of phosphorus release from sediments controlled by the reductive dissolution of iron-bound phosphorus in a deep reservoir, southwestern China. Sci Total Environ 666:39–45
Chen C, Kong M, Wang YY, Shen QS, Zhong JC, Fan CX (2020) Dredging method effects on sediment resuspension and nutrient release across the sediment-water interface in Lake Taihu, China. Environ Sci Pollut Res 27:25861–25869
Ding SM, Sun Q, Chen X, Liu Q, Wang D, Lin J, Zhang CS, Tsang DCW (2018) Synergistic adsorption of phosphorus by iron in lanthanum modified bentonite (Phoslock®): new insight into sediment phosphorus immobilization. Water Res 134:32–43
Dong S, Wang Y, Zhao Y, Zhou X, Zheng H (2017) La3+/La(OH)3 loaded magnetic cationic hydrogel composites for phosphate removal: effect of lanthanum species and mechanistic study. Water Res 126:433–441
Dong L, Wei Q, Qin W, Jiao F (2020) Selective adsorption of sodium polyacrylate on calcite surface: Implications for flotation separation of apatite from calcite. Sep Purif Technol 241:116415
Freundlich H (1926) Colloid and Capillary Chemistry. Methuen, London
Gao YL, Liang T, Tian SH, Wang LQ, Holm PE, Bruun Hansen HC (2016) High-resolution imaging of labile phosphorus and its relationship with iron redox state in lake sediments. Environ Pollut 219:466–474
Gibbs M, Özkundakci D (2011) Effects of a modified zeolite on P and N processes and fluxes across the lake sediment–water interface using core incubations. Hydrobiologia 661:21–35
Gu BW, Hong SH, Lee CG, Park SJ (2019) The feasibility of using bentonite, illite, and zeolite as capping materials to stabilize nutrients and interrupt their release from contaminated lake sediments. Chemosphere 219:217–226
Hansen J, Reitzel K, Jensen HS, Andersen FØ (2003) Effects of aluminum, iron, oxygen and nitrate additions on phosphorus release from the sediment of a Danish softwater lake. Hydrobiologia 492:139–149
Hilbrandt I, Shemer H, Ruhl AS, Semiat R, Jekel M (2019) Comparing fine particulate iron hydroxide adsorbents for the removal of phosphate in a hybrid adsorption/ultrafiltration system. Sep Purif Technol 221:23–28
Jiang L, Li Y, Shao Y, Zhang Y, Han R, Li S, Wei W (2018) Enhanced removal of humic acid from aqueous solution by novel stabilized nano-amorphous calcium phosphate: behaviors and mechanisms. Appl Surf Sci 427:965–975
Karageorgiou K, Paschalis M, Anastassakis GN (2007) Removal of phosphate species from solution by adsorption onto calcite used as natural adsorbent. J Hazard Mater 139:447–452
Kim G, Jung W (2010) Role of sand capping in phosphorus release from sediment. Ksce J Civ Eng 14:815–821
Kim LH, Choi E, Stenstrom MK (2003) Sediment characteristics, phosphorus types and phosphorus release rates between river and lake sediments. Chemosphere 50:53–61
Langmuir I (1916) The constitution and fundamental properties of solids and liquids Part I. Solid. J Am Chem Soc 38:2221–2295
Li Z, Sun X, Huang L, Liu D, Yu L, Wu H, Wei D (2017) Phosphate adsorption and precipitation on calcite under calco-carbonic equilibrium condition. Chemosphere 183:419–428
Li XC, Yang ZZ, Zhang C, Wei JJ, Zhang HQ, Li ZH, Ma C, Wang MS, Chen JQ, Hu JW (2019a) Effects of different crystalline iron oxides on immobilization and bioavailability of Cd in contaminated sediment. Chem Eng J 373:307–317
Li XD, Zhang ZY, Xie Q, Yang RJ, Guan T, Wu DY (2019b) Immobilization and release behavior of phosphorus on Phoslock-inactivated sediment under conditions simulating the photic zone in eutrophic shallow lakes. Environ Sci Technol 53:12449–12457
Lin J, Zhan Y (2012) Adsorption of humic acid from aqueous solution onto unmodified and surfactant-modified chitosan/zeolite composites. Chem Eng J 200:202–213
Lin JW, Zhan YH, Zhu ZL (2011) Evaluation of sediment capping with active barrier systems (ABS) using calcite/zeolite mixtures to simultaneously manage phosphorus and ammonium release. Sci Total Environ 409:638–646
Lin J, Sun Q, Ding S, Wang D, Wang Y, Tsang DCW (2017) First observation of labile arsenic stratification in aluminum sulfate-amended sediments using high resolution Zr-oxide DGT. Sci Total Environ 609:304–310
Lin J, He S, Zhan Y, Zhang Z, Wu X, Yu Y, Zhao Y, Wang Y (2019a) Assessment of sediment capping with zirconium-modified bentonite to intercept phosphorus release from sediments. Environ Sci Pollut Res 26:3501–3516
Lin J, Zhao Y, Zhang Z, Zhan Y, Zhang Z, Wang Y, Yu Y, Wu X (2019b) Immobilization of mobile and bioavailable phosphorus in sediments using lanthanum hydroxide and magnetite/lanthanum hydroxide composite as amendments. Sci Total Environ 687:232–243
Lin JW, He SQ, Zhang HH, Zhan YH, Zhang ZB (2019c) Effect of zirconium-modified zeolite addition on phosphorus mobilization in sediments. Sci Total Environ 646:144–157
Lin J, Zhao Y, Zhan Y, Wang Y (2020) Influence of coexisting calcium and magnesium ions on phosphate adsorption onto hydrous iron oxide. Environ Sci Pollut Res 27:11303–11319
Liu Y, Sheng X, Dong Y, Ma Y (2012) Removal of high-concentration phosphate by calcite: effect of sulfate and pH. Desalination 289:66–71
Liu Q, Ding S, Chen X, Sun Q, Chen M, Zhang C (2018) Effects of temperature on phosphorus mobilization in sediments in microcosm experiment and in the field. Appl Geochem 88:158–166
Liu E, Chen L, Dai J, Wang Y, Li C, Yan Y (2019) Fabrication of phosphate functionalized chiral nematic mesoporous silica films for the efficient and selective adsorption of lanthanum ions. J Mol Liq 277:786–793
Lu X, He J, Xie J, Zhou Y, Liu S, Zhu Q, Lu H (2020) Preparation of hydrophobic hierarchical pore carbon–silica composite and its adsorption performance toward volatile organic compounds. J Environ Sci 87:39–48
Lyngsie G, Katika K, Fabricius IL, Hansen HCB, Borggaard OK (2019) Phosphate removal by iron oxide-coated diatomite: laboratory test of a new method for cleaning drainage water. Chemosphere 222:884–890
Mahmoud ME, El-Said GF, Rashedy IRK, Abdelfattah AM (2020) Assembly and implementation of an eco-friendly marine nanosediment for adsorptive removal of heptavalent manganese: adsorption isotherm, thermodynamic and kinetics studies. Powder Technol 359:247–260
Mallet M, Barthelemy K, Ruby C, Renard A, Naille S (2013) Investigation of phosphate adsorption onto ferrihydrite by X-ray photoelectron spectroscopy. J Colloid Interface Sci 407:95–101
Meghdadi A (2018) Characterizing the capacity of hyporheic sediments to attenuate groundwater nitrate loads by adsorption. Water Res 140:364–376
Mucci M, Maliaka V, Noyma NP, Marinho MM, Lürling M (2018) Assessment of possible solid-phase phosphate sorbents to mitigate eutrophication: Influence of pH and anoxia. Sci Total Environ 619-620:1431–1440
Némery J, Garnier J (2016) The fate of phosphorus. Nature Geosci 9:343–344
Norgbey E, Li Y, Ya Z, Li R, Nwankwegu AS, Takyi-Annan GE, Luo F, Jin W, Huang Y, Sarpong L (2020) High resolution evidence of iron-phosphorus-sulfur mobility at hypoxic sediment water interface: an insight to phosphorus remobilization using DGT-induced fluxes in sediments model. Sci Total Environ 724:138204
Oldenborg KA, Steinman AD (2019) Impact of sediment dredging on sediment phosphorus flux in a restored riparian wetland. Sci Total Environ 650:1969–1979
Prepas EE, Burke JM (1997) Effects of hypolimnetic oxygenation on water quality in Amisk Lake, Alberta, a deep, eutrophic lake with high internal phosphorus loading rates. Can J Fish Aquat Sci 54:2111–2120
Qiu B, Duan F (2019) Synthesis of industrial solid wastes/biochar composites and their use for adsorption of phosphate: from surface properties to sorption mechanism. Colloid Surface A 571:86–93
Qiu H, Ni W, Zhang H, Chen K, Yu J (2020) Fabrication and evaluation of a regenerable HFO-doped agricultural waste for enhanced adsorption affinity towards phosphate. Sci Total Environ 703:135493
Schindler DW, Hecky RE, Findlay DL, Stainton MP, Parker BR, Paterson MJ, Beaty KG, Lyng M, Kasian SEM (2008) Eutrophication of lakes cannot be controlled by reducing nitrogen input: results of a 37-year whole-ecosystem experiment. Proc Natl Acad Sci USA 105:11254–11258
Sø HU, Postma D, Jakobsen R, Larsen F (2011) Sorption of phosphate onto calcite; results from batch experiments and surface complexation modeling. Geochimica et Cosmochimica Acta 75:2911–2923
Spears BM, Meis S, Anderson A, Kellou M (2013) Comparison of phosphorus (P) removal properties of materials proposed for the control of sediment P release in UK lakes. Sci Total Environ 442:103–110
Tong Y, Zhang W, Wang X, Couture R-M, Larssen T, Zhao Y, Li J, Liang H, Liu X, Bu X, He W, Zhang Q, Lin Y (2017) Decline in Chinese lake phosphorus concentration accompanied by shift in sources since 2006. Nature Geosci 10:507–511
Tu L, Jarosch KA, Schneider T, Grosjean M (2019) Phosphorus fractions in sediments and their relevance for historical lake eutrophication in the Ponte Tresa basin (Lake Lugano, Switzerland) since 1959. Sci Total Environ 685:806–817
Wang CH, Qi Y, Pei YS (2012) Laboratory investigation of phosphorus immobilization in lake sediments using water treatment residuals. Chem Eng J 209:379–385
Wang Y, Ding SM, Wang D, Sun Q, Lin J, Shi L, Chen MS, Zhang CS (2017) Static layer: a key to immobilization of phosphorus in sediments amended with lanthanum modified bentonite (Phoslock®). Chem Eng J 325:49–58
Wang J, Chen J, Chen Q, Yang H, Zeng Y, Yu P, Jin Z (2019) Assessment on the effects of aluminum-modified clay in inactivating internal phosphorus in deep eutrophic reservoirs. Chemosphere 215:657–667
Wang J, Chen J, Yu P, Yang X, Zhang L, Geng Z, He K (2020) Oxygenation and synchronous control of nitrogen and phosphorus release at the sediment-water interface using oxygen nano-bubble modified material. Sci Total Environ 725:138258
Wei Q, Dong L, Jiao F, Qin W (2019) Use of citric acid and Fe(III) mixture as depressant in calcite flotation. Colloid Surface A 578:123579
Xiong C, Wang D, Tam NF, Dai Y, Zhang X, Tang X, Yang Y (2018) Enhancement of active thin-layer capping with natural zeolite to simultaneously inhibit nutrient and heavy metal release from sediments. Ecol Eng 119:64–72
Xu X, Wang B, Tang H, Jin Z, Mao Y, Huang T (2020) Removal of phosphate from wastewater by modified bentonite entrapped in Ca-alginate beads. J Environ Manage 260:110130
Yang C, Tong L, Liu XL, Tan Q, Liu H (2019) High-resolution imaging of phosphorus mobilization and iron redox cycling in sediments from Honghu Lake, China. J Soils Sediments 19:3856–3865
Yin HB, Kong M (2015) Reduction of sediment internal P-loading from eutrophic lakes using thermally modified calcium-rich attapulgite-based thin-layer cap. J Environ Manage 151:178–185
Yin H, Ren C, Li W (2018) Introducing hydrate aluminum into porous thermally-treated calcium-rich attapulgite to enhance its phosphorus sorption capacity for sediment internal loading management. Chem Eng J 348:704–712
Yin H, Yang P, Kong M, Li W (2020) Use of lanthanum/aluminum co-modified granulated attapulgite clay as a novel phosphorus (P) sorbent to immobilize P and stabilize surface sediment in shallow eutrophic lakes. Chem Eng J 385:123395
Yu X, Grace MR, Sun G, Zou Y (2018) Application of ferrihydrite and calcite as composite sediment capping materials in a eutrophic lake. J Soils Sediments 18:1185–1193
Yuan H, Tai Z, Li Q, Liu E (2020) In-situ, high-resolution evidence from water-sediment interface for significant role of iron bound phosphorus in eutrophic lake. Sci Total Environ 706:136040
Zhan Y, Yu Y, Lin J, Wu X, Wang Y, Zhao Y (2019) Simultaneous control of nitrogen and phosphorus release from sediments using iron-modified zeolite as capping and amendment materials. J Environ Manage 249:109369
Zhan Y, Yu Y, Lin J (2020) Impact of application mode on the control of phosphorus release from sediments using zirconium-modified bentonite as geo-engineering material. Sci Total Environ 712:135633
Zhang B, Chen N, Feng C, Zhang Z (2018) Adsorption for phosphate by crosslinked/non-crosslinked-chitosan-Fe(III) complex sorbents: characteristic and mechanism. Chem Eng J 353:361–372
Zhou J, Li D, Chen S, Xu Y, Geng X, Guo C, Huang Y (2019) Sedimentary phosphorus immobilization with the addition of amended calcium peroxide material. Chem Eng J 357:288–297
Zhou J, Li D, Zhao Z, Song X, Huang Y, Yang J (2020) Phosphorus immobilization by the surface sediments under the capping with new calcium peroxide material. J Clean Prod 247:119135
Zou Y, Grace MR, Roberts KL, Yu X (2017) Thin ferrihydrite sediment capping sequestrates phosphorus experiencing redox conditions in a shallow temperate lacustrine wetland. Chemosphere 185:673–680
Funding
This research was jointly supported by the Shandong Key Scientific and Technical Innovation Project (2018YFJH0902), the National Science Foundation of China (51408354 and 50908142), the Shanghai Natural Science Foundation (15ZR1420700), and the Scientific Research Project of Shanghai Science and Technology Committee (10230502900).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Shiming Ding
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bai, X., Lin, J., Zhang, Z. et al. Interception of sedimentary phosphorus release by iron-modified calcite capping. J Soils Sediments 21, 641–657 (2021). https://doi.org/10.1007/s11368-020-02754-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-020-02754-5